
Abstract
Veterinary antibiotics (VAs) administered to livestock are introduced into the soil through livestock manure and compost. These antibiotics can run off to surface water or leach into groundwater during rainfall, causing problems such as antibiotic contamination or the occurrence of antibiotic-resistant bacteria. In this study, an analytical method for detecting four classes of VAs (penicillin, tetracyclines, macrolides, and sulfonamides) in river water and soil was developed, and the occurrence of residual antibiotics in the agricultural environment was monitored. Soil samples were extracted with the McIlvain buffer solution and pretreatment was conducted using solid-phase extraction, followed by liquid chromatography-tandem mass spectrometry to quantify target VAs. The results of this study showed that the recovery ranged from 62 to 121% in river water and 40.2–149.3% in soil. Among the other VAs, amoxicillin and spiramycin were observed to have low recoveries in all the samples. The method detection limit (MDL) was calculated in the range of 2.1–12.3 ng L−1 in river water and 1.2–13.2 ng kg−1 in soil, and the limit of quantification was 6.6–39.2 ng L−1 and 4.0–42.0 ng kg−1, respectively. This optimal method was then applied to measure the residual concentrations of VAs in river water, sediment, and soil samples around the Muhan watershed in Korea. A total of seven antibiotics were detected, and their concentrations ranged from 0.014 to 0.309 μg L−1in river water, and 1.45–9.04 μg kg−1 in sediment and arable soil. This method can be used to screen VAs in river water and soil and is expected to be used as primary data for examining the occurrence and fate of antibiotics in agricultural environments.
Similar content being viewed by others

Introduction
Veterinary antibiotics (VAs), which are widely consumed to treat and prevent bacterial infections and diseases, have played an essential role in livestock health [1]. However, these pharmaceuticals are regarded as new emerging contaminants due to their intensive use resulting in production of antibiotic resistance in the environment [2]. Moreover, the scale of the livestock industry has increased over the past few decades, owing to global population growth and increased demand for livestock products.
Antibiotics administered to livestock are not completely metabolized in the body and are excreted in feces in the form of parent compounds [3]. Un-metabolized forms of VAs remaining in manure, compost, and liquid fertilizer can be introduced into soil [4], surface water, or be leached into groundwater via rainfall [5, 6]. In addition, released VAs in the environment can be transferred and accumulated to crops or cause toxic effects, such as growth inhibition [7,8,9], reduction of soil bacterial activity or diversity [10, 11]. The most important adverse effect of VAs released into the environment is that they may cause the production of antibiotic resistance genes (ARGs) and eventually threaten human health [12, 13].
Faced with these ecological concerns, many researchers have become interested in the occurrence, fate, and distribution of residual VAs in agricultural environments. The United States Environmental Protection Agency (US EPA) developed and published Method 1694 (Pharmaceuticals and Personal Care Products in Water, Soil, Sediment, and Biosolids by HPLC/MS/MS) to determine antibiotics in the environment [14]. Many European countries developed analytical methods for identifying the presence of antibiotics in aqueous environments such as river water, wastewater, and groundwater [15]. These efforts of antibiotic management in the environment are also carried out actively in Asia countries [16, 17].
In Korea, many studies have focused on the development and distribution of residual antibiotics in agricultural and livestock products; however, there is still a lack of interest in and research on environmental media. Currently, standard analytical methods and maximum residue limits (MRLs) for VAs in agricultural, marine, and livestock products are established and managed under the supervision of the Ministry of Food and Drug Safety, and monitoring of residual antibiotics in foods has been actively conducted in various regions and institutions in Korea [18, 19]. In contrast, there are currently no standard analytical methods for antibiotics in environmental media, such as river water and soil. In addition, research on the simultaneous analysis of multiple classes of VAs was lacking.
Because environmental media are the primary sink of VAs and the main transmission route for antibiotic resistance [20], the development and monitoring of VAs is important. Liquid chromatography-tandem mass spectrometry (LC–MS/MS) is often used to analyze VAs in various samples because of its high analytical performance and low limit of detection (LOD). However, environmental samples, such as river water and soil, consisting of a very complex matrix, may be affected by various coexisting substances present in the sample during instrument analysis. These problems can reduce the sensitivity of qualitative and quantitative analyses. In addition, due to differences in the physicochemical properties of each antibiotic group, it is challenging to determine various antibiotics simultaneously through single sample preparation and instrumental analysis.
Therefore, this study aimed to develop an optimal analytical method for quantifying different classes of VAs in river water and soil. In addition, the developed analytical method was applied to measure the residual VAs in surface water and soil adjacent to confined animal feeding operations. The target analytes for this research include 12 antibiotics belonging to four different groups, penicillins, tetracyclines, macrolides, and sulfonamides, contributing to approximately 60% of the total amount of VA sales in South Korea. Although these compounds reported that they have the potential hazard of the spread of ARGs in the agricultural environment due to their higher usage than other VA classes in Korea, less research has been conducted on monitoring residual VAs in agricultural environments [21,22,23]. The results of this study can be used to monitor the residual concentrations of VAs in different environmental media and to understand the occurrence of VAs in arable areas.
Materials and methods
Chemicals and reagents
Twelve reference standards (amoxicillin, ampicillin, penicillin G sodium salt, chlortetracycline hydrochloride, oxytetracycline hydrochloride, tetracycline hydrochloride, spiramycin, tylosin tartrate, sulfadiazine, sulfamethazine, sulfamethoxazole, and sulfathiazole) were purchased from Sigma-Aldrich (St. Louis, MO, USA), and their physicochemical properties are shown in Additional file 1: Table S1. The internal standards (simeton) were obtained from AccuStandard (New Haven, CT, USA). All the organic solvents (methanol and acetonitrile) and water used for sample pretreatment and instrumental analysis were of HPLC grade (J. T. Baker, Philipsburg, NJ, USA). Sodium phosphate, citric acid, formic acid, and ethylenediaminetetraacetic acid disodium salt dihydrate (Na2-EDTA) were acquired from Sigma-Aldrich. Stock solutions (100 mg L−1) were prepared by accurately weighing each antibiotic standard in a 100 ml volumetric flask, dissolved in methanol, and stored in an amber glass bottle at − 20 °C. The working solutions were prepared by mixing and diluting each stock solution appropriately in HPLC-grade water at concentrations varying from 0.01 to 1.0 mg L−1 and were stored in an amber glass bottle at − 20 °C until analysis.
Sample preparation
River water samples were filtered through a 0.2 μm cellulose acetate membrane filter and stored at 4 °C until analysis. For antibiotic extraction in water samples, 120 mL of the filtered sample was taken into a 250 mL Erlenmeyer flask and the pH was adjusted to 2.5 ± 0.1 using 40% (v/v) sulfuric acid. Then, 500 μL of 5% (w/v) Na2-EDTA was added and the samples were shaken at 150 rpm for 15 min at room temperature (25 ℃), followed by purification by solid phase extraction (SPE). The collected soil samples were air-dried under dark conditions and sieved through a 2.0 mm mesh. For antibiotic extraction, each soil sample of 1.0 g was weighed in a centrifuge tube, and 20 mL of McIlvain buffer solution (pH 4.0) and 250 μL of 5% Na2-EDTA were added. This mixture was shaken for 15 min and centrifuged for 15 min at 4000 rpm, and the supernatant was transferred into a 250 mL Erlenmeyer flask. Soil samples were extracted twice (40 mL), to which 80 ml of ultrapure water was added. The extracted samples were filtered with a 0.2 μm cellulose acetate membrane filter and purified by SPE.
The SPE was conducted using a Visiprep SPE vacuum manifold (Supelco, Bellefonte, PA, USA). Before loading the samples, the SPE cartridge was activated by sequentially flowing methanol (0.5 M hydrochloric acid (3.0 mL) and ultrapure water (3.0 mL)) through the cartridge. The extracted samples were loaded into a Teflon tube at a flow rate of 4 ml min−1. When all the samples passed through the cartridge, the cartridge was washed with flowing ultrapure water (3.0 ml of ultrapure water in three divided portions). Further, a 15 ml glass centrifuge tube containing 50 μL of simeton (0.24 mg L−1), an internal standard, was mounted on the SPE vacuum manifold, and then 2.5 mL of methanol was passed to the cartridge twice (5.0 mL in total) to extract antibiotics. The eluate was concentrated to 50 µL at 40 °C using a nitrogen concentrator (12-position N-EVAP nitrogen evaporation system, Organomation, MA, USA). Then, 70 μL of mobile phase A was added and centrifuged using a 1.5 mL centrifuge tube containing a 0.22 μm nylon filter at 13,000 rpm for 3 min. Finally, the extract was transferred to an amber glass HPLC vial and stored at − 20 °C until analysis. Sample preparation diagram is shown in Fig. 1.

Flow diagram of analytical method for veterinary antibiotics in river water and soil
LC–MS/MS analysis
The instrument analysis for the detection and quantification of residual antibiotics was performed using HPLC (1290 Infinity II, Agilent, Santa Clara, CA, USA) coupled with triple quadrupole mass spectrometry (6500 Qtrap, SCIEX, Framingham, MA, USA) equipped with an electrospray ionization probe. All target antibiotics were analyzed using multiple reaction monitoring (MRM) in the positive ion mode. Target analytes were separated using a Zorbax Eclipse Plus-C18 column (Agilent, 4.6 × 150 mm, 3.5 μm) protected by a security guard cartridge (Phenomenex, Torrance, CA, USA) at 25 °C. The gradient elution system was set with mobile phase A (0.1% formic acid in HPLC-grade water) and mobile phase B (0.1% formic acid in acetonitrile) at a flow rate of 0.7 ml min−1, and the total runtime was set to 15 min. The detailed HPLC and mass spectrometry conditions are presented in Additional file 1: Table S2. The analyzed MRM data were processed using the Analyst software version 1.5.1 and MultiQuant™ software version 3.0.2 (SCIEX, Framingham, MA, USA).
Method validation
Method validation was performed according to the US EPA method 1694 [14], and specificity, linearity, matrix effect, accuracy, precision, method detection limit (MDL), and limit of quantification (LOQ) were evaluated in the water and soil samples. Blank river water samples were collected from Gap stream, Daejeon, South Korea. The pH and electrical conductivity (EC) were 7.10 and 0.22 dS m−1, respectively. Blank soil samples were collected from a research farm located at Chungnam National University, Daejeon, South Korea. The physicochemical properties of soil samples are as follows: a sandy loam texture (56% sand, 16% silt, and 28% clay), pH 6.22, EC 0.38 dS m−1, and organic matter content 2.87%. All these properties were determined using water pollution standard method proposed by Ministry of Environment and soil standard methods proposed by Rural Development Administration (RDA) in South Korea. Before method validation, both blank samples were analyzed using the developed method to confirm that antibiotics were not present.
Specificity
To check the specificity of the instrumental analysis of target antibiotics in water and soil, we analyzed two types of samples: blank samples and antibiotic mixture spiked samples (at low concentration levels of 0.01 mg L−1). Subsequently, specificity was verified by comparing the two types of chromatograms.
Linearity
Linearity was evaluated by constructing matrix-matched calibration curves at seven concentration levels in the range of 0.01–1.0 mg L−1.
Matrix effect
To evaluate matrix effects (ME), a matrix-matched calibration curve using blank samples and a standard solution calibration curve was constructed (in the range of 0.01–1.0 mg L−1), and the slopes between the two calibration curves were compared (Eq. 1).
Accuracy and precision
Accuracy was determined through a recovery test at two concentration levels (0.1 and 1.0 g L−1). Each sample containing the antibiotic mixture was analyzed and compared with the calculated and expected concentrations. A recovery test was performed with three duplicate samples and the relative standard deviation (RSD) was calculated to evaluate precision.
Method detection limit (MDL) and limit of quantification (LOQ)
The method detection limit (MDL) and limit of quantification (LOQ) for each analyte were evaluated using the standard deviation (SD) of seven spiked river water and soil samples. The MDL was calculated by multiplication of the SD by the Student’s t-value for six degrees of freedom at a 99% confidence level (3.143, α = 0.02). The LOQ was calculated by multiplying the SD by 10.
Analysis of agricultural environmental samples
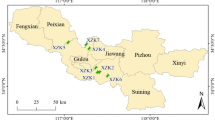
To evaluate the optimal extraction method established in this study and monitor residual antibiotics in the agricultural environment, river water, soil, and sediment samples were collected at five points around the Muhan River watershed in Yesan-gun, Chungcheongnam-do, South Korea (Fig. 2). The sampling sites for river water and sediment samples were selected as the points where the antibiotic contamination source was located, and soil samples were selected as arable soil where residual antibiotics can flow into the Muhan River during rainfall. The Yesan public sewage treatment facility and livestock manure public resource center are located at sites 2 and 3, respectively, and the effluent from these facilities is discharged into the Muhan River. Sites 4 and 5 are intensive livestock farming areas (site 4 is a mixed cattle-pig farm, and site 5 is a poultry farm), where livestock manure may directly or indirectly runoff to surface water due to rainfall. River water samples were collected in 1 L polyethylene collection bottles, placed in an icebox, transported to the laboratory, and stored at 4 °C until analysis. Sediment samples were collected using a stainless-steel shovel and topsoil layer (0–15 cm) of soil samples were collected using a hand auger after removing the organic layer from the surface. All sediment and soil samples in each location were composited to make one representative sample after collecting from 5 different locations.

Map of Muhan river with sampling sites and potential sources of antibiotics contamination
Statistical analysis
Each sample was analyzed in triplicate, and antibiotic concentrations were expressed as mean values ± standard deviation (SD). One-way ANOVA and post hoc test (Duncan's test, p < 0.05) were performed using the statistical package for social science (SPSS) version 26.0 (SPSS Inc., Chicago, IL, USA) for multiple group comparison. The Mann–Whitney U test (α = 0.05) was used to evaluate the efficiency of the antibiotic extraction method.
Results and discussion
Method development
LC–MS/MS analysis optimization
For each target analyte, the MRM conditions of the mass spectrometer were optimized to provide the best performance for VA quantification. To achieve high sensitivity, each analyte prepared as a 0.1 mg L−1 standard solution was individually injected directly into the mass spectrometer using a 1 mL Hamilton gas-tight syringe, and the mass spectrum was identified in full scan mode. In the Q1 mass spectrum, all analytes showed a high signal intensity in the form of [M + H]+, and this ion was selected as the precursor ion. To obtain the MRM conditions, the m/z value of the precursor ion was input into the tuning programs of the mass spectrometer, and the product ion was identified according to the precursor ion. Subsequently, the detection sensitivity of the product ions was increased by adjusting the declustering potential energy (DP), entrance potential energy (EP), collision energy (CE), and collision cell exit potential energy (CXP); three optimal product ions were selected. Among the three product ions, the ion with the highest signal strength was selected as the quantitative ion, and the remaining two product ions were chosen as qualitative ions (Table 1).
To get the best separation of VAs with different physicochemical properties using HPLC, a method was developed with reference to Kim and Carlson (2007) [24]. Most of the penicillin, tetracycline, macrolide, and sulfonamide group antibiotics have polar functional groups [25, 26], so a reverse phase C18 column, which is effective for analyzing polar compounds was used. For optimal gradient conditions and obtaining a good peak shape, the mobile phase consisted of HPLC-grade water (mobile A) and acetonitrile (mobile B), and 0.1% of formic acid was added as an acidic additive to increase the ionization efficiency and detection sensitivity in the mass spectrometer [27].
Validation of the separation method was confirmed by injecting a 0.1 mg L−1 antibiotic standard mixture solution into the HPLC, and all analytes were separated within 15 min of runtime (Additional file 1: Fig. S1). In addition, a calibration curve was constructed using a standard mixture in the range of 0.01–1.0 mg L−1, and coefficients of determination (R2) were calculated to be more than 0.999 for all antibiotics, indicating good linearity (Additional file 1: Table S3).
Extraction and clean-up procedure optimization
River water and soil samples are composed of a complex matrix, and most residual antibiotics in the environment are present at low concentrations [28]. Therefore, an appropriate sample preparation process is needed, including extraction of the target analyte, adjustment of the sample pH, removal of interfering substances, and sample enrichment. This study adjusted various sample preparation factors to develop an optimal extraction and cleanup procedure for target antibiotics. The selection of the extraction solvent is one of the primary parameters in the sample preparation process and affects the performance of the method for soil and sediment sample. McIlvaine [29, 30] and phosphate buffers [31, 32] are often used as extraction solvents for solid samples. In this study, the efficiency of each extraction solvent was evaluated by measuring the recovery rate of the spiked blank sample at concentrations of 0.1 mg L−1.
Extraction efficiencies using the McIlvaine buffer did not satisfy the requirements for amoxicillin (32.5%) and ampicillin (43.2%). The phosphate buffer was also tested using the same extraction process for comparison with the McIlvaine buffer. When using a phosphate buffer, the recoveries of the amoxicillin and ampicillin increased to 120.4–101.7%, respectively, compared with the McIlvaine buffer. However, sulfonamide antibiotics showed poor extraction efficiency in phosphate buffer, and the recovery of sulfamethazine showed a decrease from 81.4 to 18.9%. Furthermore, it is widely known that nonvolatile phosphate buffer contaminates the electrospray ionization source owing to its strong ionization suppression, which decreases the sensitivity of the LC–MS/MS analysis [33]. Therefore, the McIlvaine buffer was used as the extraction solvent for the analyzed soil samples.
River water and soil contain various organic and inorganic matter, as well as target analytes. These co-existing substances can block the HPLC column, and during the sample preparation process, these materials are co-extracted with the target compounds and behave as interfering substances in the ionization of the analytes in mass spectrometry. For these reasons, the SPE method was used to improve the efficiency of quantification using LC–MS/MS, and the SPE method was optimized by considering factors such as cartridge type, sample loading volume, and loading flow rate.
The selection of an adequate cartridge is the most critical step in the SPE process, and we selected the Oasis HLB (Hydrophilic-lipophilic balance) sorbent cartridge to extract target analytes based on previous literature [34,35,36]. The HLB sorbent comprises two monomers (hydrophilic N-vinylpyrrolidone and lipophilic divinylbenzene) that effectively absorb polar compounds [37, 38].
Furthermore, to adjust the sample volume parameter, the SPE loading volume was set to 120 ml and 240 ml and tested at two different final concentrations of 0.1 and 1.0 mg L−1 using blank river water and soil samples. The volume parameter was evaluated by comparing the average recovery rate (Mann–Whitney U test, n = 6), and the results showed no significant difference in the recovery rate between the sample volume factors. However, since the analysis time also increased as the sample volume increased from 120 to 240 mL, a 120 mL sample volume was adopted to achieve the effective SPE method.
To assess the efficiency of the SPE process according to the sample loading flow rate, a recovery test was performed by spiking the target analytes with two known concentration levels (0.1 and 1.0 mg L−1) into ultrapure water. During SPE extraction, the loading flow rate at which the sample passed through the cartridge was set to 2 and 4 ml min−1, and the mean recovery rate was compared (Mann–Whitney U test, n = 6). In the statistical analysis of the two different flow rates, the p-value was calculated to be higher than 0.100 for both 0.1 and 1.0 mg L−1 for all target analytes. These results indicated that the sample loading flow rate during the SPE step did not affect the extraction process. However, although there was no significant difference in the recovery rate when the sample loading flow rate was reduced from 4 mL min−1 to 2 mL min−1, the extraction time was approximately doubled. Therefore, a loading flow rate of 4 mL min−1 was used in the subsequent experiments to minimize the sample extraction run time.
Method validation
The developed analytical method was validated in terms of sensitivity, linearity, matrix effect, accuracy, precision, method detection limit (MDL), and limit of quantitation (LOQ) according to the US EPA method 1694 [39]. Validation was carried out using blank river water and soil samples with the addition of appropriate amounts of mixed antibiotic standard solution.
Specificity
Blank samples of each matrix (river water and soil) and fortified samples (low concentration level of 0.01 mg L−1) were analyzed to evaluate the presence of interference. After LC–MS/MS analysis, peaks observed near the retention time of each analyte were checked, and chromatograms of the blank and fortified samples were compared. No significant interference peaks were observed for any of the antibiotics (Additional file 1: Fig. S1). Therefore, this method was considered specific for the target analytes, and sensitivity analysis was possible even at low concentrations.
Linearity
To evaluate the linearity of the method in river water and soil samples, we prepared a matrix-matched calibration curve for each target analyte at seven different concentration levels ranging from 0.01 to 1.0 mg L−1 and obtained the correlation coefficient (R2). The linearity values in river water and soil ranged between 0.9933 and 0.9995 and 0.9916 and 0.9998, respectively, indicating good linearity (R2 more than 0.99, Additional file 1: Table S4).
Matrix effect
The slopes of these two calibration curves (matrix-matched and standard solution calibration curves) were compared to evaluate the matrix effect (ME, %). Based on the calculated percentage values, the matrix effect was classified into two categories: ion enhancement (ME > 0%) and ion suppression (ME < 0%). The percentage range of ± 0–20% is considered a soft matrix effect, but the range of 20–50% or > 50 and < − 50% is considered to suffer a medium and strong matrix effect, respectively [40]. In the river water and soil samples, the matrix effect was calculated in the ranges of − 62.9–124.9% and − 72.7–198.9%, respectively (Additional file 1: Table S5). Among penicillin antibiotics, amoxicillin and ampicillin showed insignificant matrix effects (ranging from − 11.1 to 26.1%), but penicillin G revealed medium ion suppression (ranging from − 53.4 to − 39.5%). All tetracycline antibiotics showed ion enhancement, and oxytetracycline showed a range of up to 198.9% in soil. Macrolide and sulfonamide antibiotics showed medium or strong ion suppression from -29.0 to − 72.7%. Most target antibiotics suffered medium or strong matrix effects, so we used simeton as an internal standard (adjusted ion suppression) or applied matrix-matched calibration (adjusted ion enhancement) to compensate for matrix effects and improve the sensitivity of the quantitative analysis.
MDL and LOQ
The values of MDL and LOQ were calculated according to "Method detection limit (MDL) and limit of quantification (LOQ)" and are presented in Table 2. The MDLs ranged from 2.1–12.3 ng L−1 in river water and 1.2–13.2 ng kg−1 in soil, and the LOQs ranged from 6.6–39.2 ng L−1 and 4.0 to 42.0 ng kg−1, respectively. The MDL and LOQ of all target analytes were calculated at the ng L−1 or ng kg−1 level. Thus, this method was considered sufficient to quantify trace amounts of VAs in the environment.
Accuracy and precision
The efficiency of the developed method was verified based on the recovery of the target analytes (Table 3). Most antibiotics showed moderate recovery values in river water and soil, ranging from 61.9–115.8% and 60.4–111.6%, respectively. However, amoxicillin (12.1–32.5%) and spiramycin (36.8–59.1%) showed low recoveries in all the sample matrices. The precision calculated and expressed as relative standard deviation (RSD) ranged from 0.7–12.2% in river water and 1.0–12.7% in soil. Therefore, we excluded amoxicillin and spiramycin from the target analytes in this study because these compounds were not appropriate for quantitative analysis of river water and soil using the developed method.
Applications to agricultural environmental samples
The developed method was applied to identify and quantify VAs in river water, sediment, and arable soil samples, and these environmental samples were collected from the Muhan river in Yesan-gun, Chungcheongnam-do. In this area, livestock farms and treatment facilities are adjacent to the river, so it is easy to identify the pattern of residual VAs that introduce directly or indirectly into the environment by antibiotics pollution sources.
Residual antibiotics can be introduced into river water by rainfall runoff from nearby arable soil and livestock farms, or effluents from livestock wastewater treatment plants (WWTPs) [41, 42]. In this study, seven antibiotics belonging to three classes were detected in river water samples, and the measured concentrations of the VAs ranged from 0.018 to 0.309 mg L−1 (Table 4). Tylosin was the most frequently detected compound, with values of up to 0.251 μg L−1 (site 4), whereas ampicillin and penicillin G were not detected. Ampicillin and penicillin G, which are beta-lactam antibiotics, are not frequently found in the environment because of the instability of the beta-lactam ring due to beta-lactamase or chemical hydrolysis [43]. Cha et al. also reported that almost all β-lactam antibiotics were not detected in surface water and wastewater in urban and agricultural areas in northern Colorado, USA, and ampicillin was found only once in 11 ng L−1 out of 60 samples [44].
Chlortetracycline, oxytetracycline, and tetracycline were detected in the ranges of 0.107–0.173, 0.373–0.309, and 0.080–0.135 μg L−1, respectively. In addition, sulfonamide group antibiotics were detected in the range of 0.018–0.067 μg L−1, except for sulfadiazine, and they were detected at lower concentrations than the tetracycline group antibiotics. It has been reported that sulfonamide antibiotics are detected more frequently in aquatic environments than tetracycline antibiotics because of their high water solubility and low sorption coefficient (Kd) values [45]. Dong et al. detected sulfadiazine and sulfamethoxazole in river water near arable land and livestock farms up to 1.5 and 44.1 ng L−1, respectively, but detected oxytetracycline up to 835.1 ng L−1 with a frequency higher than sulfonamide antibiotics [46]. These results indicate the high possibility of leakage into the agricultural environment due to the high sales volume and usage of tetracycline antibiotics [47].
Five antibiotics (chlortetracycline, tetracycline, tylosin, sulfamethazine, and sulfathiazole) were detected in sediment samples (Table 4). The identified antibiotics in sediment were less diverse than in river water; however, they have high concentrations ranging from 1.45 to 9.04 μg kg−1 when compared with river water samples. These results may be associated with the continuous accumulation of residual antibiotics derived from effluents and runoff from arable soils. Tetracycline antibiotics with a high adsorption capacity to soil were detected from 7.62 to 9.04 μg kg−1, whereas sulfonamide antibiotics were detected in relatively low concentrations in the range of 1.45–4.96 μg kg−1.
Sulfamethazine was the only identified target antibiotic among all the arable soil samples and was detected at a concentration of 3.66 μg kg−1 only once at site 5 (Table 4). This result suggests that the antibiotics detected in river water and sediment were not derived from arable soil.
An SPE-LC-MS/MS analytical method for the simultaneous determination of 12 VAs in river water and soil was developed, and this method was successfully applied to monitor residual antibiotics in agricultural environments. This method was validated according to the US EPA method 1694, except for amoxicillin. It satisfactorily fulfilled the criteria of linearity, accuracy, and precision for the 10 antibiotics in river water and soil. Additionally, the low values of MDL and LOQ suggest that accurate quantitative analysis of trace antibiotics in river water and soil is possible. The analysis of environmental samples showed the presence of VAs in the river water, sediment, and arable soil samples. The determined concentrations ranged between 0.013–0.309 μg L−1 in river water and 1.45–9.04 μg kg−1 in sediment and soil samples, and the highest and most diverse antibiotics were detected at sites 3 and 4, which were affected by livestock activity. The detection of antibiotics demonstrates the necessity for systematic monitoring in agricultural environments. In addition, these results are expected to be the primary data sources for developing analytical methods for VAs in livestock manure compost and liquid fertilizer.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- AMO:
-
Amoxicillin
- AMP:
-
Ampicillin
- ARGs:
-
Antibiotic resistance genes
- CE:
-
Collision energy
- CTC:
-
Chlortetracycline
- CXP:
-
Collision cell exit potential energy
- DP:
-
Declustering potential energy
- EP:
-
Entrance potential energy
- HLB:
-
Hydrophilic-lipophilic balance
- LC–MS/MS:
-
Liquid chromatography-tandem mass spectrometry
- LOD:
-
Limit of detection
- MDL:
-
Method detection limit
- ME:
-
Matrix effect
- MRLs:
-
Maximum residue limits
- MRM:
-
Multiple reaction monitoring
- OTC:
-
Oxytetracycline
- PNG:
-
Penicillin G
- RSD:
-
Relative standard deviation
- SD:
-
Standard deviation
- SDZ:
-
Sulfadiazine
- SMX:
-
Sulfamethoxazole
- SMZ:
-
Sulfamethazine
- SPE:
-
Solid phase extraction
- SPM:
-
Spiramycin
- STZ:
-
Sulfamethazine
- TC:
-
Tetracycline
- TYL:
-
Tylosin
- VAs:
-
Veterinary antibiotics
- WWTP:
-
Wastewater treatment plant
References
Hao H, Cheng G, Iqbal Z, Ai X, Hussain HI, Huang L, Dai M, Wang Y, Liu Z, Yuan Z (2014) Benefits and risks of antimicrobial use in food-producing animals. Front Microbiol 5:288
Kemper N (2008) Veterinary antibiotics in the aquatic and terrestrial environment. Ecol Indic 8:1–13
Jjemba PK (2002) The potential impact of veterinary and human therapeutic agents in manure and biosolids on plants grown on arable land: a review. Agric Ecosyst Environ 93:267–278
Larney FJ, Sullivan DM, Buckley KE, Eghball B (2006) The role of composting in recycling manure nutrients. Can J Soil Sci 86:597–611
Boy-Roura M, Mas-Pla J, Petrovic M, Gros M, Soler D, Brusi D, Menció A (2018) Towards the understanding of antibiotic occurrence and transport in groundwater: findings from the Baix Fluvià alluvial aquifer (NE Catalonia Spain). Sci Total Environ 612:1387–1406
Kümmerer K (2009) Antibiotics in the aquatic environment–a review–part I. Chemosphere 75:417–434
Hillis DG, Fletcher J, Solomon KR, Sibley PK (2011) Effects of ten antibiotics on seed germination and root elongation in three plant species. Arch Environ Contam Toxicol 60:220–232
Kong W, Zhu Y, Liang Y, Zhang J, Smith F, Yang M (2007) Uptake of oxytetracycline and its phytotoxicity to alfalfa (Medicago sativa L.). Environ Pollut 147:187–193
Pan M, Chu L (2016) Phytotoxicity of veterinary antibiotics to seed germination and root elongation of crops. Ecotoxicol Environ Saf 126:228–237
Lin H, Jin D, Freitag TE, Sun W, Yu Q, Fu J, Ma J (2016) A compositional shift in the soil microbiome induced by tetracycline sulfamonomethoxine and ciprofloxacin entering a plant-soil system. Environ Pollut 212:440–448
Liu F, Ying GG, Tao R, Zhao JL, Yang JF, Zhao LF (2009) Effects of six selected antibiotics on plant growth and soil microbial and enzymatic activities. Environ Pollut 157:1636–1642
Chee-Sanford JC, Aminov R, Krapac IJ, Garrigues-Jeanjean N, Mackie RI (2001) Occurrence and diversity of tetracycline resistance genes in lagoons and groundwater underlying two swine production facilities. Appl Environ Microbiol 67:1494–1502
Luangtongkum T, Jeon B, Han J, Plummer P, Logue CM, Zhang Q (2009) Antibiotic resistance in Campylobacter: emergence transmission and persistence. Future Microbiol 4:189–200
Englert B (2007) Method 1694: Pharmaceuticals and personal care products in water soil sediment and biosolids by HPLC/MS/MS US environmental protection agency. EPA 1–72
Carvalho IT, Santos L (2016) Antibiotics in the aquatic environments: a review of the European scenario. Environ Int 94:736–757
Murata A, Takada H, Mutoh K, Hosoda H, Harada A, Nakada N (2011) Nationwide monitoring of selected antibiotics: distribution and sources of sulfonamides, trimethoprim, and macrolides in Japanese rivers. Sci Total Environ 409:5305–5312
Lyu J, Yang L, Zhang L, Ye B, Wang L (2020) Antibiotics in soil and water in China–a systematic review and source analysis. Environ Pollut. https://doi.org/10.1016/j.envpol.2020.115147
Lee KB, Kim NW, Kim DU, Sin MH, Lee MY (2020) Monitoring of veterinary drug residues in cultured fishery products in Chungcheongnam-do Province. J Food Saf 35:447–451
Park C, Kim JH, Bang SM, Park YS, Go DS (2020) Comparison of antibiotic residues detected by the residue tests of slaughtered livestock in Korea. Korean J Vet Serv 43:17–21
Chang Q, Wang W, Regev-Yochay G, Lipsitch M, Hanage WP (2015) Antibiotics in agriculture and the risk to human health: how worried should we be? Evol Appl 8:240–247
Kim JH, Kuppusamy S, Kim SY, Kim SC, Kim HT, Lee YB (2017) Occurrence of sulfonamide class of antibiotics resistance in Korean paddy soils under long-term fertilization practices. J Soils Sediments 17:1618–1625
Seo KW, Kim JJ, Mo IP, Lee YJ (2019) Molecular characteristic of antimicrobial resistance of Salmonella Gallinarum isolates from chickens in Korea, 2014 to 2018. Poult Sci 98(11):5416–5423
Di S, Aleta P, Park M, Yoon H, Cho KH, Kim YM, Kim S (2018) Seasonal changes in antibiotic resistance genes in rivers and reservoirs in South Korea. J Environ Qual 47(5):1079–1085
Kim SC, Carlson K (2007) Quantification of human and veterinary antibiotics in water and sediment using SPE/LC/MS/MS. Anal Bioanal Chem 387:1301–1315
Thiele-Bruhn S, Seibicke T, Schulten HR, Leinweber P (2004) Sorption of sulfonamide pharmaceutical antibiotics on whole soils and particle-size fractions. J Environ Qual 33:1331–1342
Cho YJ, Choi SJ, Kim M, Kim M, Yoon SJ, Chang MI, Lee SM, Kim HJ, Jeong J, Rhee GS (2014) Simultaneous determination of aminoglycoside antibiotics in meat using liquid chromatography tandem mass spectrometry. J Food Hyg Saf 29:123–130
Seifrtová M, Nováková L, Lino C, Pena A, Solich P (2009) An overview of analytical methodologies for the determination of antibiotics in environmental waters. Anal Chim Acta 649:158–179
Kim C, Ryu HD, Chung EG, Kim Y, Rhew DH (2016) Determination of veterinary antibiotic residues: III analytical methods_a review. J Korean Soc Water Environ 32:649–669
Boscher A, Guignard C, Pellet T, Hoffmann L, Bohn T (2010) Development of a multi-class method for the quantification of veterinary drug residues in feedingstuffs by liquid chromatography-tandem mass spectrometry. J Chromatogr A 1217:6394–6404
Carmona E, Andreu V, Picó Y (2017) Multi-residue determination of 47 organic compounds in water soil sediment and fish—Turia River as case study. J Pharm Biomed Anal 146:117–125
Meng M, He Z, Xu Y, Wang L, Peng Y, Liu X (2017) Simultaneous extraction and determination of antibiotics in soils using a method based on quick easy cheap effective rugged and safe extraction and liquid chromatography with tandem mass spectrometry. J Sep Sci 40:3214–3220
Albero B, Tadeo JL, Escario M, Miguel E, Pérez RA (2018) Persistence and availability of veterinary antibiotics in soil and soil-manure systems. Sci Total Environ 643:1562–1570
Hu FY, He LM, Yang JW, Bian K, Wang ZN, Yang HC, Liu YH (2014) Determination of 26 veterinary antibiotics residues in water matrices by lyophilization in combination with LC–MS/MS. J Chromatogr B 949:79–86
Rossmann J, Schubert S, Gurke R, Oertel R, Kirch W (2014) Simultaneous determination of most prescribed antibiotics in multiple urban wastewater by SPE-LC–MS/MS. J Chromatogr B 969:162–170
Aga DS, O’Connor S, Ensley S, Payero JO, Snow D, Tarkalson D (2005) Determination of the persistence of tetracycline antibiotics and their degradates in manure-amended soil using enzyme-linked immunosorbent assay and liquid chromatography—mass spectrometry. J Agric Food Chem 53:7165–7171
Abuin S, Codony R, Compañó R, Granados M, Prat MD (2006) Analysis of macrolide antibiotics in river water by solid-phase extraction and liquid chromatography–mass spectrometry. J Chromatogr A 1114:73–81
Fontanals N, Maria Marce R, Galià M, Borrull F (2004) Synthesis of hydrophilic sorbents from N-vinylimidazole/divinylbenzene and the evaluation of their sorption properties in the solid-phase extraction of polar compounds. J Polym Sci Part A1 Polym Chem. https://doi.org/10.1002/pola.20045
Peruzzi M, Bartolucci G, Cioni F (2000) Determination of phenoxyalkanoic acids and other herbicides at the ng/ml level in water by solid-phase extraction with poly (divinylbenzene-co-N-vinylpyrrolidone) sorbent and high-performance liquid chromatography–diode-array detection. J Chromatogr A 867:169–175
EPA U (2007) EPA Method 1694: Pharmaceuticals and Personal Care Products in Water Soil Sediment and Biosolids by HPLC/MS/MS. EPA-821-R-08–002
Ferrer C, Lozano A, Agüera A, Girón AJ, Fernández-Alba A (2011) Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables. J Chromatogr A 1218:7634–7639
Le HT, Maguire RO, Xia K (2018) Method of dairy manure application and time before rainfall affect antibiotics in surface runoff. J Environ Qual 47:1310–1317
Lim JE, Kim SC, Lee HY, Kwon OK, Yang JE, Ok YS (2009) Occurrence and distribution of selected veterinary antibiotics in soils sediments and water adjacent to a cattle manure composting facility in Korea. J Korean Soc Environ Eng 31:845–854
Christian T, Schneider RJ, Färber HA, Skutlarek D, Meyer MT, Goldbach HE (2003) Determination of antibiotic residues in manure soil and surface waters. Acta Hydrochim Hydrobiol 31:36–44
Cha J, Yang S, Carlson K (2006) Trace determination of β-lactam antibiotics in surface water and urban wastewater using liquid chromatography combined with electrospray tandem mass spectrometry. J Chromatogr A 1115:46–57
Lee HY, Lim JE, Kim SC, Kim KR, Kwon OK, Yang JE, Ok YS (2009) Transport of selected veterinary antibiotics (tetracyclines and sulfonamides) in a sandy loam soil: laboratory-scale soil column experiments. J Korean Soc Environ Eng 31:1105–1112
Dong D, Zhang L, Liu S, Guo Z, Hua X (2016) Antibiotics in water and sediments from Liao River in Jilin Province China: occurrence distribution and risk assessment. Environ Earth Sci 75:1202
Ding H, Wu Y, Zhang W, Zhong J, Lou Q, Yang P, Fang Y (2017) Occurrence distribution and risk assessment of antibiotics in the surface water of Poyang Lake the largest freshwater lake in China. Chemosphere 184:137–147
Acknowledgements
This work was carried out with the support of “Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ01488502)” Rural Development Administration, Republic of Korea.
Funding
This study was funded by Grant No. PJ01488502 from the Rural Development Administration, Korea.
Author information
Authors and Affiliations
Contributions
JW wrote original draft manuscript and conducted formal analysis; YK conducted and validated formal analysis; SH gave an idea and conducted data analysis OK conducted data analysis and correction of context; YB corrected in the context and gave an idea; SC conceptualized and supervised whole manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1. Physicochemical properties of 12 antibiotics. Table S2. LC-MS/MS parameters for the analysis of antibiotics. Table S3. Standard solution calibration curves equations and coefficient of determination. Table S4. Linearity of matrix-matched calibration curves for target analytes. Table S5. Matrix effect of 12 antibiotics in river water and soil sample. Fig. S1. Extracted ion chromatogram in spiked (0.025 mg L−1) samples
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, JW., Hong, YK., Ryu, SH. et al. Development of analytical method for veterinary antibiotics and monitoring of residuals in agricultural environment. Appl Biol Chem 66, 20 (2023). https://doi.org/10.1186/s13765-023-00777-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-023-00777-3