
Abstract
Muscle atrophy, a debilitating condition characterized by loss of muscle mass and strength, is a major concern in various clinical settings. Acetyl genistin (AG), a bioactive compound, was evaluated for its role in muscle cell differentiation and its potential protective effects against dexamethasone (dexa)-induced muscle atrophy. Our study demonstrated that AG significantly promoted C2C12 myotube differentiation, as evidenced by enhanced myotube width and increased fusion index. Notably, AG treatment upregulated the expression of myogenic markers, including MHC, MyoD, and MyoG. Moreover, AG displayed protective properties by attenuating dexa-induced muscle atrophy, mainly by suppressing the expression of the atrophy-related genes MAFbx and MuRF1. AG's protective effects are mechanistically attributed to its regulation of the AMPK/FoxO-dependent signaling pathway. Our results highlighted the dual benefits of AG in fostering muscle differentiation and safeguarding against muscle atrophy, positioning it as a promising agent for muscle health and therapeutic applications.
Similar content being viewed by others


Introduction
Skeletal muscle atrophy, characterized by a progressive decline in muscle mass and strength, is a multifactorial condition with diverse etiologies [1,2,3]. Aging, termed sarcopenia when related to muscle decline, is a natural and inevitable process that results in muscle atrophy, compromised physical functionality, and increased vulnerability to falls and fractures [4, 5]. Interestingly, the molecular hallmarks underlying age-related muscle loss bear a striking resemblance to those observed in glucocorticoid-induced muscle atrophy [6].
Dexamethasone (dexa), a synthetic glucocorticoid, has been widely used to model muscle atrophy in various experimental settings [3, 6, 7]. The mechanisms driving dexa-induced atrophy, particularly the upregulation of muscle-specific ubiquitin ligases such as MAFbx and MuRF1, and the subsequent proteasomal degradation of myofibrillar proteins, mirror the catabolic pathways activated in aging muscles [8,9,10,11]. Additionally, dysregulation of the AMPK/FoxO signaling cascade, which is central to muscle protein homeostasis, is commonly observed in both dexa-treated and aged muscles [12, 13].
Recent studies have elucidated the broad spectrum of Genistein's biological effects, including its antioxidant and anti-inflammatory properties, as well as its role in angiogenesis and estrogenic effects. These mechanisms contribute to Genistein's potential therapeutic applications in cardiovascular protection and cancer prevention, particularly breast cancer, through its structural mimicry of estrogen, which allows it to modulate estrogen receptor activity [14].
Given this molecular congruence, dexa-induced muscle atrophy serves as a relevant surrogate for sarcopenia, allowing researchers to dissect intertwined molecular cascades in a controlled setting. Genistin, a notable isoflavone derivative, has recently gained significant scientific interest because of its myriad health benefits underpinned by its intricate metabolic pathways and impactful interactions with biological systems [15,16,17]. Emerging evidence, including associations between soy food consumption rich in isoflavones, such as acetyl genistin (AG), and decreased bone fracture risk in postmenopausal women, highlights the potential therapeutic applications of AG, particularly in conditions such as osteoporosis [18]. Although its impact in various biological contexts has been reported, the influence of AG on muscle differentiation and its protective role against muscle atrophy, especially in dexa-induced and aging models, are in the nascent stages of understanding. This study aimed to elucidate the modulatory effects of AG on C2C12 myotube differentiation and its potential to ameliorate dexa-induced muscle atrophy, thereby offering insights into its therapeutic potential for sarcopenia and related muscle disorders.
Materials and methods
Cell culture and maintenance
The mouse myoblast cell line, C2C12, was obtained from the American Type Culture Collection (Rockville, MD, USA). C2C12 mouse myoblasts were cultured in growth medium (GM) consisting of Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, CA, USA) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and 1% penicillin–streptomycin (Thermo Fisher Scientific). Cells were maintained in a humidified incubator at 37 °C with 5% CO2. The medium was changed every 2 days, and cells were subcultured upon reaching 80–90% confluency.
Cell viability analysis
C2C12 cells were seeded at a density of 5 × 104 cells/well in 24-well plates. Following cell attachment, the medium was treated with varying concentrations of AG (Nacalai Tesque Inc. Japan, 0, 1, 5, 10, 20, 40 μM) and incubated for 24 h at 37 °C in a humidified atmosphere with 5% CO2. After incubation, cell viability was assessed using Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan). Briefly, 10 μL of CCK-8 solution was added to each well, and the plates were further incubated at 37 °C for 1 h. The absorbance of the resulting formazan dye, which indicates cellular dehydrogenase activity, was measured at 450 nm using a microplate reader (Molecular Devices, San Jose, CA, USA). Percentage of Cell Viability = (Absorbance of the treated group/Absorbance of the control group) × 100.
Induction of myogenic differentiation and dexa-induced atrophy
Upon reaching near confluence (~ 80–90%), the growth medium (GM) of the C2C12 myoblasts was replaced with differentiation medium (DM) to initiate myogenic differentiation. The differentiation medium consisted of DMEM supplemented with 2% horse serum (Thermo Fisher Scientific) and 1% penicillin–streptomycin (Thermo Fisher Scientific). Cells were maintained in DM for up to 6 days, and the medium was changed every 2 days. During this period, the myoblasts fuse to form multinucleated myotubes.
For the induction of muscle atrophy, mature myotubes were treated with dexa at a concentration of 1 μM for 48 h. This treatment stimulates protein degradation pathways that mimic muscle atrophy [9]. After treatment, the cells were analyzed for morphological changes and markers of muscle atrophy, such as MuRF1 and MAFbx, were assessed using western blotting or quantitative reverse transcription polymerase chain reaction (qRT-PCR) to validate the induction of the atrophic state.
May–Grunwald and Giemsa staining
Differentiated myotube cells were washed with phosphate buffered saline (PBS) and fixed in 100% methanol. Cells were stained for 5 min using a 1:3 diluted May–Grunwald solution in sodium phosphate buffer (comprising 1 mM NaH2PO4·H2O and 1 mM Na2HPO4, pH 6.0). After a brief rinse with distilled water, the cells were stained with a 1:10 diluted Giemsa solution for 10 min. After staining, cells were washed, visualized, and photographed at 100 × magnification. Myotube widths were measured from randomly chosen sections using ImageJ software.
Fusion index calculation
To quantify the myogenic differentiation efficiency, random fields of view from May–Grunwald- and Giemsa-stained images were captured. The number of nuclei within the myotubes and total number of nuclei were counted. The fusion index was calculated as follows:
Western blot analysis
Cells were washed with PBS and lysed using radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with protease inhibitors (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After ultrasonication and centrifugation at 13,000 × g for 15 min at 4 °C, the protein concentration was determined using the Bicinchoninic Acid Assay (BCA) Protein Assay Kit (Pierce, Rockford, IL, USA). Proteins were electrophoresed on a 10% sodium dodecyl sulfate (SDS) gel and transferred to a nitrocellulose membrane. After blocking, the membrane was incubated with primary antibodies, including MuRF-1 (Santa Cruz Biotechnology), MAFbx (Santa Cruz Biotechnology), FoxO1 (Cell Signaling Technology, Beverly, MA, USA), FoxO3 (Cell Signaling Technology), AMPK (Cell Signaling Technology) and β-actin (Sigma-Adrich). After overnight incubation, the membrane was washed and incubated with a peroxidase-conjugated secondary antibody. Protein bands were visualized using Clarity Western ECL Substrate (Bio-Rad Laboratories, Hercules, CA, USA) and images were captured using the ChemiDocTM Touch Imaging System (Bio-Rad Laboratories).
qRT-PCR
C2C12 myoblasts were washed twice with cold PBS, and total RNA was isolated using the TRIzol reagent. The extracted RNA (2 μg) was reverse-transcribed into cDNA using an iScript cDNA Synthesis Kit (Bio-Rad Laboratories). mRNA expression was assessed using the ViiA™7 Real-Time PCR System (Applied Biosystems, Waltham, MA, USA) with TaqMan analysis. Amplification settings were: an initial denaturation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s. Primers for MuRF1 (Mm01185221 m1), MAFbx (Mm00499523 m1), MyoD (Mm00440387 m1), MyoG (Mm00446194 m1), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Mm99999915 g1) were used.
Statistical analysis
Statistical evaluations were conducted using the GraphPad Prism software (version 5.01; GraphPad Software, San Diego, CA, USA). Experiments were performed thrice, with results presented as mean ± standard deviation (SD). The significance between two groups was assessed using Student’s t-test, with statistical significance set at *p < 0.05.
Results
Acetyl genistin facilitates C2C12 myotube differentiation
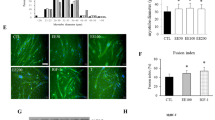
To elucidate the potential effects of AG on myoblast differentiation and its molecular attributes, we designed and executed a comprehensive experimental approach involving molecular characterization, viability assays, morphological assessments, and fusion efficiency evaluations. The molecular architecture of AG was delineated, highlighting its unique chemical configuration (Fig. 1a). Viability assays of C2C12 cells across a gradient of AG concentrations revealed robust cell survival, with an upward trend in viability discernible at elevated AG concentrations (Fig. 1b). Morphological assessments over a temporal gradient post-AG exposure revealed enhanced myotube differentiation, particularly pronounced on day six (Fig. 1c), an effect that persisted even in the presence of dexa, a known myogenesis antagonist. Quantification of myotube width substantiated the potentiation of myotube differentiation by AG, with evidently augmented widths relative to the controls (Fig. 1d). Concurrently, the Fusion Index, a quantitative metric gauging myoblast fusion efficiency, showed an enhanced fusion profile in AG-treated cohorts, suggesting superior nuclear integration within emergent myotubes compared to that in controls (Fig. 1e). Collectively, these findings underscore the potent role of AG in augmenting C2C12 myotube differentiation, a phenomenon that is robust against dexa-mediated adverse effects.

Acetyl genistin facilitates C2C12 myotube differentiation. a Chemical structure of acetyl genistin. b C2C12 cells were cultured in a medium containing different concentrations (0, 1, 5, 10, 20, and 40 μM) of acetyl genistin for 24 h. Cell viability was measured using a Cell Counting Kit-8 (CCK-8) assay. The data is shown as a percentage of viability relative to the control, and the bars in the graph indicate the mean ± standard deviation (SD) (n = 4 per group). c The cells were stained using the May–Grunwald and Giemsa staining methods on day 2, day 4, and day 6. They were incubated with 40 μM acetyl genistin in the presence or absence of 1 μM dexamethasone, according to the respective time points. d Measurement of myotube widths based on images stained with May–Grunwald and Giemsa. The presented data represent the mean ± SD (n = 100). *** p < 0.001 vs. CTL. e Fusion Index. We counted the total number of nuclei incorporated into myotubes and the overall number of nuclei. The fusion index was then determined as the percentage of total nuclei incorporated into myotubes. * p < 0.05 vs. CTL. CTL control, AG acetyl genistin
AG modulates the expression of key myogenic markers
To investigate the pro-myogenic effects of AG, we focused on the canonical myogenic markers MHC, MyoD, and MyoG. Historically significant in skeletal muscle biology, MyoD signifies early commitment to the myogenic lineage [19], MyoG governs the terminal differentiation of myoblasts [20], and MHC confirms mature myotube formation [21]. Examination of the modulatory effects of AG on crucial myogenic markers revealed distinct transcriptional and translational alterations. Sequential evaluations on days 2, 4, and 6 post-treatment showed robust upregulation of MyoD and MyoG gene expression in AG-treated cells relative to controls, with GAPDH serving as a normalization control. In the case of MyoD, the AG treatment group showed a 1.3-fold increase on day 2, a 0.8-fold increase on day 4, and a 0.6-fold increase on day 6 compared to the control group. Similarly, MyoG expression also tended to increase with AG treatment, specifically by 1.2-fold on day 2, 0.7-fold on day 4, and 0.9-fold on day 6 compared to the control group (Fig. 2a). Concordantly, western blot analysis corroborated these findings at the protein level, demonstrating enhanced expression of MHC, MyoD, and MyoG in C2C12 myotubes treated with AG, with β-actin used to ensure equal loading. Specifically, MHC expression in AG-treated cells showed increases of 1.1-fold on day 2, 2.3-fold on day 4, and 3.5-fold on day 6, compared to controls. For MyoD, expression increased to 1.2-fold on day 2, 0.4-fold on day 4, and 0.4-fold on day 6 post-AG treatment. Similarly, MyoG expression was enhanced to 1.4-fold on day 2, 0.7-fold on day 4, and 0.8-fold on day 6 (Fig. 2b). These results underscore AG's potent role in augmenting the expression of key myogenic markers—MHC, MyoD, and MyoG—at both the mRNA and protein levels, further supporting its positive impact on muscle differentiation and growth.

Acetyl genistin enhances the expression of MHC, MyoD and MyoG. a Quantitative evaluation of myogenic marker genes (MyoD and MyoG) over 3 time points (day 2, day 4, and day 6) in cells exposed to either the control or acetyl genistin treatment. Glyceraldehyde 3-phosphate dehydrogenase was used as a control. Data are shown as mean ± standard deviation (SD) (n = 4 per group). ** p < 0.01 and *** p < 0.001 vs. CTL. b The expression of MHC, MyoD and MyoG protein in C2C12 myotubes was estimated by western blot analysis. β-actin was used as a control for protein loading. Data are shown as mean ± SD. (n = 3 per group). * p < 0.05, ** p < 0.01, and *** p < 0.001 vs. CTL. CTL control, AG acetyl genistin
AG mitigates dexa-triggered muscle atrophy via downregulation of MAFbx and MuRF1
MAFbx and MuRF1 are the key E3 ubiquitin ligases involved in skeletal muscle protein degradation. Their elevation is linked to muscle atrophy, notably from glucocorticoids, such as dexa [8, 9]. Assessing their modulation offers insight into potential therapies for muscle wasting. As shown in Fig. 3, meticulous analyses highlighted the protective effects of AG against the deleterious effects of dexa on skeletal muscle cells. Histological examinations using the May–Grunwald and Giemsa staining revealed the profound muscle-preserving effect of AG. Specifically, while dexamethasone exposure precipitated discernible muscle atrophy, concomitant treatment with AG remarkably curbed this atrophic response, as evidenced by the denser myotube structures (Fig. 3a). Molecular investigations provided mechanistic insights by corroborating these morphological observations. Quantitative PCR revealed pronounced upregulation of MAFbx and MuRF1 mRNA levels upon dexamethasone exposure, both of which are essential markers of muscle degradation. Interestingly, AG treatment significantly attenuated the expression of these atrophy-related genes, suggesting its direct involvement in curbing muscle protein degradation (Fig. 3b). Western blot analysis confirmed these observations. A stark increase in the protein levels of MAFbx and MuRF1 was observed in dexamethasone-treated cells. However, the co-administration of AG markedly suppressed the expression of these catabolic markers, reflecting its potential to counteract dexamethasone-induced protein degradation (Fig. 3c). AG has emerged as a promising therapeutic agent with potent muscle-sparing effects, particularly against glucocorticoid-induced muscle atrophy, by modulating the expression of pivotal muscle degradation markers.

AG attenuates dexamethasone-induced muscle atrophy in C2C12 myotubes by suppressing MAFbx and MuRF1 expression. a May–Grunwald and Giemsa staining. The cells were incubated with 40 μM acetyl genistin for 48 h in the presence or absence of 1 μM dexamethasone for 48 h. b MAFbx and MuRF1 mRNA levels were analyzed by quantitative polymerase chain reaction. Glyceraldehyde 3-phosphate dehydrogenase was used as a control. Data are shown as mean ± standard deviation (SD) (n = 4 per group). ** p < 0.01 and *** p < 0.001 vs. CTL; ## p < 0.01 vs. Dexa. c The expression of MAFbx and MuRF1 protein in C2C12 myotubes was estimated by western blot analysis. β-actin was used as a control for protein loading. Data are shown as mean ± SD. (n = 3 per group). *** p < 0.001 vs. CTL, ## p < 0.01 vs. Dexa. CTL control, Dexa dexamethasone, AG acetyl genistin, DAG dexamethasone + acetyl genistin
AG counters dexa-mediated atrophy through the modulation of AMPK/FoxO signaling
AMP-activated protein kinase (AMPK) and forkhead box O (FoxO) signaling pathways are central to muscle metabolism and atrophy. AMPK, known as the “master regulator” of cellular energy, is crucial for muscle energy balance, while the FoxO transcription factors, especially FoxO1 and FoxO3, govern genes associated with muscle atrophy under stress conditions [9, 12, 22]. In Fig. 4, we discerned the intricate molecular underpinnings of the action of AG against dexa-induced skeletal muscle atrophy. Western blotting revealed that dexa exposure curtailed the activation of AMPK while concurrently upregulating the expression of FoxO1 and FoxO3 transcription factors, key orchestrators of muscle catabolism. Strikingly, AG administration not only restored AMPK activity but also attenuated the elevated levels of both FoxO1 and FoxO3 (Fig. 4a). The accompanying quantification further substantiated these observations, underscoring the significant inhibitory effects of AG on the atrophic effects of dexa via the AMPK/FoxO axis. Collectively, these findings suggest that AG is a potent modulator of muscle metabolism, acting primarily through the AMPK/FoxO-dependent pathway to combat glucocorticoid-induced muscle wasting.

Acetyl genistin inhibits dexamethasone-induced atrophy in C2C12 myotube via AMPK/FoxO-dependent signaling. a Representative images of the western blot analyses for AMPK, FoxO1, and FoxO3 in C2C12 myotubes. b Quantification of the indicated proteins. β-actin was used as a control for protein loading. Data shown are mean ± standard deviation (SD) (n = 3 per group). * p < 0.05, ** p < 0.01, and *** p < 0.001 vs. CTL; ## p < 0.01 and ### p < 0.001 vs. Dexa. CTL control, Dexa dexamethasone, AG acetyl genistin, DAG dexamethasone + acetyl genistin
In this study, the therapeutic potential of AG on muscle differentiation and its protective efficacy against muscle atrophy were elucidated. Upon exposure to AG, enhanced myoblast differentiation was observed, as indicated by the upregulation of key markers, such as MHC, MyoD, and MyoG. Furthermore, in the presence of dexa, a known inducer of muscle atrophy, AG demonstrated protective effects, mainly through modulation of the AMPK/FoxO signaling pathway and suppression of the atrogenes MAFbx and MuRF1. The presented schematic provides comprehensive insight into the molecular mechanisms underlying these effects (Fig. 5).

The overall acetyl genistin mechanisms related to dexamethasone-induced muscle atrophy. Acetyl genistin exerts its effects on muscle physiology. Acetyl genistin, a potent compound, plays a pivotal role in promoting myoblast differentiation into myotubes by increasing the expression of key markers such as MHC, MyoD, and MyoG. Acetyl genistin encourages myoblast differentiation into myotubes and acts as a natural defense against dexamethasone-induced muscle atrophy. This defense involves the inherent ability of acetyl genistin to regulate AMPK, a pivotal upstream regulator, leading to a subsequent reduction in FoxO1/3 activity and the consequent suppression of MAFbx expression. This dual functionality highlights the potential of acetyl genistin in both facilitating muscle differentiation and protecting against muscle atrophy, contributing to overall muscle health and function. Figure created using Biorender (https://biorender.com/)
Discussion
Muscle atrophy, either due to natural processes such as aging or pharmacological induction such as dexa, has been the subject of extensive research in recent years [1,2,3,4,5,6,7]. Age-associated muscle atrophy, termed sarcopenia, has direct implications on physical functionality and poses increased susceptibility to falls and fractures [4, 5]. Dexamethasone, a synthetic glucocorticoid, serves as a model for inducing muscle atrophy in experimental settings, providing significant insights into the molecular pathways akin to aging muscles [3, 6, 7].
Genistin and genistein, the predominant isoflavones found in soy, exhibit distinct structural and bioactive properties. Genistein, the aglycone form, is directly involved in its biological activities, which are attributed to the absence of a sugar moiety. Conversely, genistin, a glycoside, possesses a sugar molecule that must be removed by intestinal bacteria for conversion to genistein before exerting similar effects. This conversion process underlies the bioavailability and absorption differences observed between these two compounds [23, 24]. The therapeutic potential of isoflavones and their derivatives, especially genistin and its derivative, AG, has gained increasing scientific attention. Not only are they associated with a reduced risk of bone fractures in postmenopausal women, but there is also burgeoning evidence hinting at their role in mitigating oxidative stress and inflammatory responses [15,16,17,18]. The antioxidant properties of genistein have been reported to counteract the effects of oxidative stress, which is a crucial factor in muscle degeneration [14]. Given the close interplay between muscle and bone health, the role of genistin in promoting bone health also resonates with its potential role in muscle differentiation, given the close interplay between muscle and bone health [18, 25].
The present study investigated the influence of AG on muscle health, more specifically on C2C12 myotube differentiation, and its protective effects against dexa-induced muscle atrophy. The observed augmentation of myotube differentiation and modulation of myogenic markers upon AG treatment is consistent with the findings of Liu et al., wherein isoflavones enhanced myogenesis and muscle regeneration in mice [26]. MyoD, the master regulator of myogenic commitment, and MyoG, essential for terminal differentiation, are critical for muscle fiber formation. MHC is indicative of mature myotube formation. AG may enhance MyoD and MyoG expression via the Wnt/β-catenin pathway, known for its role in myoblast commitment, and the PI3K/Akt pathway, crucial for MyoG activity during myotube formation [27,28,29]. AG’s potential suppression of myostatin could upregulate myogenic factors, promoting myotube formation [29]. These pathways highlight AG’s therapeutic promise for muscle regeneration and atrophy conditions, suggesting a need for further investigation into its molecular interactions within myogenic machinery.
Our findings elucidate AG’s role in mitigating dexamethasone-induced muscle degradation, highlighting its interference with the ubiquitin–proteasome system (UPS), a key player in protein catabolism within muscle cells. Dexamethasone activates E3 ubiquitin ligases, such as MAFbx/atrogin-1 and MuRF1, marking proteins for degradation and leading to muscle atrophy [30]. AG’s modulation of these ligases suggests its potential to disrupt dexamethasone signaling. Similarly, the dampening of the muscle-specific ubiquitin ligases MAFbx and MuRF1 by AG resonated with the findings of Bodine and Baehr, emphasizing the pivotal role of these ligases in muscle atrophy [31]. Our study expands on these findings by demonstrating that AG treatment can mitigate the deleterious effects of dexamethasone on muscle cells. AG's protective actions suggest an interaction with the UPS, downregulating the expression of key muscle-specific E3 ubiquitin ligases, MAFbx and MuRF1, which are typically upregulated by dexamethasone and associated with increased protein degradation.
The modulatory effect of AG on the AMPK/FoxO signaling pathway, which was elucidated in our study, adds another layer to its therapeutic potential. The AMPK pathway, previously highlighted by Hardie and Mammucari et al., plays a central role in maintaining cellular energy homeostasis and muscle protein balance [22, 32]. Mechanistic insights into the activation of atrogin-1 and MuRF1 by dexamethasone highlight the involvement of the Akt/FOXO1 pathway. Dexamethasone impairs Akt signaling, leading to the activation of FOXO1, which in turn upregulates atrogin-1 and MuRF1, promoting protein degradation and muscle atrophy [33]. AG’s effect on the AMPK/FoxO signaling axis, crucial for cellular energy homeostasis and the regulation of catabolic genes, underscores its therapeutic potential against muscle wasting. This complex interplay between dexamethasone, the UPS, and AMPK/FoxO pathways, revealed by our study, opens new avenues for targeted interventions in muscle atrophy.
In conclusion, although our study sheds light on the promising role of AG in muscle differentiation and its protective efficacy against muscle atrophy, it is essential to situate these findings in the broader context of the existing literature. Our results, juxtaposed with those of prior research, underscore the multifaceted benefits of AG, ranging from bone health to muscle differentiation and protection against atrophy. However, as with any scientific investigation, more exhaustive and comprehensive studies are warranted to fully understand the spectrum of therapeutic actions of AG.
Availability of data and materials
All data is available in the main text.
Abbreviations
- AG:
-
Acetyl genistin
- Dexa:
-
Dexamethasone
- GM:
-
Growth medium
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FBS:
-
Fetal bovine serum
- CCK-8:
-
Cell Counting Kit-8
- DM:
-
Differentiation medium
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
- PBS:
-
Phosphate buffered saline
- RIPA:
-
Radioimmunoprecipitation assay
- BCA:
-
Bicinchoninic acid assay
- SDS:
-
Sodium dodecyl sulfate
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- SD:
-
Standard deviation
References
Ryall JG, Schertzer JD, Lynch GS (2008) Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology 9:213–228
Narici MV, Maffulli N (2010) Sarcopenia: characteristics, mechanisms and functional significance. Br Med Bull 95:139–159
Bonaldo P, Sandri M (2013) Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6:25–39
Cruz-Jentoft AJ, Bahat G, Bauer J et al (2019) Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing 48:16–31
Frontera WR, Ochala J (2015) Skeletal muscle: a brief review of structure and function. Calcif Tissue Int 9:183–195
Schakman O, Gilson H, Thissen JP (2008) Mechanisms of glucocorticoid-induced myopathy. J Endocrinol 197:1–10
Menconi M, Gonnella P, Petkova V, Lecker S, Hasselgren PO (2008) Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J Cell Biochem 105:353–364
Bodine SC, Latres E, Baumhueter S et al (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294:1704–1708
Sandri M, Sandri C, Gilbert A et al (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117:399–412
Kandarian SC, Jackman RW (2006) Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 33:155–165
Lecker SH, Jagoe RT, Gilbert A et al (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18:39–51
Greer EL, Oskoui PR, Banko MR et al (2007) The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem 282:30107–30119
Mammucari C, Milan G, Romanello V et al (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6:458–471
Sharifi-Rad J, Quispe C, Imran M et al (2021) Genistein: an integrative overview of its mode of action, pharmacological properties, and health benefits. Oxid Med Cell Longev 2021:3268136
Bhat SS, Prasad SK, Shivamallu C et al (2021) Genistein: a potent anti-breast cancer agent. Curr Issues Mol Biol. https://doi.org/10.3390/cimb43030106
Islam A, Islam MS, Uddin MN, Hasan MMI, Akanda MR (2020) The potential health benefits of the isoflavone glycoside genistin. Arch Pharm Res 43:395–408
Hwang ST, Yang MH, Baek SH, Um JY, Ahn KS (2020) Genistin attenuates cellular growth and promotes apoptotic cell death breast cancer cells through modulation of ERalpha signaling pathway. Life Sci 263:118594
Zhang X, Shu XO, Li H et al (2005) Prospective cohort study of soy food consumption and risk of bone fracture among postmenopausal women. Arch Intern Med 165:1890–1895
Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R (1993) MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75:1351–1359
Hasty P, Bradley A, Morris JH et al (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364:501–506
Schiaffino S, Reggiani C (2011) Fiber types in mammalian skeletal muscles. Physiol Rev 91:1447–1531
Hardie DG (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8:774–785
Xu X, Harris KS, Wang HJ, Murphy PA, Hendrich S (1995) Bioavailability of soybean isoflavones depends upon gut microflora in women. J Nutr 125:2307–2315
Setchell KD, Brown NM, Desai P et al (2001) Bioavailability of pure isoflavones in healthy humans and analysis of commercial soy isoflavone supplements. J Nutr 131:1362S-1375S
Laurent MR, Dubois V, Claessens F et al (2016) Muscle-bone interactions: from experimental models to the clinic? A critical update. Mol Cell Endocrinol 432:14–36
Zhang Z, Jin F, Lian X et al (2018) Genistein promotes ionizing radiation-induced cell death by reducing cytoplasmic Bcl-xL levels in non-small cell lung cancer. Sci Rep 8:328
Zammit PS (2017) Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol 72:19–32
Matheny RW Jr, Geddis AV, Abdalla MN et al (2018) AKT2 is the predominant AKT isoform expressed in human skeletal muscle. Physiol Rep 6:e13652
Wang G, Zhu H, Situ C et al (2018) p110α of PI3K is necessary and sufficient for quiescence exit in adult muscle satellite cells. EMBO J 37:e98239
Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL (2004) IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab 287:E591–E601
Bodine SC, Baehr LM (2014) Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab 307:E469-484
Mammucari C, Gherardi G, Zamparo I et al (2015) The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep 10:1269–1279
Yamamoto D, Ikeshita N, Matsubara T et al (2008) GHRP-2, a GHS-R agonist, directly acts on myocytes to attenuate the dexamethasone-induced expressions of muscle-specific ubiquitin ligases, Atrogin-1 and MuRF1. Life Sci 82:460–466
Acknowledgements
Not applicable.
Funding
This study was conducted with support from the Rural Development Administration (Project No. PJ0141550402023), Republic of Korea.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Y-SH, SGK. Performed the experiments: Y-IC, S-JL. Analyzed the data: WMJ, SJK, JYH, DYL, DKJ, S-JL, Y-SH. Contributed reagents/materials/analysis tools: S-JK, SGK. Wrote the paper: WMJ, S-JK, JYH, Y-SH, SGK. All authors participated in manuscript preparation, read, and approved the final version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jeong, W.M., Kwag, SJ., Ha, J.Y. et al. Acetyl genistin modulates myotube differentiation and attenuates dexamethasone-induced muscle atrophy through the FoxO1/3 signaling pathway in C2C12 myotubes. Appl Biol Chem 67, 31 (2024). https://doi.org/10.1186/s13765-024-00885-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-024-00885-8