
Abstract
Background
Enterobacter cloacae complex is a group of common opportunistic pathogens on neonatal intensive care units. Active microbiological screening to guide empirical antimicrobial treatment or to detect transmission events is recommended in high-risk preterm neonates. A rise in colonization with E. cloacae complex was observed in a German perinatal centre. The aim of this study was to evaluate the performance of different typing techniques using whole genome sequencing (WGS) as a reference.
Methods
Enterobacter cloacae complex isolates from clinical and screening specimens with an epidemiological link to the neonatal intensive care units were further assessed. Identification and antibiotic susceptibility testing was performed by a combination of VITEK2 (bioMérieux) and MALDI-TOF (Bruker Daltonics), followed by RAPD/rep-PCR and PFGE (XbaI). Retrospectively, all isolates were analyzed by Fourier-transform infrared (FTIR) spectroscopy (IR Biotyper, Bruker Daltonics). Whole genome sequencing with SNP-based clustering was used as the reference method. Furthermore, resistome analysis, sequence type and species identification were derived from the WGS data. Transmission analysis was based on epidemiological and typing data.
Results
Between September 2017 and March 2018 32 mostly preterm neonates were found to be colonized with E. cloacae complex and 32 isolates from 24 patients were available for further typing. RAPD/rep-PCR and PFGE showed good concordance with WGS whereas FTIR displayed mediocre results [adjusted rand index (ARI) = 0.436]. A polyclonal increase and two dominant and overlapping clonal clusters of two different E. hormaechei subspecies were detected. Overall, four different species were identified. Genotyping confirmed third-generation cephalosporin resistance development in isolates of the same patient. During the six-month period several infection prevention interventions were performed and no E. cloacae complex isolates were observed during the following months.
Conclusions
Interpretation of the microbiological results alone to detect transmission events is often challenging and bacterial typing is of utmost importance to implement targeted infection control measures in an epidemic occurrence of E. cloacae complex. WGS is the most discriminatory method. However, traditional methods such as PFGE or RAPD/rep-PCR can provide reliable and quicker results in many settings. Furthermore, research is needed to quickly identify E. cloacae complex to the species level in the microbiological laboratory.
Similar content being viewed by others


Background
Enterobacter cloacae complex comprises several closely related Enterobacter species that are difficult to differentiate by standard microbiological methods such as biochemical profiling or mass spectroscopy. Members of this genetically diverse group are described in the environment or as commensals of the animal and human gut. Moreover, E. cloacae complex has emerged as an important facultative pathogen and significant cause of hospital-acquired infections. Special attention has been paid to infections with multidrug-resistant E. cloacae complex. Patient-to-patient or environment-to-patient transmissions can lead to outbreaks in the hospital setting [1,2,3,4].
In preterm neonates, early gut colonization with E. cloacae complex is common [5, 6]. At the same time, E. cloacae complex is one of the most common causes of outbreaks in this specialized setting and can cause severe infections [7]. For this reason, microbiological colonization screening for E. cloacae complex is officially recommended in high-risk neonates with a gestational weight below 1500 g in Germany (irrespective of the antibiotic susceptibility profile and amongst other relevant Gram-negative and Gram-positive bacteria) [8, 9]. However, interpretation of this data is often challenging. If transmission is suspected on the basis of spatio-temporal relationship of the isolates/patients, quick, reliable and discriminatory genotyping is of utmost importance to implement targeted infection control measures [10].
Here we report of a sudden increase of patients colonized with E. cloacae complex on a neonatal intensive care unit and highlight the challenges and pitfalls related to different typing techniques applied.
Methods
Setting and screening strategy
The perinatal centre of the Children's Hospital of Cologne (neonatal level III care according to international classification [11]) includes a 10-bed neonatal intensive care unit (NICU) and another 16-bed paediatric (and neonatal) intensive care unit (PICU). The two units are housed in two separate hospitals; the NICU is located next to the maternity ward, whereas the PICU is located in the Children’s Hospital. In general, preterm neonates are hospitalized first on the NICU and thereafter released to the neonatal general ward in the Children’s Hospital. If surgery or other highly-specialized treatment is indicated, patients are transferred to the Children’s Hospital. The protocol of the German healthcare-associated infection surveillance for very low birthweight infants (NEO-KISS, < 1.500 g) and on intensive care units (ITS-KISS, PICU) was followed during the study period [12, 13]. A weekly microbiological screening (perianal and combined nasal/oropharyngeal swabs) was performed on all infants of the NICU and on the preterm neonates of the PICU and the neonatal general ward according to the German infection control guideline that includes screening for E. cloacae complex [8]. The number of patients colonized/infected with E. cloacae complex was assessed using the laboratory surveillance information system HyBASE® v.6 (epiNET AG, Bochum, Germany).
Identification and susceptibility testing
Screening swabs were inoculated on Columbia blood agar, chocolate agar, McConkey agar (all Becton Dickinson, Heidelberg, Germany) and chromogenic chromID® ESBL (bioMérieux, Marcy l’Etoile, France) media and incubated at 35 ± 1 °C at least 48 h. All isolates were identified with standard microbiological procedures using the VITEK 2 system (Vitek GN-ID, bioMérieux) or MALDI-TOF (Bruker Daltonics, Bremen, Germany). Susceptibility testing was performed with the VITEK 2 system (Vitek AST-N248). EUCAST breakpoints were used for interpretation (v. 7.1 in 2017 and v. 8.0 in 2018). At least one isolate per patient and per phenotype (antibiotic susceptibility testing) was stored in a 30%-glycerol stock at − 20 °C. During the study period, E. cloacae complex isolates from screening and clinical specimens were taken into account.
Band-based genotyping
Strain relatedness was assessed by a combination of random amplification of polymorphic DNA (RAPD) using the primer ST272 and repetitive-element PCR (rep-PCR) using two primers: ERIC-1 and ERIC-2 [14, 15]. In the text this method is referred to as RAPD. Isolates differing by one or more bands in at least one primer assay were assigned to distinct types (RAPD type). Every single new RAPD genotype was included in every new run.
Genotyping was additionally carried out by pulsed-field gel electrophoresis (PFGE) on all isolates after XbaI (New England BioLabs, Ipswich, MA, USA) restriction under the following conditions: 6 V/cm for 20 h with pulse times of 5–50 s at 14 °C. To overcome degradation bacteria were heated in EDTA and thiourea was added to the buffer [16]. The strain relatedness was calculated with GelCompar II version 6.0 software (Applied Maths NV, Sint-Martens-Latem, Belgium) and in accordance with the Tenover et al. criteria [17].
Fourier-transform infrared (FTIR) spectroscopy
Retrospectively, all isolates were analysed by FTIR spectroscopy. FTIR spectrum acquisition and analysis was performed using an IR Biotyper System (Bruker Daltonik, Bremen, Germany) running the IR Biotyper software (version 1.5) as previously described [18]. A cut-off value of 0.77 was chosen for cluster attribution. Additionally, to improve the discrimination and clustering of FTIR spectra in our dataset we used an artificial neural network (ANN) established by Vogt et al. [18] The concordance of FTIR clustering in comparison to WGS genotyping was determined by calculation of the adjusted rand index (ARI) [19] using the online tool at www.comparingpartitions.info.
Genome sequencing, assembly and analysis
Genomic DNA was extracted for WGS analysis, using the UltraClean Microbial DNA isolation kit (MOBIO Laboratories Inc., Carlsbad, CA, United States) or DNeasy Ultraclean Microbial Kit (Qiagen, Venlo, Netherlands). DNA library preparation was performed with the TruSeqNano DNA LT or HT Kit (Illumina, San Diego, CA, United States) and sequenced on an Illumina MiSeq (Illumina, San Diego, CA, United States) or Illumina Nextseq (Illumina, San Diego, CA, United States) sequencer. Assembly of sequence reads was performed as previously described by Vogt et al. using the A5 pipeline (version 20,140,604) and SPAdes (version 3.7.0) [18, 20, 21]. Core genomes for phylogenetic analysis were calculated using Spine (version 0.1.2) [22]. Prophage regions were investigated using PHASTER2 and removed using a customized script of the A5 pipeline [23]. SNP calling was performed by mapping high-quality sequencing reads previously generated by Trimmomatic (version 0.35) to the core genome using BioNumerics 7.6 (Applied Maths/bioMérieux, Sint-Martens-Latem, Belgium) with default settings [24].
The traditional multilocus sequence type was assessed by the CGE MLST platform (software version 2.0.4, database version 2.0.0) [25]. Acquired resistance genes on assembled sequences were identified by ResFinder (version 4.1; threshold of 98% identity and minimum length of 60%) [26]. Afterwards WGS-based species identification with the assembled genomes was performed using JSpeciesWS (version 3.6.1) [27]. Sequence reads of the strains have been deposited as a project at the European Nucleotide Archive under the project accession number PRJEB46479 (samples ERS6676481 through ERS6676512).
Infection prevention and control (IPC) analysis
Bacterial colonizations and infections were considered as community-acquired (including acquisition at birth), if the collection of the specimen or the start of infection occurred on or before the 2nd day after admission/birth. Thereafter, bacterial colonizations and infections were defined as hospital-acquired. Transmission analysis was based on epidemiological data (direct room or ward contact, and/or documented care by the same staff) and genetic data. Transmission events were defined as proven if isolation of genetically-related isolates from two patients who were on the same ward at the same time (patient-to-patient transmission) or in the same room with a maximum time interval of one week (room-to-patient transmission). Hospital-acquired infections were classified according to the CDC/NHSN definitions [28]. Patients without related signs of infection were considered to be colonized. Standard and contact precautions (barrier nursing, use of gowns and gloves) were applied for every patient colonized with a third-generation cephalosporin-resistant E. cloacae complex (3GCR-EC). In case of additional ciprofloxacin or carbapenem resistance, patients were preferably isolated in a single room. Alternatively, strict contact precautions were applied. Additional interventions were performed throughout the outbreak period (training of the healthcare workers, hand hygiene compliance observations, extensive environmental sampling (surfaces, medical devices etc.), and intensified twice daily cleaning and disinfection). During the outbreak period clinicians were advised to use carbapenems in case of neonatal sepsis as this is the optimal choice to treat E. cloacae complex.
Results
Isolate and patient characteristics
Between September 2017 and March 2018, 32 patients were found to be colonized with E. cloacae complex. Out of these patients eight infants were twins (four sets). All except one patient were preterm neonates (gestational age < 37 weeks) with a median (range) gestation age of 26 + 0 (23 + 1–38 + 2) weeks and a median (range) birth weight of 905 (355–3000) g. 22 patients were of very low birth weight (< 1500 g). The median (range) age and length of stay at first isolation were 18 (4–109) days and 18 (1–67) days, respectively. All isolates but one were hospital-acquired. One patient developed a primary bloodstream infection with E. cloacae complex that was successfully treated (empirically with cefotaxime and definite therapy with meropenem); all other patients were considered as colonized.
Overall, 18 patients carried a third-generation cephalosporin-susceptible E. cloacae complex (3GCS-EC) isolate, five patients a 3GCR-EC and nine patients E. cloacae complex isolates displaying both resistance types. 24 first isolates and 8 follow-up isolates were available for further genotyping analysis. One follow-up isolate was collected after the study period from patient ID27. Relevant microbiological, epidemiological, resistome, and genotyping data are displayed in Table 1.
Genotyping and transmission analysis
Based on conventional genotyping by RAPD and PFGE, we were able to show two main clonal clusters of E. cloacae complex isolates: PFGE type A/RAPD type 1 cluster containing eight patients (including one set of twins) and PFGE type B/RAPD type 2 cluster containing 10 patients (including two sets of twins). The other clusters contained isolates from the same patient (PFGE type C/RAPD type 3 and PFGE type E/RAPD type 5) or from twins (PFGE type G/RAPD type 7). Isolates 10,938 and 11,183 were the sole members of PFGE type D/RAPD type 4 and PFGE type F/RAPD type 6, respectively. All RAPD/PFGE clusters were confirmed by whole genome sequencing using a single-nucleotide polymorphism (SNP)-based phylogeny (Table 1). Intra-cluster variability was at most one SNP, whereas inter-cluster SNP difference was at least 180 SNPs (Fig. 2). Isolate 11,183 exhibited the largest genetic distance to all other isolates. Overall, all twins were colonized with the same clone and all patients from whom two resistance types were recovered (3GCR-EC and 3GCS-EC) carried one clone.
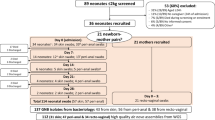
The epidemiological and WGS typing data of the E. cloacae complex surveillance are displayed in Fig. 1.

Epidemiological curve of new cases with E. cloacae complex per calendar week (only first isolate of E. cloacae complex from each patient). Only clusters containing more than one patient are shown in different colours, all other isolates are “non clonal” (no clonal relationship to other patients)
FTIR spectroscopy-based clustering displayed relatively poor correlation with the SNP-based clustering WGS reference (Fig. 2). Infrared spectroscopy was not able to differentiate WGS clonal clusters 1 and 2. First and follow-up isolates from Patient ID13 and ID27 were not classified as being clonally related. The ARI value for comparison of FTIR spectrum-based clustering (using a similarity cut-off value of 0.77) and SNP-based clustering was 0.436 (when a value of 1 represents complete concordance of the resulting clusters by each method). The spectrum-based clustering did not change, when an Artificial Neural Network was employed for assessing the spectral similarity.

Genomic and spectral clustering of 32 E. cloacae complex isolates from 24 patients. SNP-based clustering of E. cloacae complex isolates. Values on the branches indicate the number of SNPs with logarithmic scaling of the branch length. Numbers on the right show the assigned SNP cluster types. The colour coding indicates the assigned FTIR spectroscopy cluster of the isolates
Analysing spatiotemporal links, we were not able to identify index patients admitted with the clonal clusters. By conventional epidemiology, all other transmission events within clonal cluster 1, 2 and 7 were confirmed as “proven”, except for patient ID04 (isolate 10,506) from cluster 2 who had no epidemiological link to the other patients. Although most proven transmissions occurred within the NICU and all patients stayed on the NICU, three patients in clonal cluster 1 must have acquired the respective clone afterwards on the PICU from a patient transferred from the NICU. Analysing the epidemiology of the eight non-genotyped isolates from the study period only one patient could be linked to the clonal cluster 1 and another one to the clonal cluster 2.
Further whole genome sequencing analysis
In-silico species identification revealed four different (sub-)species of E. cloacae complex: E. hormaechei subsp. steigerwaltii, E. hormaechei subsp. oharae, E. hormaechei subsp. hoffmannii and E. cloacae subsp. cloacae respectively (Table 1). Genetic in silico search for bla genes causing third-generation cephalosporin-resistance displayed AmpC beta-lactamase genes in all isolates encoding different ACT-types in 31 isolates and CMH-3 in one isolate. All isolates except two carried the fosA gene (Table 1).
Discussion
In this study, we report on a molecular surveillance of E. cloacae complex in a perinatal centre of a tertiary care center. During the six-month study period, an increase in colonization with E. cloacae complex was observed in a group of preterm infants of which more than half had a very low birth weight; at first two dominant clones emerged followed by an increase of polyclonal isolates. Throughout the hospital stay preterm neonates are a vulnerable group prone to colonization with common Gram-negative organisms such as E. cloacae complex. Actually, Enterobacter species are one of the dominant bacterial organisms of the intestinal microbiome development in preterm low birth weight infants compared to term infants [5]. In a study, colonization with E. cloacae, the most common mucosal colonizer, was associated with the gestational age [6]. However, colonization with E. cloacae complex can serve as a source for infections or transmissions. Various outbreaks on neonatal intensive care units are described in the literature [29,30,31,32,33]. Thus, active routine microbiological surveillance screening can guide the antimicrobial empirical treatment and infection control strategies.
Whether hospital-acquired E. cloacae complex colonizations in two or more patients are independent acquisitions of different strains or transmission events of one clone is difficult to assess based on the microbiological report alone. It is known that E. hormaechei and E. cloacae are the most frequently isolated species from this group in human clinical specimen [1]. However, species identification of the different E. cloacae complex species cannot be routinely performed by most laboratories and demands more laborious molecular analysis. First reports that identification of different E. cloacae complex species with MALDI-TOF is feasible are promising [34]. Hence, if direct and reliable identification to the species or subspecies level is possible, this will lead to a better epidemiological understanding. Moreover, if different species are directly detected, workload and arising expenses of bacterial typing by the IPC team can be reduced. By demonstrating the molecular detection of four different species our study supports the need for research and development especially of MALDI-TOF mass spectrometry, which is used in most German laboratories nowadays. Moreover, differentiation based on phenotypes such as the antibiotic susceptibility pattern to rule out transmission events can be misleading, as clonal outbreaks with one clone displaying different phenotypes are quite common in E. claoacae complex [32]. The most important intrinsic mechanism of third-generation cephalosporin resistance in E. cloacae complex is de-repression of AmpC β–lactamases (e.g., of the ACT-type, found in nearly all isolates of this study) [1, 4]. When a patient was colonized with bacterial isolates exhibiting different resistance patterns (third-generation cephalosporin resistance or susceptibility) over time in this study, genotyping showed that the isolates were genetically highly-related, suggesting mutational evolution probably by antibiotic selection pressure.
Overall, typing methods are crucial to confirm or rule out transmission events. Low-discriminatory methods can lead to an overestimation of an outbreak and to unnecessary IPC interventions [33]. The preferred method should be selected based on its discriminatory power, its turnaround time and techniques available on site. Other constraints are reproducibility, costs or hands-on time [35, 36]. In this study, we applied typing methods of moderate to high discriminatory power (RAPD/rep-PCR, FTIR spectroscopy, MLST and PFGE) and compared those with WGS, which serves as the current gold standard of bacterial typing. All methods are well described for E. cloacae complex except for FTIR spectroscopy. This technique was only recently studied on a selection of E. cloacae complex isolates with overall satisfying results [18, 37]. Vogt et al. showed good concordance with SNP-based clustering (ARI = 0.818) [18]. Distinct bacterial cell structures are targeted by the typing methods applied in this study: unspecified genomic sequences (RAPD), repetitive genetic elements (rep-PCR), genomic restriction sites (PFGE), (mostly) polysaccharides (FITR), several housekeeping genes (MLST) or the whole genome (WGS). Comparative analysis of the isolate collection of this study showed good concordance between the conventional methods (RAPD/rep-PCR, PFGE and MLST) and WGS whereas the discriminatory power of FTIR was only moderate. Other studies showed more discriminatory results using FTIR for outbreak investigation [18, 37]. FTIR depends much more on the bacterial phenotype, the overall cellular composition and growth conditions than the other methods [18]. We also experienced that mucoid bacteria might be difficult to evaluate by this technique (e.g. patient ID13 carrying one mucoid and one non-mucoid type).
RAPD and rep-PCR both banding patterns typing methods can differ in discriminatory power. For example, Steffen et al. showed little discriminatory power compared to all other method used, but applied only one arbitrary primer variant [33]. However, these methods have the advantage that they are inexpensive and quick and can be a reliable tool if the information of several primers is combined and if reference strains are run as shown with different species and settings [38, 39]. Primers, cyclers and gel chambers needed for this method are available in most laboratories. Difficulties can arise from varying band intensities and may thus result in interpretation errors. These methods have their limits in case of complex outbreaks with many different strains involved as gels are less comparable compared to PFGE [33]. PFGE on the other hand is very labor intensive, but leads to good results in many cases [35].
WGS as the new gold standard is cost intensive and needs advanced skills of interpretation. Usage of WGS was previously described in E. cloacae outbreaks [40, 41]. A worldwide application of WGS is neither affordable nor necessary in most circumstances, especially to rule out transmission. At the moment, its application in real-time surveillance is limited due to costs and turnaround time. However, other information than the genotype can be obtained from the WGS data such as (sub-)species, plasmid structures or resistome.
In the clinical setting of nosocomial acquisition, reservoirs such as other humans, mostly patients, or the environment have to be identified. In general, the most common cause of nosocomial acquisition is person-to-person transmission via contaminated hands by health-care workers. Nevertheless, colonized healthcare workers are rarely described as a source of an outbreak [42]. Another source is the environment. However, we were not able to find an environmental source despite extensive efforts. No more E. cloacae complex isolates were detected on the NICU and PICU in the colonization screening during the three months following the study period (April–June 2018).
There are a few limitations to this study. First, we only analyzed one isolate per resistance pattern per patient. Patients colonized/infected with more than one strain of the same antibiotic resistance pattern might have remained undetected. Secondly, our conclusions cannot be generalized due to the limited number and the specific epidemic setting. Thirdly, rectal or vaginal screening of mothers recommended in the likelihood of a preterm delivery was not performed on a regular basis. Thus mother-to-child transmission could not be assessed.
Conclusions
A molecular and infection surveillance of hospital-acquired E. cloacae complex based on periodic screening, conventional epidemiology, genotyping and identification to the species level revealed simultaneously occurring independent transmission events and clusters and four different species. This underlines the importance of such an extensive surveillance methodology in IPC programs especially in vulnerable patient populations such as preterm neonates.
Availability of data and materials
Sequence reads have been deposited at the nucleotide accession number GenBank PRJEB46479. All other data generated or analysed during this study are included in this published article.
Abbreviations
- ANN:
-
Artificial neural network
- ARI:
-
Adjusted rand index
- CDC:
-
Centers for Disease Control and Prevention
- cgMLST:
-
Core genome multilocus sequence type
- EUCAST:
-
European Committee on Antimicrobial Susceptibility Testing
- FTIR:
-
Fourier-transform infrared
- ICU:
-
Intensive care unit
- IPC:
-
Infection prevention and control
- ITS-KISS:
-
Intensivstation-Krankenhaus-Infektions-Surveillance-System = German national nosocomial infections surveillance on intensive care units
- MALDI-TOF:
-
Matrix-assisted laser desorption/ionization-time-of-flight mass spectrometer
- MDR:
-
Multidrug-resistant
- MLST:
-
Multilocus sequence type
- NEO-KISS:
-
Surveillance System nosokomialer Infektionen für Frühgeborene auf Intensivstationen = German national neonatal infection surveillance system (for very low birth weight infants)
- NHSN:
-
National Healthcare Safety Network
- NICU:
-
Neonatal intensive care unit
- PFGE:
-
Pulsed-field gel electrophoresis
- PICU:
-
Paediatric (and neonatal) intensive care unit
- RAPD:
-
Random amplification of polymorphic DNA
- rep-PCR:
-
Repetitive-element PCR
- SNP:
-
Single-nucleotide polymorphism
- WGS:
-
Whole genome sequencing
- 3GCS-EC:
-
Third-generation cephalosporin-susceptible E. cloacae complex
- 3GCR-EC:
-
Third-generation cephalosporin-resistant E. cloacae complex
References
Davin-Regli A, Lavigne JP, Pages JM. Enterobacter spp.: update on taxonomy, clinical aspects, and emerging antimicrobial resistance. Clin Microbiol Rev. 2019;32:e00002-19.
Chavda KD, Chen L, Fouts DE, Sutton G, Brinkac L, Jenkins SG, et al. Comprehensive genome analysis of carbapenemse-producing Enterobacter spp.: new insights into phylogeny, population structure, and resistance mechanisms. mBio. 2016;7:e02093-16.
Kremer A, Hoffmann H. Prevalences of the Enterobacter cloacae complex and its phylogenetic derivatives in the nosocomial environment. Eur J Clin Microbiol Infect Dis. 2012;31:2951–5.
Mezzatesta ML, Gona F, Stefani S. Enterobacter cloacae complex: clinical impact and emerging antibiotic resistance. Future Microbiol. 2012;7:887–902.
Korpela K, Blakstad EW, Moltu SJ, Strommen K, Nakstad B, Ronnestad AE, et al. Intestinal microbiota development and gestational age in preterm neonates. Sci Rep. 2018;8:2453.
Parm U, Metsvaht T, Sepp E, Ilmoja ML, Pisarev H, Pauskar M, et al. Risk factors associated with gut and nasopharyngeal colonization by common Gram-negative species and yeasts in neonatal intensive care units patients. Early Hum Dev. 2011;87:391–9.
Schwab F, Geffers C, Piening B, Haller S, Eckmanns T, Gastmeier P. How many outbreaks of nosocomial infections occur in German neonatal intensive care units annually? Infection. 2014;42:73–8.
KRINKO. Praktische Umsetzung sowie krankenhaushygienische und infektionspräventive Konsequenzen des mikrobiellen Kolonisationsscreenings bei intensivmedizinisch behandelten Früh- und Neugeborenen. Epidemiol Bull 2013;2013:421–31.
Simon A, Tenenbaum T. Surveillance of multidrug-resistant Gram-negative pathogens in high-risk neonates—does it make a difference? Pediatr Infect Dis J. 2013;32:407–9.
Grundmann H, Barwolff S, Tami A, Behnke M, Schwab F, Geffers C, et al. How many infections are caused by patient-to-patient transmission in intensive care units? Crit Care Med. 2005;33:946–51.
American Academy of Pediatrics Committee on Fetus and Newborn. Levels of neonatal care. Pediatrics. 2012;130:587–97.
Leistner R, Piening B, Gastmeier P, Geffers C, Schwab F. Nosocomial infections in very low birthweight infants in Germany: current data from the National Surveillance System NEO-KISS. Klin Padiatr. 2013;225:75–80.
Schroder C, Schwab F, Behnke M, Breier AC, Maechler F, Piening B, et al. Epidemiology of healthcare associated infections in Germany: nearly 20 years of surveillance. Int J Med Microbiol. 2015;305:799–806.
Versalovic J, Koeuth T, Lupski JR. Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res. 1991;19:6823–31.
Mahenthiralingam E, Campbell ME, Foster J, Lam JS, Speert DP. Random amplified polymorphic DNA typing of Pseudomonas aeruginosa isolates recovered from patients with cystic fibrosis. J Clin Microbiol. 1996;34:1129–35.
O’Reilly LC. A method for overcoming DNA degradation during PFGE for Serratia marcescens. J Microbiol Methods. 2011;85:173–4.
Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, et al. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol. 1995;33:2233–9.
Vogt S, Loffler K, Dinkelacker AG, Bader B, Autenrieth IB, Peter S, et al. Fourier-transform infrared (FTIR) spectroscopy for typing of clinical enterobacter cloacae complex isolates. Front Microbiol. 2019;10:2582.
Carrico JA, Silva-Costa C, Melo-Cristino J, Pinto FR, de Lencastre H, Almeida JS, et al. Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J Clin Microbiol. 2006;44:2524–32.
Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol. 2013;20:714–37.
Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 2015;31:587–9.
Ozer EA, Allen JP, Hauser AR. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools spine and AGEnt. BMC Genomics. 2014;15:737.
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16-21.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114–20.
Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–61.
Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–4.
Richter M, Rossello-Mora R, Oliver Glockner F, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32:929–31.
Horan TC, Andrus M, Dudeck MA. CDC/NHSN surveillance definition of health care-associated infection and criteria for specific types of infections in the acute care setting. Am J Infect Control. 2008;36:309–32.
Dalben M, Varkulja G, Basso M, Krebs VL, Gibelli MA, van der Heijden I, et al. Investigation of an outbreak of Enterobacter cloacae in a neonatal unit and review of the literature. J Hosp Infect. 2008;70:7–14.
Ferry A, Plaisant F, Ginevra C, Dumont Y, Grando J, Claris O, et al. Enterobacter cloacae colonisation and infection in a neonatal intensive care unit: retrospective investigation of preventive measures implemented after a multiclonal outbreak. BMC Infect Dis. 2020;20:682.
Oteo J, Cercenado E, Vindel A, Bautista V, Fernandez-Romero S, Saez D, et al. Outbreak of multidrug-resistant CTX-M-15-producing Enterobacter cloacae in a neonatal intensive care unit. J Med Microbiol. 2013;62:571–5.
Pestourie N, Garnier F, Barraud O, Bedu A, Ploy MC, Mounier M. Outbreak of AmpC beta-lactamase-hyper-producing Enterobacter cloacae in a neonatal intensive care unit in a French teaching hospital. Am J Infect Control. 2014;42:456–8.
Steffen G, Pietsch M, Kaase M, Gatermann S, Werner G, Fuchs S, et al. Overestimation of an outbreak of enterobacter cloacae in a neonatal intensive care unit in Germany, 2015. Pediatr Infect Dis J. 2019;38:631–7.
Welker M, Van Belkum A, Girard V, Charrier JP, Pincus D. An update on the routine application of MALDI-TOF MS in clinical microbiology. Expert Rev Proteomics. 2019;16:695–710.
Sabat AJ, Budimir A, Nashev D, Sa-Leao R, van Dijl J, Laurent F, et al. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill. 2013;18:20380.
Novais A, Freitas AR, Rodrigues C, Peixe L. Fourier transform infrared spectroscopy: unlocking fundamentals and prospects for bacterial strain typing. Eur J Clin Microbiol Infect Dis. 2019;38:427–48.
Martak D, Valot B, Sauget M, Cholley P, Thouverez M, Bertrand X, et al. Fourier-transform infrared spectroscopy can quickly type gram-negative bacilli responsible for hospital outbreaks. Front Microbiol. 2019;10:1440.
Schafer E, Malecki M, Tellez-Castillo CJ, Pfennigwerth N, Marlinghaus L, Higgins PG, et al. Molecular surveillance of carbapenemase-producing Pseudomonas aeruginosa at three medical centres in Cologne, Germany. Antimicrob Resist Infect Control. 2019;8:208.
Wendel AF, Malecki M, Otchwemah R, Tellez-Castillo CJ, Sakka SG, Mattner F. One-year molecular surveillance of carbapenem-susceptible A. baumannii on a German intensive care unit: diversity or clonality. Antimicrob Resist Infect Control. 2018;7:145.
Stoesser N, Sheppard AE, Shakya M, Sthapit B, Thorson S, Giess A, et al. Dynamics of MDR Enterobacter cloacae outbreaks in a neonatal unit in Nepal: insights using wider sampling frames and next-generation sequencing. J Antimicrob Chemother. 2015;70:1008–15.
Wendel AF, Meyer S, Deenen R, Kohrer K, Kolbe-Busch S, Pfeffer K, et al. Long-term, low-frequency cluster of a german-imipenemase-1-producing Enterobacter hormaechei ssp. steigerwaltii ST89 in a Tertiary Care Hospital in Germany. Microb Drug Resist. 2018;24:1305–15.
Ulrich N, Gastmeier P, Vonberg RP. Effectiveness of healthcare worker screening in hospital outbreaks with gram-negative pathogens: a systematic review. Antimicrob Resist Infect Control. 2018;7:36.
Acknowledgements
We thank Ingo Winterfeld and the infection control nurses Stefanie Maczewski, Bilgen Pehlivan and Eva Ellers (all from the Institute of Hygiene, Cologne) for technical help. We also want to thank the team of the private microbiology laboratory MVZ synlab Leverkusen for routine microbiological analysis.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
AFW participated in design and coordination of the study, interpretation of the genotyping and surveillance data and drafted the manuscript. DP conceived of the study, and participated in its design and coordination, interpretation of the surveillance data and helped to draft the manuscript. FM conceived of the study and participated in its design. MW participated in the coordination and acquisition of clinical data. MH participated in the coordination and acquisition of clinical data. SW carried out the infrared spectroscopy analysis and participated in the interpretation. BB carried out the whole genome sequencing and bioinformatic analysis. SP supervised the whole genome sequencing and participated in the bioinformatic analysis. JL participated in the analysis and interpretation of infrared spectroscopy and whole genome sequencing data and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the recommendations for surveillance and cluster detections of nosocomial infections of the legally assigned institute for infection control and prevention (Robert Koch Institute). Formal consent was therefore not required.
Consent for publication
Not applicable.
Competing interests
All authors have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wendel, A.F., Peter, D., Mattner, F. et al. Surveillance of Enterobacter cloacae complex colonization and comparative analysis of different typing methods on a neonatal intensive care unit in Germany. Antimicrob Resist Infect Control 11, 54 (2022). https://doi.org/10.1186/s13756-022-01094-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13756-022-01094-y