
Abstract
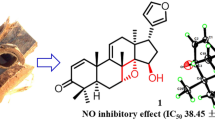
Two novel phragmalin type limonoids, Tabularisin Q (1) and Chuktabularin Y (18), together with 22 known limonoids have been separated from bark of Chukrasia tabularis. The structures of 1–24 were identified with spectroscopic method (NMR, IR and mass spectrometry) and comparison with literature. The anti-inflammatory activity of all limonoids was assayed in RAW264.7 cells in vitro by evaluating the production of nitric oxide induced by lipopolysaccharide. Limonoids 8, 9, 1, and 18 showed significant anti-inflammatory activity with the inhibitory rates of 4.32, 11.28, 13.13, and 10.40 μm, respectively, and likely to be useful for the development as therapeutic agents for inflammatory diseases.
Similar content being viewed by others

Introduction
Meliaceae plants are famous for their various limonoid compounds with structural diversity and bioactivity [1]. Through the chemical study of Chukrasia, a series of limonoid compounds of phragmalin type were isolated [2]. Chukrasia tabularis belongs to the family Meliaceae and mainly distributed in Southern and Eastern Asia [3]. Its barks are traditionally used in India and southern China for astringent, antidiarrheal and anti-flu properties [4]. Previous chemical study of the genus has led to the separation of several interesting carbon skeleton phragmalin limonoids, including the normal phragmalin limonoid and ortho ester derivative [5], 16 norphragmalin limonoid with a 13,14,18 cyclopropane ring [6]. Research have shown that phragmalin limonoid has a variety of bioactivities such as antifeeding [7], antibacterial [8], blocking potassium channels [9], anti-inflammatory [10], etc. As part of our ongoing study project to separate new limonoids from Meliaceae family, a novel phragmalin limonoid with 13/14/18-cyclopropane ring (1) and a novel 16-norphragmalin limonoid (18) were separated from the bark of C. tabularis, together with 16 known phragmalin orthoesters (2–17) and 6 known norphragmalin limonoids (19–24). In addition, the anti-tumor and antiinflammatory activity of these limonoids was also investigated. In vitro anti-inflammatory activity data show that compound 18 can effectively inhibit the inflammation of LPS-stimulated RAW164.7 cells, which is mediated by inhibiting the activation of JAK2/STAT3 and NF-κB signaling pathways. We reported herein on the separation, structural analysis and bioactivities of isolated limonoids.
Materials and methods
Plant material
The stem barks of Chukrasia tabularis A. Juss were collected in Fuzhou, Fujian, China, on July 2019, and identified by Dr. Yonghong Zhang from Department of Pharmacy, Fujian Medical University, where a voucher specimen (ZYH20190703) was preserved at that department.
General experimental procedures
The optical rotation was determined with JASCOP-1020 polarimeter. The IR spectrum were determined in Nicolet 170SX FT-IR spectrometer, while UV data was determined on UV-210A spectra. NMR spectra were acquired on Bruker AM-400 spectrometer. ESI–MS and HRESI-MS were recorded in LCMS-2020 instrument and Bruker APEXII mass instrument. Preparative HPLC was carried out Waters column (250 × 10 mm, Waters). Silica gel (230–300 mesh; Qingdao Ocean Chemical Factory) was used as adsorbent for column chromatography. MCI gel CHP20P (75–150 μm; Mitsubishi Chemical Company) and ODS (50 μm, Quebec, Canada). TLC plate was precoated with SiO2 gel GF254 (Qingdao Ocean Chemical Factory). The enzyme immunoassay kit of NF-κB, TNF-α and IL-6 were provided by R&D System (MN, USA). Lipopolysaccharide (LPS) was from Sigma (Chemical Company in St. Louis, USA). Nitric oxide (NO) was from the Nan-jing Jian-cheng Bioengineering Institute (Jiangsu, China). Antibodies against p-JAK2, p-STAT3, NF-κB p-P65, P65 p-IKBα and p-IKKα/β were from Cell Signaling Technology (MA, USA). The ECD spectra were determined in MeOH with Jasco J 1500 spectropolarimeter (Jasco, Tokyo, Japan).
Extraction and isolation of C. tabularis
The chipped dried stem barks of Chukrasia tabularis (17.5 kg) were extracted four times at RT with MeOH (35 L × 4). The mixed MeOH extract were distilled in vacuo to obtain a MeOH residue (1980 g), which were suspended in H2O and separated by petroleum ether, dichloromethane, EtOAc and n-BuOH. Furthermore, the CH2Cl2 extract (386.8 g) was applied to MCI gel column and washed with 10% (FrA), 30% (FrB), 50% (FrC), 70% (FrD), 90% (FrE) and 100%MeOH (FrF). FrC (108.2 g) was applied to silica gel column and washed with PE-EtOAc (10:0, 8:2, 6:4, 4:6, 2:8 and 0:10, each 8 L) to obtain thirteen fractions (Frs C1-C13). FrC6 (36.7 g) was fractionated to Sephadex LH-20 (CH2Cl2-MeOH) to obtain four fractions (FrsC6-1-C6-4). Then FrC6-2 (16.7 g) was subjected to HPLC (MeCN:H2O = 13:7) to obtain compound 20 (55.6 mg, tR15.0 min), 14 (14.5 mg, tR9.8 min), 11 (26.7 mg, tR15.7 min), 15 (19.8 mg, tR17.6 min), and 17 (33.5 mg, tR12.0 min). Fr D (121.4 g) was applied to silica gel column and washed with PE-EtOAc (8:2, 6:4, 4:6, 2:8 and 0:10, each 8 L) to obtain fifteen fractions (Frs D1-D15). FrD5 (32.9 g) was fractionated to Sephadex LH-20 (CH2Cl2–MeOH) to gain three fractions (Frs D5-1-D5-3). FrD5-2 (14.8 g) was applied to C18 column chromatography (ODS) (MeOH/H2O) and subjected to HPLC (MeCN–H2O = 13:7) to obtain compounds 19 (33.7 mg, tR20.0 min), 1 (28.6 mg, tR15.1 min), 7 (35.1 mg, tR15.9 min), 16 (11.9 mg, tR18.9 min), 13 (37.9 mg, tR14.3 min), and 21 (9.2 mg, tR22.0 min). FrD6 (12.9 g) was fractionated to Sephadex LH-20 (CH2Cl2–MeOH) and subjected to HPLC (MeCN:H2O = 3:1) for compounds 18 (31.6 mg, tR15.2 min), 6 (27.5 mg, tR19.0 min), 5 (8.2 mg, tR11.7 min), and 4 (15.7 mg, tR20.3 min). FrD7 (14.6 g) was applied to C18 column chromatography (MeOH:H2O), Sephadex LH-20 (CH2Cl2:MeOH), and subjected to HPLC (MeCN:H2O = 7:3) for compounds 10 (19.6 mg, tR13.3 min), 12 (13.2 mg, tR14.7 min), 24 (25.6 mg, tR12.6 min), and 22 (8.7 mg, tR17.5 min). FrD9 (26.1 g) was fractionated to Sephadex LH-20 (CH2Cl2:MeOH) and then subjected to HPLC (MeCN:H2O = 6:4) to obtain compounds 8 (17.2 mg, tR12.9 min), 9 (35.1 mg, tR16.8 min), 2 (16.8 mg, tR20.1 min), 3 (21.4 mg, tR7.9 min), and 23 (13.8 mg, tR24.6 min) (Additional file 1: Fig. S15).
Tabularisin Q (1): White amorphous powder; [α]28D-0.33 (c 0.45, CHCl3), The 1H and 13C NMR see Table 1. IR (KBr): νmax3489, 2975, 1753, 1423, 1367, 1219, 1025, 891 cm−1; HR-ESI–MS m/z: 883.2625 [M + Na]+ (calcd for C41H48O20 Na, 883.2637).
Chuktabularin U (18): Colourless crystals; [α]28D + 0.32 (c 1.91, CHCl3), The 1H and 13C NMR see Table 1. IR (KBr): νmax3465, 2941, 1751, 1420, 1354, 1232, 1045, 886 cm−1; HRESI–MS m/z: 833.2867[M + H]+(calcd for C40H49O19, 833.2867).
Anti-inflammation activity assay
Male ICR mice (about 18 ± 2 g each), obtained from Laboratory Animal Center, Fujian Medical University. The extract was suspended in a 0.5% CMC-Na, and Aspirin was used as positive control. After intragastric administering the extract or control for 1 h, each mouse right ear was treated with 40 μL xylene solution, and the left ear was taken as control. One hour after xylene treatment, the mice were sacrificed for cervical dislocation. The round part of each ear with a diameter of 6 mm was weighed by an electronic analytical balance with an accuracy of 0.1 mg, and its inhibitory effect on ear edema was calculated. The weight difference between the two plugs is used to measure the edema response (Additional file 1: Table S1). Animal care and use comply with institutional guidelines, and the program was affirmed by Experimental Animal Management Committee of Fujian Medical University.
Anti-infammatory activity in vitro
The antiinflammatory effects of compound 1–24 were measured by its inhibitory activity against lipopolysaccharide-induced mouse macrophage RAW264.7 using the Griess reaction. The contents of NO were detected with the NO kit based on a preciously described protocol [11].
Detections of cytokines
The level of TNF-α, NO and IL-6 in the supernatant of RAW264.7 cells of compound 18 was determined by ELISA.
Cytotoxicity assay
HepG2 (hepatocellular), KB (oral epithelial), Hela (cervical), MCF-7 (human breast) and A-549 (lung) cancer cell lines (provided by Shanghai Cell Bank) were maintained in RPMI 1640 at 37 °C supplemented with 10% FBS. Cytotoxicity was determined by the SRB method [12], with 5-fluorouracil (5-FU) as positive control. The IC50 value is calculated using the GraphPad 7.00 prism.
Western blot analysis
Western blotting experiment was performed to investigate effect of compound 18 on protein level of p-IKBα, p-IKKα/β, NF-κB p-P65, P65, p-JAK2 and p-STAT3. RAW264.7 cells were dispensed in 6 well plates for 24–48 h. Cells were treated with HFPS and induced by LPS for 24–48 h. Cells were collected after treatment and lysed. The Western blot analysis were determined based on method described of Li et al. [13].
Results and discussion
Twenty four limonoids 1–24 were isolated from the C. tabularis barks through repeated silica gel, C18 and Sephadex LH-20 column chromatography. 1–17 were identified as phragmalin limonoid orthoesters and 19–24 were norphragmalin limonoids from the spectroscopic data and confirmed with literature data.
Tabularisin Q (1) was found as white amorphous powder, having molecular formula of C41H48O20 according to its HRESI-MS ion at m/z 883.2625 [M + Na]+ (calcd for C41H48O20Na, 883.2637) which indicated eighteen degrees of unsaturation. IR spectra indicated absorption bands of hydroxy and ester moieties at 3489 and 1753 cm−1. 1H-NMR spectrum implied two singlet methyls (δH 0.89, 1.60), four acetyls (δH 2.22, 2.05, 2.07, 2.15), one methoxy (δH 3.81), a typical β substituted furan ring (δH 6.66, s; 7.71, s; 7.61, s), and one isobutyroyl [δH 1.11 (d, J = 4.3 Hz, 3H), 1.22 (d, J = 4.3 Hz, 3H) (Table 1). 13C-NMR spectra of 1 confirmed the existence of nine methyls and one methoxy, two methenes, six oxygenated and three olefinic methines, five oxygenated and one olefinic quaternary carbons, and six carbonyls (Table 1). One furan ring, one isobutyryl, and six esters are 10 degrees unsaturated, and remaining 8 degrees unsaturated require an octacyclic core. The carbon signal at δC 119.3 (C-31) in 13C NMR spectra demonstrated that 1 was phragmalin limonoid orthoester [14, 15]. The 13, 14, 18 cyclopropane ring was noticed in HMBC spectra of H-18, H-17, and H-11 with C-13; H-17, H-30, H-18, and H-15 to C-14; and H-17, H-15, and H-12 to C-18, and was supported by the quarterly carbon signal of upfield displacement at δC 33.6 (C-13) and 31.1 (C-14) and methylene at δC 16.2 (C-18) compared to that of phragmalin limonoids (Fig. 1A) [5]. The comprehensive analysis of 1, especially HMBC spectra confirmed the skeleton structure of phragmalin limonoid, whose methoxy group is connected to C-7 (Fig. 1). NMR analysis showed presence of three acetoxy groups (δH2.15, 2.22, 2.05; δC 21.8, 171.7; 21.0, 169.3; 19.9, 171.7) at C-2, C-6 and C-15 based on the low-field judgment of C-2 at δC 82.7, C-6 at δC 70.6 and C-15 at δC 69.9 compared to 2 (Table 1). The HMBC correlation of H-3 and OAc-3, H-6 and OAc-6, and H-15 and OAc-15 confirmed this allocation (Fig. 1A). The remaining acetoxy moiety was then classified to C-2 based on its downfield displacement carbon resonance at δC 82.7 (for 2-OH, C-2 carbon resonance usually occurs near δC 78.0), which is further verified by ROESY cross-peak of H-32 with OAc-2 (Fig. 1B). The existence of 1,8,9 ortho acetate unit was determined by the chemical shift of C-1, C-9, C-8 and HMBC correlation between H-32 to C-31, H-11 to C-8 and C-9, and H3-19 to C-1. According to the HMBC correlation of the carbonyl resonance at 174.6 and H-30 at δH 5.15, the isobutyryloxy group was situated at C-30. Thus, the plane structure of 1 was finally demonstrated. The ROESY cross-peak of H-17 to H-30, H-15, H-11, and H-12, of H-30 to H-5 revealed that these H atoms were in β-orientations (Fig. 1B) [16]. The ROESY correlations between H-5 with H-6, H-12, H-11, H-30, H-12 with H-17, H-30, H-11 with H-12, H-17 with H-15, and H-17 with H-30 revealed that H-5, H-6, H-11, H-12, H-15, H-17, and H-30 were in β-orientations (Fig. 1B) [16]. Thence, the ROESY correlations between H2-18 to OAc-15 and H3-32 to OAc-2 indicated that H2-18, OAc-15, 1, 8, 9 orthoacetate, and OAc-2 were α-oriented [17]. Furthermore, based on the ROESY correlation between Me-19 to Hb-29, Me-19 and H2-29 were designated as α-orientation. In addition, the ROESY correlation between H-29 to H-3 and H-28 revealed that H-3 was in α-orientation, which was persistent with ROESY cross-peak between H-21 with 3-OAc. Based on the above result, the relative configuration of 1 is completed as shown in Fig. 2. By comparing experimental and calculated ECD data, the absolute configuration of 1 is finally demonstrated, which is a suitable method for solving absolute configuration of natural product [18, 19]. The ECD spectrum was calculated by using gaussian-16 software through the system conformation search, geometric optimization and time dependent density functional theory (TDDFT) calculation. The calculated ECD spectrum is very constant with the experimental spectra, pointing to the absolute configuration of 1R, 2S, 3S, 4R, 5S, 6R, 8R, 9S, 10R, 11R, 12S, 13R, 14S, 15S, 17R, and 30R for 1 (Fig. 3A). Thus, Compound 1 was demonstrated and named as tabularisin Q.

Key correlations observed in the HMBC and NOESY spectra of 1

Structures of the identified compounds 1–24

Calculated and experimental ECD spectra of 1 and 18
Chuktabularin Y (18) was isolated as amorphous powder, and its molecular formula was demonstrated to C40H48O19 by HR-ESI–MS ion at m/z 833.2867 [M + H]+ (calcd 833.2867). IR spectra exhibited absorption bands of hydroxy and ester moieties at 3465 and 1751 cm−1. 1H- and 13C-NMR spectrum as well as information from 2D NMR studies indicated that 18 was phragmalin limonoid (Table 1) and suggested the existence of β substituted furan ring [δH 6.73, 7.80, 7.80; δC 124.1, 111.9, 141.7, 144.3], two methyls [δH 0.91 (s, 3H), 0.81 (s, 3H); δC 16.8, 18.8], and four acetyls. A pair of germinal doublet of H-29 protons at δH 2.15 (d, J = 13.7 Hz) and 1.99 (d, J = 13.7 Hz) indicated a phragmalin 4/29/1 ring bridge (Fig. 2), were determined by HMBC correlation of H-29 and quaternary carbon at δC 85.8 (C-1), 84.7 (C-3), 42.8 (C-5), 46.5 (C-4), and 55.1 (C-10). The two oxygenated methylene groups at δH 4.58 and 4.49 (d, J = 12.8 Hz) correspond to the 13C-NMR at δC 67.0 (C-19) revealed that they are related to the carbon at δC 77.7 (C-9), 55.1 (C-10), and 42.8 (C-5) and showed that 19-methyl unit has been oxygenated [17]. HMBC correlation of C-7 (δC 175.0) to H-6 (δH 2.16) and oxygenated C-19 methylene at δH 4.58 (H-19) suggested existence of C-6, C-7 phragmalin limonoid and C-7, C-19 δ lactone ring appendage [16].
Information from 1 and 2D NMR spectrum showed that 18 was 16 norphragmalin limonoid with ketal group of limonoid framework and extended acetyl moiety at C-15, which was verified by HMBC correlation between ketal resonances at δC 110.9 (C-31) with H-15 (δH 1.82, dd, J = 11.3, 2.0 Hz), a methyl at δH1.68, and H-30 at δH 4.77 (Fig. 4). The HMBC cross-peak of C-2′ (δC 171.0) and H-17 (δH 2.46, m) and methoxy at δH 3.70 (s, 3H) proved C-1′-C-2′ appendage characteristic of phragmalin limonoid. In 1H NMR spectrum of 18, the downfield shifted H-1′(δH5.16, m) indicated that an acetyl was connected to C-1′(δC73.1), which was sustained by HMBC cross-peak between H-1′ with acetyl carbonyl at 1′-OAc (δC170.9). Furthermore, three acetyl moieties were allocated in C-3 (δC 84.7), C-11 (δC 72.9), and C-12 (δC 73.9) based on their corresponding HMBC correlation. According to chemical shift of C-2 at δC 83.7 and H-30 at δH 4.77, the remaining acetyl group was connected to C-2 [20]. The NOESY spectra of 18 revealed that all asymmetric carbon adopted same relative configurations as 1 (Fig. 4B). The strong correlations of H-11 with H-30, H-17, and H-5, and of H-17 with H-1′, H-30 and H-12 revealed that these protons have an β direction. The NOESY cross-peak between H-14 with H-18, and H-3 with H-29, revealed an α-orientation of these protons (Fig. 4B). After the relative configuration was demonstrated, the experimental ECD spectra of 18 was recorded, and the calculated ECD spectrum was calculated by TDDFT (Fig. 3B) [21]. The good agreement between experimental and calculated spectrum showed the absolute configuration of 18 (1R, 2S, 3S, 4R, 5S, 8S, 9S, 10S, 11R, 12R, 13S, 14S, 17R, 30R, 31R, 1′R). Therefore, the absolute configuration of 18 named chuktabularin Y was demonstrated as shown (Fig. 2).

Key correlations observed in the HMBC and NOESY spectra of 18
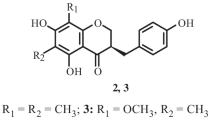
In addition to limonoids 1 and 18, 22 known compounds namely tabularisin J (2) [21], tabularisin K (3) [21], tabularisin A (4) [5], tabularisin B (5) [5], tabularisin C (6) [5], tabularisin D (7) [5], Encandollen A (8) [22, 23], procerin (9) [24], tabulalide G (10) [25], busseine H (11) [26], 3,30-diacetylphragmalin (12) [24], 3, 30- diisobutyroyl- 12- acetylphragmalin (13) [24], 3-diisobutyroyl-30-propanoyl-12-acetylphragmalin (14) [24], tabulalide C (15) [2], tabulain (16) [24], tabulalide H (17) [25], Chuktabularoid C (19) [6], Chuktabularin B (20) [17], tabulalide A (21) [25], swietemahonin A (22) [27], swietemahonin B (23) [27], humilin B (24) [28], have been separated and their structures were depicted in Fig. 2.
In conclusion, we have determined 17 phragmalin ortho esters (1–17), based on the position of the orthoacetate group, these limonoids included 1, 8, 9- (1, 8, 11–15, 17), 8, 9, 11- (2–5), 8, 9, 14- (9, 16), 8, 9, 30- (6–7, 10) phragmalin ortho esters, and 7 norphragmalin limonoids (18–24) from the bark of Chukrasia tabularis.
Anti-inflammatory assays
The antiinflammatory activities of the extract and fraction were studied against ear edema induced by xylene in mice. The results indicated that the dichloromethane extract of the plant showed effective antiinflammatory activity with inhibitory rate of 39.68% (800 mg/kg). The subfractions of dichloromethane extract FrC and FrD showed notable antiinflammatory activity with inhibitory value of 40.40% and 34.34% (400 mg/kg), respectively (Additional file 1: Table S1), so the isolation and purification focus on these two fractions.
Anti-inflammatory activity
The anti-infammatory activity of the limonoids (1–24) was determined in vitro in RAW 264.7 cells by evaluating production of NO induced by LPS. Cell viability determination indicated that limonoids (1–24) have no toxicity to RAW 264.7 cell at concentration of 100 μm. To determine whether limonoids (1–24) inhibited NO production in macrophages stimulated by LPS, the concentration of NO in the medium containing these limonoids was evaluated. Indomethacin (INM) was used as the positive control. As shown in Table 2, 24 limonoids showed antiinfammatory activities at the tested concentration. Limonoids 1, 4–6, 8–9, and 18–19 could significant inhibit NO production with the inhibition rate between 4.32 and 19.21 μm. Limonoids 2–3, 7, 10–17, and 20–24 exhibited potent anti-infammatory activities with the inhibition rate between 20.23 and 41.55 μm. Compound 18 showed the significant anti-inflammatory activity with IC50 value of 10.40 μm. Therefore, the potential antiinflammatory activities and molecular mechanism of compound 18 were further studied.
Effect of compound 18 on LPS-stimulated production of TNF-α, IL-6 and NO
NO, IL-6 and TNF-α are important inflammatory regulators produced in the process of inflammatory response, which can be produced and released in large quantities under condition of infection, injury and immune response [29]. Therefore, they are often used as indicators for evaluating antiinflammatory effect of compound. The antiinflammatory effect of compound 18 on LPS stimulated TNF-α, IL-6 and NO were determined. As shown in Fig. 5A–C, the addition of compound 18 notably inhibited production of TNF-α, IL-6 and NO with concentration dependent manner. The result showed that compound 18 could inhibit the expressions of NO, IL-6 and TNF-α in LPS-induced macrophages, and achieved antiinflammatory effect. The regulation of anti-inflammatory effect on macrophages may be partly involved in the protective effect of Chukrasia tabularis on inflammatory diseases.

Effects of compound 18 on the production of TNF-α, NO and IL-6 in LPS-induced macrophages. The macrophages were incubated with LPS (1 μg/mL) and treated with compound 18 (5.2 and 20.8 μmol/L) for 24 h. A The levels of TNF-α, B NO, and C IL-6 in the supernatant were assayed using ELISA kits. All values given are the mean ± SD. **p < 0.01 compared to the LPS group
Compound 18 blocked LPS-stimulated NF-κB activation
NF-κB signaling cascades play a key role in inflammation regulation [30, 31]. In order to explore the anti-inflammatory mechanism of compound 18, the expression of key molecules including NF-κB P65, p-P65, p-IKKα/β, and IKBα were detected by Western blot analysis. The result show that compound 18 down-regulates the expression of NF-κB P65 and p-P65 (Fig. 6A–C). In addition, compound 18 can also inhibit the phosphorylation and degradation of p-IKBα, and p-IKKα/β (Fig. 6A, D and E). These data demonstrate that compound 18 blocks the activation of the NF-κB signaling pathway induced by LPS.

Effect of compound 18 on inhibiting activation of the IKKβ/NF-κB signaling pathway induced by LPS. A Representative result of Western blot. Relative expression levels of p-P65 (B), P65 (C), p-IKBα (D), and p-IKKα/β (E). *p < 0.05, **p < 0.01 compared to model cells that treated by LPS. Statistical differences were analyzed by unpaired t-test
Compound 18 blocked LPS-stimulated STAT3 activation
The JAK2/STAT3 signaling pathway is involved in the transmission of multiple cytokines in inflammatory responses and is activated by interleukin and interferon [32]. To study the effects of compound 18 on the JAK2 and STAT3 signaling cascades in LPS-induced RAW264.7 cells, the levels of p-JAK2 and p- STAT3 were detected. As shown in Fig. 7A–C, compared to blank group, the expression level of p-STAT3 and p-JAK2 in the RAW264.7 cells was notably increased, while the expression in compound 18 group was notably reduced.

Effect of compound 18 on inhibiting activation of the JAK2/STAT3 signaling pathway induced by LPS. A Representative result of Western blot. Relative expression levels of p-JAK2 (B) and p-STAT3 (C). *p < 0.05, **p < 0.01 compared to model cells that treated by LPS. Statistical differences were analyzed by unpaired t-test
Cytotoxic activity
The cytotoxicity of all compounds on five human tumor cell lines MCF-7, HepG2, KB, Hela, and A-549 was evaluated by the SRB method (Additional file 1: Table S2). The phragmalin limonoid ortho esters, 1–2, 4–9, and 15 showed weak cytotoxicities against these tumor cell lines. While limonoids 3, 10–14, 16–22, and 24 exhibited no cytotoxicities against these tumor cell lines.
The results suggest that the ring-D-seco-phragmalin limonoid orthoester (limonoids 8 and 9) showed significant anti-inflammatory activity, phragmalin-type limonoids orthoesters with 13/14/18-cyclopropane rings (limonoids 1–7) exhibited potent anti-infammatory activity, norphragmalin limonoids (limonoids 20–24) had weaker effects than phragmalin limonoid ortho esters which indicated that the orthoester group plays a crucial role in the activity of phragmalin limonoids.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
References
Hay AE, Ioset JR, Ahua KM, Diallo D, Brun R, Hostettmann K (2007) Limonoid orthoacetates and antiprotozoal compounds from the roots of Pseudocedrela kotschyi. J Nat Prod 70(1):9–13
Nakatani M, Abdelgaleil SAM, Saad MMG, Huang RC, Iwagawa T (2005) Phragmalin limonoids from Chukrasia tabularis. Phytochemistry 65(20):2833–2841
Kaur R, Arora S (2009) Chemical constituents and biological activities of Chukrasia tabularis A. Juss.-A review. J Med Plants Res 3(4):196–216
Ogbole OO, Saka YA, Fasinu PS, Fadare AA, Ajaiyeoba EO (2016) Antimalarial and cytotoxic properties of Chukrasia tabularis A. Juss and Turraea vogelii Hook F. Ex. Benth. Parasitol Res 115:1667–1674
Fan CQ, Wang XN, Yin S, Zhang CR, Wang FD, Yue JM (2007) Tabularisins A-D, phragmalin ortho esters with new skeleton isolated from the seeds of Chukrasia tabularis. Tetrahedron 63(43):6741–6747
Wang CC, Li Y, Xu R, Zhang PP, Zhang WY, Wei SH, Li Y, Luo J, Kong LY (2019) Phragmalin-type limonoids with structural diversity at D-ring from the fruit shells of Chukrasia tabularis. Fitoterapia 134:188–195
Wu J, Xiao Q, Zhang S, Li X, Xiao Z, Ding H, Li Q (2005) Xyloccensins Q-V, six new 8,9,30-phragmalin ortho ester antifeedants from the Chinese mangrove Xylocarpus granatum. Tetrahedron 61(35):8382–8389
Liu JQ, Peng XR, Zhang WM, Shi L, Li XY, Chen JC, Qiu MH (2013) Swietemahalactone, a rearranged phragmalin-type limonoid with anti-bacterial effect, from Swietenia mahagoni. RSC Adv 3(15):4890–4893
Liu HB, Zhang H, Li P, Gao ZB, Yue JM (2012) Chukrasones A and B: potential Kv1.2 potassium channel blockers with new skeletons from Chukrasia tabularis. Org Lett 14(17):4438–4441
Luo J, Wang JS, Luo JG, Wang XB, Kong LY (2011) Velutabularins A-J, phragmalin-type limonoids with novel cyclic moiety from Chukrasia tabularis var. velutina. Tetrahedron 67(16):2942–2948
Senthil Kumar KJ, Wang SY (2009) Lucidone inhibits iNOS and COX-2 expression in LPS-induced RAW 264.7 murine macrophage cells via NF-κB and MAPKs signaling pathways. Planta Med 75(5):494–500
Vichai V, Kirtikara K (2006) Sulforhodamine B colorimetric assay for cytoxicity screening. Nat Protoc 1(3):1112–1116
Li LC, Pan ZH, Ning DS, Fu YX (2020) Anti-inflammatory effect of simonsinol on lipopolysaccharide stimulated RAW264.7 cells through inactivation of NF-κB signaling pathway. Molecules 25(16):3573
Wu CC, Lu YH, Wei BL, Yang SC, Won SJ, Lin CN (2008) Phloroglucinols with prooxidant activity from Garcinia subelliptica. J Nat Prod 71(2):246–250
Winkelmann K, Heilmann J, Zerbe O, Rali T, Sticher O (2000) New phloroglucinol derivatives from Hypericum papuanum. J Nat Prod 63(1):104–108
Luo J, Wang JS, Wang XB, Huang XF, Luo JG, Kong LY (2009) Chukvelutilides A–F, phragmalin limonoids from the stem barks of Chukrasia tabularis var. velutina. Tetrahedron 65(17):3425–3431
Zhang CR, Yang SP, Liao SG, Fan CQ, Wu Y, Yue JM (2008) Chuktabularins A-D, four new limonoids with unprecedented carbon skeletons from the stem bark of Chukrasia tabularis. Org Lett 9(17):3383–3386
Zhao S, Yan X, Zhao Y, Wen J, Zhao Z, Liu H (2018) Dihydroisocoumarins from Radix glycyrrhizae. BMC Chem 12(58):1–6
Michalska K, Bednarek E, Gruba E, Lewandowska K, Mizera M, Cielecka-Piontek J (2017) Comprehensive spectral identification of key intermediates to the final product of the chiral pool synthesis of radezolid. BMC Chem 11(82):1–16
Luo J, Wang JS, Luo JG, Wang XB, Kong LY (2009) Chukvelutins A-C, 16-norphragmalin limonoids with unprecedented skeletons from Chukrasia tabularis var. velutina. Org Lett 11(11):2281–2284
Kitagawa T, Matsumoto T, Imahori D, Kobayashi M, Okayama M, Ohta T, Yoshida T, Watanabe T (2021) Limonoids isolated from the fortunella crassifolia and the citrus junos with their cell death-inducing activity on adriamycin-treated cancer cell. J Nat Med 65(1):1–7
Zhang CR, Yang SP, Chen XQ, Wu Y, Zhen XC, Yue JM (2008) Limonoids from the twigs and leaves of Chukrasia tabularis. Helv Chim Acta 91(12):2338–2350
Quasie O, Li H, Luo J, Kong LY (2017) Two new phragmalin-type limonoids orthoesters from Entandrophragma candollei. Chin J Nat Med 15(9):680–683
Narender T, Khaliq T, Shweta, (2008) 13C NMR spectroscopy of D and B, D-ring seco-limonoids of Meliaceae family. Nat Prod Res 22(9):763–800
Luo J, Wang JS, Wang XB, Luo JG, Kong LY (2011) Phragmalin-type limonoid orthoesters from Chukrasia tabularis var. velutina. Chem Pharm Bull 59(2):225–230
Guex M, Tamm C (1984) Die Busseine C, D, E, F, G, H, J, K, L und M, zehn neue Tetranortriterpene aus Entandrophragma bussei Harms. Helv Chim Acta 67(3):885–901
Kadota S, Marpaung L, Kikuchi T, Ekimoto H (1990) Constituents of the seeds of Swietenia mahagoni JACQ. II. Structures of swietemahonin A, B, C, D, E, F, and G and swietemahonolide. Chem Pharm Bull 38(4):894–901
Jimenez A, Villarreal C, Toscano RA, Cook M, Mata R (1998) Limonoids from Swietenia humilis and Guarea grandiflora (Meliaceae). Phytochemistry 49(7):1981–1988
Schuerwegh A, Dombrecht E, Stevens W, Van J, Bridts C, De L (2003) Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthr Cartilage 11(9):681–687
Staal J, Bekaert T, Beyaert R (2011) Regulation of NF-κB signaling by caspases and MALT1 paracaspase. Cell Res 21:40–54
Fallahi-Sichani M, Kirschner D, Linderman J (2012) NF-κB signaling dynamics play a key role in infection control in tuberculosis. Front Physiol 3:170
Xiong X, Huang C, Wang F, Dong J, Zhang D, Jiang J, Feng Y, Wu B, Xie T, Cheng L (2020) Qingxue jiedu formulation ameliorated DNFB-induced atopic dermatitis by inhibiting STAT3/MAPK/NF-κB signaling pathways. J Ethnopharmacol 270:113773
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (41806173), the Natural Science Foundation of Fujian Province (2020J01619, 2016J01369 and 2018J01847), the Open Project of National Marine Bureau Key Laboratory of Marine Biogenetic Resources (HY201807, HY201604), and the Science and Technology Innovation Platform Project of Fujian Province (2018Y2001).
Author information
Authors and Affiliations
Contributions
JS, YZ, NY designed the experiments. YZ, NY, XM completed the isolation and elucidation the structures. JS, YZ, NY tested cytotoxicity and anti-inflammatory effects of the compounds. JS, YZ interpreted the data and wrote the paper. YZ, NY, XM, TZ revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experiments involving the use of animals have been approved by the Institutional Animal Protection and Use Committee of Fujian Medical University (Approval No. 2017-0102).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Inhibitory effect of Chukrasia tabularis on ear edema induced by xylene in mice. Table S2. Cytotoxicity of 1–24 against tested cell lines. Figure S1. 1H NMR spectrum (400 MHz) of in DMSO-d6. Figure S2. 13C NMR spectrum (100 MHz) of 1 in DMSO-d6. Figure S3. HMQC spectrum of 1. Figure S4. HMBC spectrum of 1. Figure S5. 1H-1H COSY spectrum of 1. Figure S6. NOESY spectrum of 1. Figure S7. HRESIMS spectrum of 1. Figure S8. 1H NMR spectrum (400 MHz) of 18 in CD3OD. Figure S9. 13C NMR spectrum (100 MHz) of 18 in CD3OD. Figure S10. HMQC spectrum of 18. Figure S11. HMBC spectrum of 18. Figure S12. 1H-1H COSY spectrum of 18. Figure S13. NOESY spectrum of 18. Figure S14. HRESIMS spectrum of 18. Figure S15. HPLC analysis of FrC6-2, FrD5-2, FrD6, FrD7 and FrD9 (at 210 nm).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, J., Zhang, Y., Yang, N. et al. Limonoids from the barks of Chukrasia tabularis and their anti-inflammatory activity. Appl Biol Chem 65, 7 (2022). https://doi.org/10.1186/s13765-022-00674-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-022-00674-1