
Abstract
Background
Antimicrobial resistance (AMR) is a critical threat to human health. Escherichia coli and Klebsiella pneumoniae are clinically the most important species associated with AMR and are the most common carbapenemase-producing (CP) Enterobacterales detected in human specimens in Finland. Wastewater surveillance has emerged as a potential approach for population-level surveillance of AMR, as wastewater could offer a reflection from a larger population with one sample and minimal recognized ethical issues. In this study, we investigated the potential of wastewater surveillance to detect CP E. coli and K. pneumoniae strains similar to those detected in human specimens.
Methods
Altogether, 89 composite samples of untreated community wastewater were collected from 10 wastewater treatment plants across Finland in 2021–2022. CP E. coli and K. pneumoniae were isolated using selective culture media and identified using MALDI-TOF MS. Antimicrobial susceptibility testing was performed using disk diffusion test and broth microdilution method, and a subset of isolates was characterized using whole-genome sequencing.
Results
CP E. coli was detected in 26 (29.2%) and K. pneumoniae in 25 (28.1%) samples. Among E. coli, the most common sequence type (ST) was ST410 (n = 7/26, 26.9%), while ST359 (n = 4/25, 16.0%) predominated among K. pneumoniae. Globally successful STs were detected in both E. coli (ST410, ST1284, ST167, and ST405) and K. pneumoniae (ST512, ST101, and ST307). K. pneumoniae carbapenemases (KPC) were the most common carbapenemases in both E. coli (n = 11/26, 42.3%) and K. pneumoniae (n = 13/25, 52.0%), yet also other carbapenemases, such as blaNDM-5, blaOXA-48, and blaOXA-181, were detected. We detected isolates harboring similar ST and enzyme type combinations previously linked to clusters in Finland, such as E. coli ST410 with blaKPC-2 and K. pneumoniae ST512 with blaKPC-3.
Conclusions
Our study highlights the presence of clinically relevant strains of CP E. coli and K. pneumoniae in community wastewater. The results indicate that wastewater surveillance could serve as a monitoring tool for CP Enterobacterales. However, the specificity and sensitivity of the methods should be improved, and technologies, like advanced sequencing methods, should be utilized to distinguish data with public health relevance, harness the full potential of wastewater surveillance, and implement the data in public health surveillance.
Similar content being viewed by others

Background
Antimicrobial resistance (AMR) is a significant threat to global health [1]. In 2019 alone, AMR was estimated to be responsible for approximately 4.95 million deaths [2]. Escherichia coli and Klebsiella pneumoniae, among the leading pathogens associated with AMR, are of particular concern. These species belonged to the top five bacterial pathogens responsible for infection-related deaths in 2019, each responsible for over 500 000 deaths globally [2, 3]. They both rank also in the top three bacterial species causing the largest burden of disease estimated by the European Centre for Disease Prevention and Control (ECDC) [4]. E. coli and K. pneumoniae are Gram-negative opportunistic pathogens belonging to Enterobacterales and are part of the normal microbiota in human and animal gastrointestinal tracts [5, 6]. Additionally, they are found in fecally contaminated environmental sources like soil and water [6, 7].
Carbapenem resistance, emerging in Enterobacterales [8], is a critical threat to human health [9]. Generally, the resistance percentage in all bacterial species in Finland is low (in 2021: 6.4%) [10], and none of the E. coli or K. pneumoniae isolated from blood was resistant to meropenem in 2021 [11]. However, the number of detected carbapenem-resistant isolates from different sample types has increased in recent years [12], and the emergence of carbapenem resistance is a worrisome threat also in Finland. As in the global situation, E. coli and K. pneumoniae are the most common carbapenemase-producing Enterobacterales (CPE) in Finland, comprising 48% and 45% of CPE isolates in human specimens in 2022, respectively [12]. Certain sequence types (ST) of E. coli and K. pneumoniae, known as globally dominant STs, have considerable clinical relevance, i.e., are more frequently detected in clinical samples. Globally dominant STs of Carbapenemase-producing (CP) E. coli (e.g., ST410, ST131, ST1284, ST167, and ST405) and CP K. pneumoniae (e.g., ST512, ST437, ST258, ST11, ST15, ST101, ST307, and ST147) [13,14,15] have been detected in human specimens in Finland [12, 16, 17].
The dissemination of carbapenemase-encoding genes is a major concern, as they have the potential to rapidly spread within and between bacterial species due to their frequent location on plasmids [18]. K. pneumoniae carbapenemases (KPC) are particularly successful in this regard [19]. In Finland, KPC are the most common carbapenemases found in human specimens [12], with blaKPC-3 being the most prevalent type in 2012–2018 [16]. However, plasmid-mediated New Delhi metallo-β-lactamases (NDM) and oxacillinase-48-type carbapenemases (OXA-48-like) are also widespread and common in Northern Europe, including Finland [12, 20].
The ongoing but evolving nature of AMR and the lack of clear epidemic peaks, as detected in viral pandemics, often lead to its oversight. Hence, AMR is commonly referred to as the silent pandemic [21]. Current AMR surveillance primarily focuses on bacteria that cause healthcare-associated infections and aims to detect the potential threats these bacteria could create to the population. Consequently, this could result in poorly monitored and understood AMR prevalence in the healthy population [22], and relying only on current AMR surveillance data could lead to potential biases from the population surveillance perspective [23]. To address this gap, alternative options for AMR surveillance at the population level, such as wastewater surveillance (WWS), have been explored [23]. WWS offers the potential for population-level assessment and survey of AMR while avoiding the ethical issues associated with sampling of individuals [24]. However, it is important to understand that AMR and AMR-related genes, both intrinsic and acquired, occur in multiple bacterial species, including clinically less relevant species that are abundant in wastewater [25].
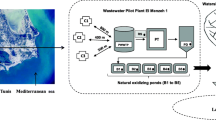
In this study, we focused on evaluating the occurrence and characteristics of carbapenemase-producing (CP) E. coli and K. pneumoniae in Finnish community wastewater influent. There has been an increasing recognition of various CP organisms globally. However, CP E. coli and K. pneumoniae are the most prevalent CP organisms in clinical samples in Finland, having a high clinical relevance. With a combination of culture-based and molecular methods, we investigated the phenotypical and genotypical features of CP E. coli and K. pneumoniae in 89 wastewater samples collected before any treatment from ten wastewater treatment plants (WWTP) across Finland during 2021–2022 (Fig. 1).

Illustration of the locations of the included wastewater treatment plants (WWTP), the timeline of the sample collection, and the workflow of the study. CP, carbapenemase-producing. WGS, whole-genome sequencing. 1One sample missing.2If applicable
Methods
Sample collection, bacterial isolation, and quantitation of preliminary CP E. coli
A total of 89 samples of community wastewater influent were collected from 10 different WWTP across Finland between February 2021 and February 2022. The included WWTP serve around 40% of the Finnish population and are distributed across the country [26]. Among these samples, 86 were 24-h composite samples, while, due to sporadic issues with composite collectors, one was a 4-h and one 6-h composite sample, and one was a grab sample (Table 1). The wastewater collection was conducted as part of the “WastPan” consortium project [26]. A 1 L sample was delivered to the laboratory in a cold container with ice packs and processed within 24 h of collection.
In the laboratory, a serial dilution in buffered peptone water (BPW) (Oxoid, Basingstoke, Hampshire, United Kingdom) was prepared using dilutions of 10–1, 10–2, and in summer months also 10–3. An aliquot of 100 µL from the undiluted sample and each dilution was plated on individual CHROMagar mSuperCARBA (CHROMagar, Paris, France) plates and incubated aerobically for 18–24 h at 37 °C. The colony morphology was observed following the manufacturer’s instructions, and up to 12 colonies showing different colony morphology were selected. The main objective was to identify E. coli and K. pneumoniae, typically showing dark pink to reddish and blue colonies, respectively. An additional objective was to identify other bacteria belonging to ESKAPE-E [27], including Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter cloacae, showing typically cream, translucent, and blue colonies, respectively. The colonies were subcultured with a 1 µL sterile loop on CHROMagar mSuperCARBA and incubated aerobically for 18–24 h at 37 °C until a pure culture was obtained. Lastly, isolates were subcultured on bovine blood agar plates (Columbia Blood Agar Base, Oxoid Ltd., Basingstoke, United Kingdom) and incubated aerobically for 18–24 h at 37 °C for further characterization.
The quantitation of preliminary CP E. coli was conducted by counting the colonies showing the characteristic appearance of E. coli (dark pink to reddish) on plates containing 10–100 typical colonies. From each sample, five typical colonies were selected and subcultured to achieve pure cultures, following the procedure described above (Fig. 1).
Bacterial species identification
Isolates were identified with a matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) -based Bruker Microflex LT/SH (Bruker Daltonics GmbH & Co. KG, Bremen, Germany). A score value of > 2.0 was considered high confidence, as per the manufacturer’s instructions, and set as the criterion.
Antimicrobial susceptibility testing
Susceptibility to carbapenems was tested for all presumptive CP E. coli and K. pneumoniae isolates with meropenem (10 µg) (Abtek Biologicals Ltd, Liverpool, United Kingdom) and ertapenem (10 µg) (Oxoid, Basingstoke, United Kingdom) with a disk diffusion test according to the EUCAST (European Committee of Antimicrobial Susceptibility Testing) standard [28]. Furthermore, antimicrobial susceptibility testing was performed with the broth microdilution method using Sensititre EURGNCOL plates (Thermo Fischer Scientific, East Grinstead, United Kingdom) to determine the minimum inhibitory concentration (MIC) of colistin, piperacillin/tazobactam, ceftazidime/avibactam, ceftolozane/tazobactam, and meropenem. The method was performed according to the manufacturer’s instructions, except for using 0.9% saline instead of sterile water. E. coli ATCC 25922 was included as a quality control for each patch of Müeller-Hinton agars. The results were interpreted according to EUCAST epidemiological cut-off values (ECOFFs) [29].
DNA extraction and whole-genome sequencing (WGS)
In total, 26 E. coli and 25 K. pneumoniae isolates were subjected to whole-genome sequencing (WGS). One isolate of each species from each city on each sampling month was chosen, if applicable. If multiple isolates were detected, the selection criteria were as follows: (1) isolate with the highest MIC value for meropenem, (2) isolate with the smallest inhibition zone for meropenem, and (3) isolate that was first selected from the primary agar plate. Strains were grown in Tryptone Soya Broth (Oxoid, Basingstoke, United Kingdom) at 37 °C for 16 h and the DNA was extracted from cells harvested from 1 mL of culture by using QIAcube Connect instrument (QIAGEN, Hilden, Germany) with DNeasy Blood & Tissue kit (QIAGEN, Valencia, CA, USA). The quality of DNA was assessed by using a NanoDrop ND-1000 spectrophotometer (Thermo Fischer Scientific, Wilmington, DE, USA) based on a 260/280 ratio. DNA quantity was measured using a Qubit 2.0 fluorometer (Invitrogen, Life Technologies, Carlsbad, CA, USA). Library preparation was performed with a NEBNext Ultra DNA Library Prep Kit for Illumina with 300 bp fragment length. Sequencing was performed with Illumina NovaSeq 6000 (outsourced to Novogene, Cambridge, United Kingdom) with targeted genomic coverage of 100 × and 2 × 150 bp read length.
Bioinformatical analyses
All (n = 51) sequenced isolates were analyzed with Ridom SeqSphere + software v7.7.5 (Ridom GmbH, Germany) [30]. Quality analysis of the sequences was performed with FastQC v0.1.1.7 [31] and adapters were removed with Trimmomatic v0.36 [32]. Raw reads were assembled with SKESA v2.3.0 using default settings [33], and quality trimming was performed with an average quality of ≥ 30 and a window of 20 bases. Remapping and polishing were performed with the BWA-MEM mapping algorithm. Sequencing statistics are presented in Additional file 1. Acquired AMR genes were identified from assembled genomes with NCBI AMRFinderPlus 3.2.3 [34], using 100% alignment and > 90% identity. STs were analyzed by using multilocus sequence types (MLST) [35] in Ridom SeqSphere + (Ridom, Munster, Germany). Warwick MLST scheme was chosen for E. coli isolates. E. coli isolates with novel STs were submitted to Enterobase [36] and K. pneumoniae isolates to Institut Pasteur [37, 38] to assign new STs. Phylogenetic analysis was conducted for all E. coli and K. pneumoniae isolates with core genome multilocus sequence typing (cgMLST) by comparing 2513 and 2365 alleles with pairwise missing values, respectively. A cluster threshold was determined by 10 allelic differences [39].
Results
Detection of E. coli and K. pneumoniae with reduced susceptibility to carbapenems
In total, 50 E. coli isolates from 26 wastewater samples (n = 26/89, 29.2%) and 44 K. pneumoniae isolates from 25 wastewater samples (n = 25/89, 28.1%) were recovered from CHROMagar mSuperCARBA during the study period (Table 1). Up to five E. coli and four K. pneumoniae isolates were recovered in each sample. An additional 52 species were identified, including A. baumannii and E. cloacae, but not P. aeruginosa (Additional file 2).
Quantity of preliminary CP E. coli was under the detection limit in 62 samples (n = 62/89, 69.7%) and peaked at 6.0 × 102 colony forming units/mL (Additional file 3).
Antimicrobial susceptibility
Disk diffusion test and broth microdilution were performed for all E. coli and K. pneumoniae isolates. In total, 44 E. coli (n = 44/50, 88.0%) and 43 K. pneumoniae (n = 43/44, 97.7%) isolates were considered resistant against meropenem according to the broth microdilution method. Phenotypical resistance against colistin was expressed by four E. coli (n = 4/50, 8.0%) and eleven K. pneumoniae (n = 11/44, 25.0%) isolates. Distributions of MICs are presented in Table 2. MIC values and inhibition zones for individual isolates are presented in Additional file 4.
Multilocus sequence types, antimicrobial resistance genes, and phylogenetics
In total, 14 different STs of E. coli and 14 of K. pneumoniae were identified. In E. coli, the most prevalent was ST410 (n = 7/26, 26.9%), followed by ST401 (n = 3/26, 11.5%) and ST607 (n = 3/26, 11.5%). In K. pneumoniae, the most prevalent was ST359 (n = 4/25, 16.0%), followed by ST512 (n = 3/25, 12.0%) and ST307 (n = 3/25, 12.0%) (Fig. 2A and 2B).

Distribution of sequence types and carbapenemase genes in carbapenemase-producing (CP) Escherichia coli (n = 26) and Klebsiella pneumoniae (n = 25) isolates. Geographical distribution of sequence types (ST) across the studied wastewater treatment plants (WWTP), distribution of carbapenemase genes in different STs and detection time of Carbapenemase-producing (A) Escherichia coli and (B) Klebsiella pneumoniae. The size of the circle reflects the number of isolates. Location tick marks on the map indicate that no isolates were detected in the corresponding WWTP. Isolate identification numbers (ID) are indicated (e.g., E2). ST, sequence type
In total, 23 E. coli (n = 23/26, 88.5%) and 18 K. pneumoniae (n = 18, 72.0%) isolates were confirmed to carry carbapenemase-encoding genes. In E. coli, the most prevalent was blaKPC-2 (n = 6/26, 23.1%), followed by blaKPC-3 (n = 5/26, 19.2%) and blaNDM-5 (n = 4/26, 17.4%). In K. pneumoniae, the most prevalent was blaKPC-3 (n = 11/25, 44.0%), followed by blaOXA-48 (n = 4/25, 16.0%) (Fig. 2A and 2B).
All isolates carried at least one additional beta-lactamase gene, including Extended-Spectrum Beta-Lactamases such as blaCTX-M-15, blaCTX-M-14, and blaSHV-27 (Fig. 3). All K. pneumoniae isolates and 22 (84.6%) of the E. coli isolates were multidrug resistant (i.e., harbored resistance genes to at least one agent in three or more antimicrobial categories [40]). Known genes related to colistin resistance (mcr) were not found.

Heatmap of the presence of antimicrobial resistance genes (dark blue) in whole-genome sequenced isolates of carbapenemase-producing Escherichia coli (n = 26, indicated with Isolate IDs E2–E50) and Klebsiella pneumoniae isolates (n = 25, indicated with Isolate IDs K1–K44) from wastewater treatment plants across Finland (n = 10). The dendrogram is based on the similarity of resistance gene profiles between the isolates
CgMLST revealed closely related strains (< 10 allele difference [39]) in both E. coli and K. pneumoniae. Closely related strains were detected only in samples from the same WWTP (Additional file 5).
Discussion
In this study, we describe the phenotypic and genomic characteristics of CP E. coli and K. pneumoniae isolated from community wastewater influent in ten cities across Finland. We demonstrate the presence of clinically relevant STs and enzyme types of CP E. coli and K. pneumoniae known to be carried in the population. The results indicate that WWS has the potential to monitor CPE in the population.
In total, 14 different STs of both E. coli and K. pneumoniae were identified in wastewater. Notably, we detected dominant global STs of CP E. coli, such as ST410, ST1284, ST167, and ST405 [15], and CP K. pneumoniae, such as ST512, ST15, ST101, and ST307 [14]. Furthermore, we identified several other STs of both E. coli and K. pneumoniae, some of which have been previously detected in human specimens in Finland (e.g., E. coli ST345, ST540, ST617, ST744, and ST1485 and K. pneumoniae ST37) (Kati Räisänen, personal communication). The carbapenemases identified in wastewater isolates closely resembled those found in human specimens in Finland, with blaKPC-2 and blaKPC-3 being the most prevalent in wastewater. We also detected ST and enzyme type combinations previously linked to clusters in Finland, including E. coli ST410 with blaKPC-2 and K. pneumoniae ST512 with blaKPC-3 [12, 16]. Notably, certain ST and enzyme type combinations found in wastewater, such as E. coli ST1284 with blaNDM-5 in Turku and K. pneumoniae ST101 with blaOXA-48 in Kuopio, have earlier been identified in human specimens from the respective regions (Kati Räisänen, personal communication). Some ST and enzyme type combinations were recurrent in the same WWTP, and some globally dominant STs, E. coli ST410 and K. pneumoniae ST512 and ST307, were detected in multiple locations. Only some of the recurrent STs in the same WWTP were closely related (< 10 allele difference), whereas in some cases, the isolates belonging to the same ST were phylogenetically distinct. Closely related strains in the same WWTP could originate from one source that is persistently excreting the strain to the wastewater, be a result of persisting strain in the wastewater, or be related to an undetected, potentially local, outbreak in the population. The globally dominant STs between multiple WWTP were genetically distinct, and their occurrence in multiple WWTP could be a consequence of their prevalence in the population. Strains belonging to the epidemiological clusters of E. coli ST410 and K. pneumoniae ST512 in Finland and an additional E. coli ST410 strain distinct from the cluster, were reported in human specimens during the wastewater sampling in 2021–2022 (Kati Räisänen, personal communication). The wastewater strains of E. coli ST410 and K. pneumoniae ST512 are unlikely linked to at least a single outbreak, as the strains were genetically distinct. However, these strains may represent the diversity of strains circulating in the population. The diversity of strains can be a result of a distinct epidemiological origin of the strains or a natural genetical shift happening in the bacterial population over time [39].
We did not identify known carbapenemases in three E. coli and seven K. pneumoniae isolates. Resistance mechanisms other than carbapenemase production, such as the loss of outer membrane porins and increased expression of efflux pumps, could contribute to the reduced susceptibility to carbapenems [41]. Furthermore, some isolates may have expressed novel carbapenemases. The limited number of sequenced isolates does not reveal the full diversity of possible STs. Hence, the representation of ST distribution across WWTP may have been biased. Furthermore, strains expressing weaker carbapenemases may be more susceptible to meropenem, and as certain carbapenemases co-occur more commonly with designated STs [15, 42], the selection criteria for sequencing (phenotypical resistance to meropenem) may have influenced the results.
Understanding AMR at the population level and the potential of WWS to act as an early warning tool are the key possibilities of WWS from the public health perspective [23]. Clinical AMR surveillance is crucial for prevention measures in healthcare settings and guiding the treatment of patients. However, clinical surveillance is not particularly suitable for providing an unbiased picture of AMR in the healthy population since the samples are gained from a limited number of individuals who are usually attributed to healthcare and may have a higher probability of carrying AMR bacteria. WWS aims to provide a population-level view with a sample from a larger and more heterogeneous population [23]. Optimally, WWS could assess the incidence and prevalence of CPE in the population and produce descriptive data about the isolates. Moreover, longitudinal and continuous quantitation could reveal the potential trends of CPE occurrence in the population. Here, quantitation was performed with the colony-forming unit (CFU) method, which has limitations, especially when the number of bacteria is low. The method could be further optimized in the future, for example, by accompanying it with molecular methods like qPCR [43]. Describing the STs and enzyme types of the CPE isolates and establishing a phylogenetic comparison scheme (e.g., cgMLST) with CPE isolates from clinical samples and wastewater could reveal the fluctuation of different STs and potential epidemics, especially in long-term surveillance. Quantitative and descriptive WWS could offer valuable early-warning data for healthcare operatives and act as a rationale to increase the clinical surveillance or infection control measures in healthcare settings on a local level. CP E. coli and K. pneumoniae were present in approximately one-third of the wastewater samples. The prevalence of CPE in the healthy Finnish population is currently unknown, but as CPE is rarely detected in clinical samples, the prevalence is presumably low [12, 44]. Estimating the CPE carriage in the Finnish population with our limited wastewater data is challenging, and the proportion of positive wastewater samples and prevalence in humans in Finland are not directly comparable. For example, some of the detected isolates may have been originally carbapenem-sensitive and have received resistance genes in wastewater through horizontal gene transfer [45]. Furthermore, the dynamics, e.g., the persistence and survival, of clinically relevant AMR bacteria in sewerage systems are not well known, and various factors, such as method sensitivity, unique microbial communities in sewerage systems, and WWTP, as well as conditions in wastewater, such as fluctuating temperature, limited nutrient availability, and chemicals [46], can affect the presence and abundance of CPE in wastewater. These factors impede the assessment of the prevalence of AMR carriage in the community through WWS. The assessment may be less complicated if surveillance is recurrent and long-term, as the identification of trends and epidemiological spikes could be more straightforward [21, 47].
Wastewater is a complex material containing bacteria from various sources [48], and the microbiome can hinder the sensitivity of WWS to detect clinically relevant AMR bacteria. In addition to STs related to humans, we described E. coli STs linked both in human and non-human reservoirs (ST137, ST540, ST617, and ST744), animal reservoirs (ST345), environment (ST607), and wastewater systems (ST746 and ST401) [46, 49,50,51]. Differentiating the bacteria originating from human and non-human sources and further discriminating isolates that have public health relevance is one of the challenges of WWS. Future research should address this challenge by exploring different methods, for example, the potential of utilizing the quantitation of crAssphage, a bacteriophage abundant in the human gut and used in the interpretation of data from WWS of viruses [52]. Furthermore, we identified a diverse array of species in the wastewater, reflecting the complex microbiome of wastewater, which can hamper the identification of the targeted or relevant species. While the culture-based approach provides in-depth knowledge of bacterial STs in wastewater, it requires extensive culturing and utilization of molecular methods (PCR or WGS) to identify all relevant bacterial STs and AMR genes. Continually evolving methodologies, such as culture-enriched metagenomics, HI-C ligation, deep- and long-read sequencing, and single-cell metagenomics, offer potential solutions for these challenges [53,54,55,56]. These methodologies, accompanied by artificial intelligence tools, could help to produce population-level data on key infectious agents and their resistance profiles and help to enhance public health security. However, implementing these methodologies requires specialized expertise and extensive resources that may not yet be readily available at the local level, thereby limiting their accessibility. In contrast, the culture-based approach is widely accessible and comparatively low-cost [57], making it a noteworthy option for AMR surveillance, particularly in resource-limited settings.
The selection of approaches and methods in WWS should be guided by specific study objectives, as different approaches provide distinct information about AMR genes, species taxonomy, or bacterial STs and their characteristics. For instance, a gene-based approach may not be optimal for estimating CPE occurrence in the community but could be well-suited for evaluating AMR gene reservoirs or assessing wastewater treatment efficiency. While a culture-based approach alone may not be able to uncover trends in short-term surveillance, it provides information about the epidemiology, abundance, and strain characteristics that could be valuable for health officials or researchers.
Conclusions
In conclusion, WWS has the potential to monitor CPE in the population. WWS could provide valuable information about the key infectious agents and their resistance profiles at a population level, which offers valuable data from a public health perspective. Furthermore, WWS could identify the geographical hotspots of AMR and guide potential interventions. However, the interpretation of WWS data and the estimations of how well wastewater samples reflect CPE occurrence in the community require further improvements, including enhanced methodologies and optimally also broad screening of CPE in the healthy Finnish population.
Availability of data and materials
The data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB64775 (https://www.ebi.ac.uk/ena/browser/view/PRJEB64775). Isolate accession numbers are provided in Additional file 1.
Abbreviations
- AMR:
-
Antimicrobial resistance
- BPW:
-
Buffered peptone water
- CFU:
-
Colony-forming unit
- cgMLST:
-
Core genome multilocus sequence typing
- CP:
-
Carbapenemase-producing
- CPE:
-
Carbapenemase-producing Enterobacterales
- ECDC:
-
European centre for disease prevention and control
- KPC:
-
Klebsiella pneumoniae carbapanemases
- MALDI-TOF MS:
-
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- MIC:
-
Minimum inhibitory concentration
- MLST:
-
Multilocus sequence typing
- NDM:
-
New Delhi metallo-β-lactamases
- OXA-48-like:
-
Oxacillinase-48-type carbapenemases
- ST:
-
Sequence type
- WGS:
-
Whole-genome sequencing
- WWS:
-
Wastewater surveillance
- WWTP:
-
Wastewater treatment plants
References
WHO. WHO: Antimicrobial resistance. https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance. Accessed 1 Sept 2023.
Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet. 2022;399(10325):629–55.
Ikuta KS, Swetschinski LR, Robles Aguilar G, Sharara F, Mestrovic T, Gray AP, et al. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. The Lancet. 2022 Nov;
European Centre for Disease Prevention and Control. Assessing the health burden of infections with antibiotic-resistant bacteria in the EU/EEA, 2016–2020. Stockholm, 2022. Doi: https://doi.org/10.2900/73460. Accessed 1 Sep 2023.
Conlan S, Kong HH, Segre JA. Species-Level Analysis of DNA Sequence Data from the NIH Human Microbiome Project. PLoS One. 2012;7(10).
Van Elsas JD, Semenov A V., Costa R, Trevors JT. Survival of Escherichia coli in the environment: Fundamental and public health aspects. Vol. 5, ISME J. 2011.
Melo-Nascimento AO dos S, Treumann C, Neves C, Andrade E, Andrade AC, Edwards R, et al. Functional characterization of ligninolytic Klebsiella spp. strains associated with soil and freshwater. Arch Microbiol. 2018;200(8).
Tilahun M, Kassa Y, Gedefie A, Ashagire M. Emerging carbapenem-resistant enterobacteriaceae infection, its epidemiology and novel treatment options: A review. Vol. 14, Infection and Drug Resistance. 2021.
Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18(3).
THL. Bakteerien mikrobilääkeresistenssi Suomessa – Finres 2021. Helsinki, 2022. https://urn.fi/URN:ISBN:978-952-343-920-7. Accessed 25 Sep 2023.
European Centre for Disease Prevention and Control and World Health Organization. Antimicrobial resistance surveillance in Europe 2023 - 2021 data. Stockholm, 2023. Doi: https://doi.org/10.2900/63495. Accessed 25 Sep 2023.
THL. CPE-esiintyvyys Suomessa. 2023. https://thl.fi/fi/web/infektiotaudit-ja-rokotukset/taudit-ja-torjunta/taudit-ja-taudinaiheuttajat-a-o/cpe/cpe-esiintyvyys-suomessa. Accessed 24 May 2023.
Peirano G, Chen L, Kreiswirth BN, Pitout JDD. Emerging Antimicrobial-Resistant High-Risk Klebsiella pneumoniae Clones ST307 and ST147. Antimicrob Agents Chemother. 2020 Sep 21;64(10).
Wyres KL, Holt KE. Klebsiella pneumoniae population genomics and antimicrobial-resistant clones. Trends Microbiol. 2016;24(12):944–56.
Peirano G, Chen L, Nobrega D, Finn TJ, Kreiswirth BN, DeVinney R, et al. Genomic epidemiology of global carbapenemase-producing Escherichia coli, 2015–2017. Emerg Infect Dis. 2022 May;28(5).
Räisänen K, Lyytikäinen O, Kauranen J, Tarkka E, Forsblom-Helander B, Grönroos JO, et al. Molecular epidemiology of carbapenemase-producing Enterobacterales in Finland, 2012–2018. European Journal of Clinical Microbiology and Infectious Diseases. 2020;39(9).
Linkevicius M, Bonnin RA, Alm E, Svartström O, Apfalter P, Hartl R, et al. Rapid cross-border emergence of NDM-5-producing Escherichia coli in the European Union/European Economic Area, 2012 to June 2022. Eurosurveillance. 2023 May 11;28(19).
Andrade LN, Curiao T, Ferreira JC, Longo JM, Clímaco EC, Martinez R, et al. Dissemination of bla KPC-2 by the Spread of Klebsiella pneumoniae Clonal Complex 258 Clones (ST258, ST11, ST437) and Plasmids (IncFII, IncN, IncL/M) among Enterobacteriaceae Species in Brazil. Antimicrob Agents Chemother. 2011;55(7):3579–83.
Queenan AM, Bush K. Carbapenemases: the versatile β-lactamases. Vol. 20, Clinical Microbiology Reviews. 2007.
Logan LK, Weinstein RA. The epidemiology of carbapenem-resistant enterobacteriaceae: the impact and evolution of a global menace. J Infect Dis. 2017;215(1):S28-36.
Laxminarayan R. The overlooked pandemic of antimicrobial resistance. The Lancet. 2022;399(10325):606–7.
Reinthaler FF, Galler H, Feierl G, Haas D, Leitner E, Mascher F, et al. Resistance patterns of Escherichia coli isolated from sewage sludge in comparison with those isolated from human patients in 2000 and 2009. J Water Health. 2013;11(1):13–20.
Chau KK, Barker L, Budgell EP, Vihta KD, Sims N, Kasprzyk-Hordern B, et al. Systematic review of wastewater surveillance of antimicrobial resistance in human populations. Vol. 162, Environment International. 2022.
Hendriksen RS, Munk P, Njage P, van Bunnik B, McNally L, Lukjancenko O, et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat Commun. 2019;10(1).
Hultman J, Tamminen M, Pärnänen K, Cairns J, Karkman A, Virta M. Host range of antibiotic resistance genes in wastewater treatment plant influent and effluent. FEMS Microbiol Ecol. 2018 Apr 1;94(4).
Lehto KM, Hyder R, Länsivaara A, Luomala O, Lipponen Anssi, Hokajärvi AM, et al. Wastewater-based surveillance is an efficient monitoring tool for tracking influenza A virus in the community. 2023. https://doi.org/10.1101/2023.08.28.23294723. Accessed 9 Jan 2024.
Ayobami O, Brinkwirth S, Eckmanns T, Markwart R. Antibiotic resistance in hospital-acquired ESKAPE-E infections in low- and lower-middle-income countries: a systematic review and meta-analysis. Emerg Microbes Infect. 2022;11(1):443–51.
EUCAST. Antimicrobial susceptibility testing EUCAST disk diffusion method Version 10.0. 2022. https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/2022_manuals/Manual_v_10.0_EUCAST_Disk_Test_2022.pdf. Accessed 22 Sep 2022.
EUCAST. MIC and zone diameter distributions and ECOFFs. 2023. Available from: https://www.eucast.org/mic_distributions_and_ecoffs/. Accessed 9 Jan 2024.
Jünemann S, Sedlazeck FJ, Prior K, Albersmeier A, John U, Kalinowski J, et al. Updating benchtop sequencing performance comparison. Vol. 31, Nature Biotechnology. 2013.
Babraham Institute. Babraham Bioinformatics–FastQC A Quality Control Tool for High Throughput Sequence Data. 2021. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 26 Jul 2022.
Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15).
Souvorov A, Agarwala R, Lipman DJ. SKESA: Strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018;19(1).
Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, Tolstoy I, et al. Validating the AMRFINder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob Agents Chemother. 2019;63(11).
Becker L, Kaase M, Pfeifer Y, Fuchs S, Reuss A, von Laer A, et al. Genome-based analysis of Carbapenemase-producing Klebsiella pneumoniae isolates from German hospital patients, 2008–2014. Antimicrob Resist Infect Control. 2018;7(1).
Zhou Z, Alikhan NF, Mohamed K, Fan Y, Achtman M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020;30(1):138–52.
Brisse S, Fevre C, Passet V, Issenhuth-Jeanjean S, Tournebize R, Diancourt L, et al. Virulent clones of Klebsiella pneumoniae: Identification and evolutionary scenario based on genomic and phenotypic characterization. PLoS One. 2009;4(3).
Diancourt L, Passet V, Verhoef J, Grimont PAD, Brisse S. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J Clin Microbiol. 2005;43(8).
Jamin C, de Koster S, van Koeveringe S, de Coninck D, Mensaert K, de Bruyne K, et al. Harmonization of whole-genome sequencing for outbreak surveillance of enterobacteriaceae and enterococci. Microb Genom. 2021;7(7).
Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18(3):268–81.
Bradford PA, Urban C, Mariano N, Projan SJ, Rahal JJ, Bush K. Imipenem resistance in Klebsiella pneumoniae is associated with the combination of ACT-1, a plasmid-mediated AmpC beta-lactamase, and the foss of an outer membrane protein. Antimicrob Agents Chemother. 1997;41(3):563–9.
Boyd SE, Holmes A, Peck R, Livermore DM, Hope W. OXA-48-Like β-Lactamases: global epidemiology, treatment options, and development pipeline. Antimicrob Agents Chemother. 2022;66(8).
Abramova A, Berendonk TU, Bengtsson-Palme J. A global baseline for qPCR-determined antimicrobial resistance gene prevalence across environments. Environ Int. 2023;178: 108084.
Osterblad M, Kirveskari J, Hakanen AJ, Tissari P, Vaara M, Jalava J. Carbapenemase-producing Enterobacteriaceae in Finland: the first years (2008–11). J Antimicrob Chemother. 2012;67(12):2860–4.
Majewski P, Wieczorek P, Łapuć I, Ojdana D, Sieńko A, Sacha P, et al. Emergence of a multidrug-resistant Citrobacter freundii ST8 harboring an unusual VIM-4 gene cassette in Poland. Int J Infect Dis. 2017;61.
Behruznia M, Gordon DM. Molecular and metabolic characteristics of wastewater associated Escherichia coli strains. Environ Microbiol Rep. 2022;14(4):646–54.
Saguti F, Magnil E, Enache L, Churqui MP, Johansson A, Lumley D, et al. Surveillance of wastewater revealed peaks of SARS-CoV-2 preceding those of hospitalized patients with COVID-19. Water Res. 2021;189: 116620.
Tiwari A, Kurittu P, Al-Mustapha AI, Heljanko V, Johansson V, Thakali O, et al. Wastewater surveillance of antibiotic-resistant bacterial pathogens: a systematic review. Front Microbiol. 2022;15:13.
Haenni M, Beyrouthy R, Lupo A, Châtre P, Madec JY, Bonnet R. Epidemic spread of Escherichia coli ST744 isolates carrying mcr-3 and blaCTX-M-55 in cattle in France. J Antimicrob Chemother. 2018;73(2):533–6.
Delgado-Blas JF, Ovejero CM, David S, Montero N, Calero-Caceres W, Garcillan-Barcia MP, et al. Population genomics and antimicrobial resistance dynamics of Escherichia coli in wastewater and river environments. Commun Biol. 2021;4(1):457.
Grönthal T, Österblad M, Eklund M, Jalava J, Nykäsenoja S, Pekkanen K, et al. Sharing more than friendship – transmission of NDM-5 ST167 and CTX-M-9 ST69 Escherichia coli between dogs and humans in a family, Finland, 2015. Eurosurveillance. 2018 Jul 5;23(27).
Wilder ML, Middleton F, Larsen DA, Du Q, Fenty A, Zeng T, et al. Co-quantification of crAssphage increases confidence in wastewater-based epidemiology for SARS-CoV-2 in low prevalence areas. Water Res X. 2021;11: 100100.
Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36(4):338–45.
Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods. 2012;58(3):268–76.
Arikawa K, Ide K, Kogawa M, Saeki T, Yoda T, Endoh T, et al. Recovery of strain-resolved genomes from human microbiome through an integration framework of single-cell genomics and metagenomics. Microbiome. 2021;9(1):202.
Zhang Z, Zhang G, Ju F. Using culture-enriched phenotypic metagenomics for targeted high-throughput monitoring of the clinically important fraction of the β-Lactam resistome. Environ Sci Technol. 2022;56(16):11429–39.
Pruden A, Vikesland PJ, Davis BC, de Roda Husman AM. Seizing the moment: now is the time for integrated global surveillance of antimicrobial resistance in wastewater environments. Vol. 64, Current Opinion in Microbiology. 2021.
Acknowledgements
The authors warmly thank Kirsi Ristkari for assistance in the laboratory and Kristiina Valkama for help with sample transportation arrangements. The personnel of the wastewater treatment facilities in all WastPan surveillance locations in Finland are gratefully acknowledged for their support and timely efforts in wastewater composite sampling and sample transportation arrangements.
WastPan Study Group: Ahmad Al-Mustapha and Paula Kurittu (University of Helsinki, Finland); Annika Länsivaara, Rafiqul Hyder, and Erja Janhonen (Tampere University, Finland); and Ananda Tiwari, Anna-Maria Hokajärvi, Aleksi Kolehmainen, Teemu Möttönen, Oskari Luomala, Aapo Juutinen, Soile Blomqvist, Carita Savolainen-Kopra (Finnish Institute for Health and Welfare, Finland), and Anniina Sarekoski (University of Helsinki and Finnish Institute for Health and Welfare, Finland).
Funding
Open Access funding provided by University of Helsinki (including Helsinki University Central Hospital). This work was funded by the Academy of Finland (Grant No. 339417) and the Doctoral Program in Food Chain and Health, Faculty of Veterinary Medicine, University of Helsinki, Finland. Open access funded by Helsinki University Library.
Author information
Authors and Affiliations
Consortia
Contributions
V. H.: Conceptualization, Methodology, Formal analysis, Investigation, Data curation, Writing—original draft, Writing—review & editing, Visualization. O. T.: Methodology, Formal analysis, Investigation, Writing—review & editing. V. J.: Conceptualization, Methodology, Investigation, Writing—review & editing. J-P. V.: Investigations, Writing—review & editing. K. R.: Investigations, Writing—review & editing. K-M. L.: Writing—review & editing, Project administration. A. L.: Writing—review & editing, Project administration. S. O.: Writing—review & editing, Project administration. T. P.: Writing—review & editing, Project administration. W. S. G.: Resources, Project administration. A. H.: Conceptualization, Writing—review & editing, Supervision, Project administration, Funding acquisition.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare the following financial interests or personal relationships related to the subject matter but not directly to this manuscript: S. O. reports a relationship with Greenseq Ltd. that includes board membership. K-M. L. reports a relationship with Greenseq Ltd. that includes board membership and travel reimbursement.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
All authors in the wastpan study group are listed in the acknowledgements.
Supplementary Information
Additional file 1.
Accession numbers for a study deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB64775 (https://www.ebi.ac.uk/ena/browser/view/PRJEB64775) and sequencing statistics for whole genome sequenced isolates of Escherichia coli (n = 26) and Klebsiella pneumoniae (n = 25) from 10 wastewater treatment plants across Finland in 2021–2022.
Additional file 2.
Additional bacterial species (n = 52) isolated from community wastewater influent samples using ChromAgar mSuperCARBA. Bacterial species were identified using MALDI-TOF MS.
Additional file 3: A.
Quantity of preliminary* carbapenemase-producing Escherichia coli from Espoo, Helsinki, Kuopio, and Lappeenranta wastewater treatment plants in 2021–2022. Nine samplings indicated by mm/yyyy. No visible bar indicates that the quantity was below the detection limit. CFU, colony-forming unit. B. Quantity of preliminary* carbapenemase-producing Escherichia coli from Oulu,Pietarsaari, and Rovaniemi wastewater treatment plants in 2021–2022. Nine samplings indicated bymm/yyyy. No visible bar indicates that the quantity was below the detection limit. CFU, colony-forming unit. C. Quantity of preliminary* carbapenemase-producing Escherichia coli from Seinäjoki, Tampere, and Turku wastewater treatment plants in 2021–2022. Nine samplings indicated by mm/yyyy. No visible bar indicates that the quantity was below the detection limit. CFU, colony-forming unit. *Preliminary, as not all isolates were confirmed to carry carbapenemase-encoding genes.
Additional file 4.
Minimum inhibitory concentration and zone of inhibition of antimicrobials for 50 Escherichia coli and 44 Klebsiella pneumoniae isolates from 10 wastewater treatment plants across Finland in 2021–2022. Epidemiological cut-off values (ECOFFs) (mg/L and mm) are indicated. ECOFFs in brackets for K. pneumoniae differing from ECOFFs for E. coli. Isolate ID (identification number) with bold lettering indicates that the isolate was subjected to sequencing. I/D displays insufficient data. COL, Colistin. P/T4, Piperacillin/Tazobactam constant 4. C/T, Ceftolozane/Tazobactam 4. CZA, Ceftazidime/Avibactam. MRP, Meropenem. MRP10 Meropenem (10μg), ERT10 Ertapenem (10μg).
Additional file 5.
Minimum spanning trees of core genome multilocus sequence typing (cgMLST) of (A) 26 carbapenemase-producing Escherichia coli isolates and (B) 25 carbapenemase-producing Klebsiella pneumoniae isolates. Each circle represents one or multiple identical sequences, and the numbers between the circles indicate the allele differences. Text in the circle indicates the isolate identification number, sample month/year, and city; colors indicate sequence type (ST). A gray background indicates closely related isolates (<10 allele difference). (A) cgMLSTwas based on 2513 columns, pairwise ignoring missing values. (B) cgMLSTwas based on 2365 columns, pairwise ignoring missing values, logarithmic scale.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Heljanko, V., Tyni, O., Johansson, V. et al. Clinically relevant sequence types of carbapenemase-producing Escherichia coli and Klebsiella pneumoniae detected in Finnish wastewater in 2021–2022. Antimicrob Resist Infect Control 13, 14 (2024). https://doi.org/10.1186/s13756-024-01370-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13756-024-01370-z