
Abstract
The burden of multidrug resistance in Gram-negative bacilli (GNB) now represents a daily issue for the management of antimicrobial therapy in intensive care unit (ICU) patients. In Enterobacteriaceae, the dramatic increase in the rates of resistance to third-generation cephalosporins mainly results from the spread of plasmid-borne extended-spectrum beta-lactamase (ESBL), especially those belonging to the CTX-M family. The efficacy of beta-lactam/beta-lactamase inhibitor associations for severe infections due to ESBL-producing Enterobacteriaceae has not been adequately evaluated in critically ill patients, and carbapenems still stands as the first-line choice in this situation. However, carbapenemase-producing strains have emerged worldwide over the past decade. VIM- and NDM-type metallo-beta-lactamases, OXA-48 and KPC appear as the most successful enzymes and may threaten the efficacy of carbapenems in the near future. ESBL- and carbapenemase-encoding plasmids frequently bear resistance determinants for other antimicrobial classes, including aminoglycosides (aminoglycoside-modifying enzymes or 16S rRNA methylases) and fluoroquinolones (Qnr, AAC(6′)-Ib-cr or efflux pumps), a key feature that fosters the spread of multidrug resistance in Enterobacteriaceae. In non-fermenting GNB such as Pseudomonas aeruginosa, Acinetobacter baumannii and Stenotrophomonas maltophilia, multidrug resistance may emerge following the sole occurrence of sequential chromosomal mutations, which may lead to the overproduction of intrinsic beta-lactamases, hyper-expression of efflux pumps, target modifications and permeability alterations. P. aeruginosa and A. baumannii also have the ability to acquire mobile genetic elements encoding resistance determinants, including carbapenemases. Available options for the treatment of ICU-acquired infections due to carbapenem-resistant GNB are currently scarce, and recent reports emphasizing the spread of colistin resistance in environments with high volume of polymyxins use elicit major concern.
Similar content being viewed by others

Background
The burden of antimicrobial resistance in Gram-negative bacilli (GNB) is a daily challenge to face for intensive care unit (ICU) physicians. Indeed, GNB are responsible for 45–70% of ventilator-associated pneumonia (VAP) [1], 20–30% of catheter-related bloodstream infections [2], and commonly cause other ICU-acquired sepsis such as surgical site or urinary tract infections (UTI) [3]. In such situations, the timely administration of adequate antibiotic coverage is a crucial determinant of patient outcome, especially when criteria for severe sepsis are present [4]. Nevertheless, alarming resistance rates are now reported worldwide, and rising trends may elicit concerns for the coming years [2, 3, 5–9]. Almost exclusively restricted to the hospital setting till the beginning of the century, this issue increasingly applies for patients with healthcare-associated [10, 11] and even community-acquired infections [12–14]. Enterobacteriaceae and non-fermenting GNB (Pseudomonas aeruginosa, Acinetobacter baumannii and Stenotrophomonas maltophilia) account for the major part of the problem [15].
Antimicrobial resistance in GNB results from the expression of antibiotic-inactivating enzymes and non-enzymatic mechanisms [16]. Both may be intrinsically expressed by a given species (chromosomal genes), or acquired by a subset of strains as a consequence of two distinct albeit not mutually exclusive genetic events:
-
1.
Mutations in chromosomal genes resulting in an increase in the expression of intrinsic resistance mechanisms (either antibiotic-inactivating enzymes or efflux pumps), permeability alterations by loss of outer membrane porins, or target modifications;
-
2.
Horizontal transfers of mobile genetic elements (MGEs) carrying resistance genes, most notably plasmid-encoding beta-lactamases, aminoglycosides-modifying enzymes (AMEs), or non-enzymatic mechanisms such as Qnr for fluoroquinolone resistance in Enterobacteriaceae. Since these plasmids commonly bear multiple resistance determinants, a single plasmid conjugation may suffice to confer a multidrug resistance phenotype to the recipient strain.
The mechanisms of antimicrobial resistance in GNB may interfere with several facets of antibiotic stewardship algorithms in critically ill patients, including the choice of empirical regimen, available options for de-escalation, and the management of clinical failure due to the emergence of resistance under therapy [17, 18]. In this concise review, we sought to summarize the current knowledge on resistance mechanisms and epidemiologic trends in the main clinically relevant species belonging to Enterobacteriaceae and non-fermenting GNB, and make the connection with the use of antimicrobial therapy in the ICU.
Review
Current trends in the global epidemiology of multidrug-resistant GNB
Each given ICU has its own bacterial ecology, which may fluctuate owing to antibiotic use policies, patient recruitment and sporadic outbreaks. Yet, data from large surveillance networks yield a general overview of resistance rates in GNB causing ICU-acquired infections (Table 1). Following a decade of steady rise [19], rates of resistance to third-generation cephalosporins (3GC) in Enterobacteriaceae are now constantly above 10% and may reach 70% in certain settings [3, 5, 9]. This situation mainly results from the rapid spread of extended-spectrum beta-lactamase (ESBL)-producing strains, which currently account for 15–25% of Enterobacteriaceae isolated from clinical samples in critically ill patients [2, 9]. Far more worrying is the on-going dissemination of carbapenem-resistant Enterobacteriaceae (CRE), with an overall prevalence of 2–7% in ICUs in Europe, Asia and the United States [3, 5, 8, 9]. This issue appears especially critical for Klebsiella pneumoniae, with carbapenem resistance rates above 25% in several Southern European countries such as Italy or Greece [5]. Current rates of ceftazidime and carbapenem resistance in P. aeruginosa range from 20 to 40%. Multidrug resistance (i.e., resistance to at least three antimicrobial classes out of piperacillin–tazobactam, ceftazidime, fluoroquinolones, aminoglycosides and carbapenems) and extensive drug resistance (i.e., resistance to the five classes mentioned above) accounted for, respectively, 13 and 4% of P. aeruginosa isolates reported to the European Center for Disease Prevention and Control in 2013 [5]. Resistance rates are equally on the rise in A. baumannii, with 40 to 70% of isolates responsible for ICU-acquired infections being carbapenem resistant [2, 3, 8, 9].
Antimicrobial resistance in Enterobacteriaceae
Resistance to beta-lactams
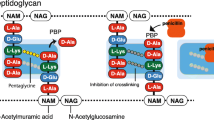
Beta-lactamase production is the main mechanism of beta-lactam resistance in Enterobacteriaceae (Fig. 1) [20]. These highly diversified enzymes hydrolyze beta-lactams in the periplasmic space, thus preventing penicillin-binding protein inhibition. Enterobacteriaceae are usually classified with regard to their intrinsic beta-lactamase content (Additional file 1: Table S1). A peculiar phenotype is observed in species that produce an inducible, chromosome-encoded AmpC cephalosporinase, notably Enterobacter sp., Citrobacter freundii, Hafnia alvei, Morganella morganii, Serratia marcescens and Providencia sp. [21]. AmpC is strongly induced by amoxicillin, clavulanic acid, cefoxitin and first-generation cephalosporins (1GC), which results in intrinsic resistance. Carbapenems are also potent inducers but remain active due to lack of significant AmpC-mediated hydrolysis, whereas other beta-lactams are weaker inducers. Infections caused by wild-type-inducible AmpC producers should be preferably treated by ticarcillin or piperacillin: 3GC, while active, must be avoided owing to a higher risk for selecting resistant mutants (see below) and a larger ecological impact [22–24]. Mutations in the induction system may permanently lead to AmpC overexpression (i.e., derepression). Of note, these mutations happen spontaneously and are only selected—but not prompted—by beta-lactams [25]. AmpC-hyperproducing mutants are resistant to penicillins, aztreonam, 3GC and even ertapenem when the enzyme is massively expressed [26] : imipenem and meropenem remain the most active beta-lactams, although cefepime stands as a valuable carbapenem-sparing option when tested susceptible and provided that the source of infection is controlled [27, 28]. The selection of a resistant mutant must be ruled out when a VAP or another infection due to wild-type-inducible AmpC producers does not improve, or relapse, under a first-line beta-lactam: in this case, a new sample should be obtained for susceptibility control [29]. Overall, AmpC hyperproducers account for 50–65% of 3GC-resistant Enterobacteriaceae recovered from carriage or clinical specimen in ICU with high prevalence of ESBL-producing strains [2, 30, 31]. Besides, the genome of Escherichia coli holds a very low-level expressed chromosomal AmpC not regulated by the induction system mentioned above [21]. Hyperproducing mutants may occasionally emerge; however, this mechanism of 3GC resistance remains anecdotal in E. coli when compared to ESBL [32].

Intrinsic and acquired beta-lactamases in Enterobacteriaceae.
The dissemination of plasmid-borne beta-lactamases constitutes by far the most critical resistance issue in Enterobacteriaceae. Inhibitor-susceptible TEM and SHV penicillinases emerged first in the 1960s, and spread rapidly afterwards. Then, mutations in the catalytic site enabled several TEM and SHV variants to resist to clavulanate or tazobactam, without hydrolyzing cephalosporins (Fig. 1) [33]. Besides, other mutations extended their hydrolysis spectrum to 3GC: these ESBL variants of TEM and SHV were described in the 1980s, soon after the introduction of 3GC [34]. They spread successfully in healthcare-associated strains of K. pneumoniae, Enterobacter sp. and, in a lesser extent, E. coli, causing major hospital outbreaks in the 1990s [35–37]. Nowadays, TEM-type and SHV-type ESBL are still endemic in many hospitals around the world; nevertheless, they tend to be outnumbered by another ESBL class, referred as CTX-M and first described in the early 1990s [38–40]. A key epidemiological aspect of CTX-M-type ESBL is to be mostly found in E. coli colonizing subjects with no medical condition, antibiotic exposure, or previous contact with the healthcare setting [41, 42]. This community reservoir fuels a continuous influx of ESBL into the hospital system [43, 44]. The diffusion of CTX-M-producing Enterobacteriaceae has been particularly massive in Southeast Asia and Eastern Mediterranean countries (estimated rates of intestinal carriage, ~60% and ~30%, respectively), and traveling in these areas is a major risk factor for carriage acquisition [42, 45, 46]. Carriage rates in the community are now above 5–10% in many other geographic areas [42], and concerns may logically be raised by reports of ESBL-producing Enterobacteriaceae (ESBL-PE) in community-acquired UTI [47], intra-abdominal sepsis [13], or even pneumonia [14]. In Europe, at present, 5–15% of critically ill patients are colonized with ESBL-PE at ICU admission [12, 30, 48]; however, very few among them are admitted for a community-acquired ESBL-PE infection [12].
ESBL-PE are resistant to most beta-lactams except cefoxitin, carbapenems and, for a subset of strains, temocillin [49]. Cefoxitin and temocillin have proven efficacy in murine models of urinary sepsis [50, 51], yet clinical data are still pending [52]. Meanwhile, co-resistances to fluoroquinolones, cotrimoxazole and aminoglycosides are commonly observed in ESBL-PE [7], leaving few alternatives to carbapenems for the treatment of severe infections. However, TEM, SHV and CTX-M are all class A beta-lactamases, and many ESBL variants remain susceptible in vitro to beta-lactamase inhibitors. Consequently, the careful use of certain beta-lactam/beta-lactamase inhibitor (BLBLI) associations (namely, amoxicillin–clavulanate, ticarcillin–clavulanate and piperacillin–tazobactam) to treat ESBL-PE with minimal inhibitory concentrations (MIC) ≤8 mg/L is now approved by the European Committee for Antimicrobial Susceptibility Testing (EUCAST), in an attempt to reduce carbapenem consumption and slow down the spread of carbapenem-resistant GNB [53]. Amoxicillin–clavulanate is most frequently inactive (many ESBL-PE coproduce clavulanate-resistant beta-lactamases such as AmpC or OXA-1), and clinical data are lacking for the ticarcillin–clavulanate association, thereby restricting the issue to the use of piperacillin–tazobactam for ESBL-PE with MIC below the aforementioned breakpoint. A meta-analysis published in 2012 reported no statistically significant difference in mortality between carbapenems and BLBLIs administered as either empirical or definite therapy; nevertheless, as underlined by the authors, included studies were heterogeneous and non-randomized, and most of severe patients were initially treated with carbapenems [54]. Piperacillin–tazobactam is probably a suitable carbapenem-sparing option in bacteraemic UTI [55], and shall be safely used in non-urinary sepsis when MICs are ≤2 mg/L [55, 56]. Conversely, an increase in 14-day mortality has been recently reported in patients with ESBL-PE bacteremia (MIC of piperacillin–tazobactam ranging from 4 to 16 mg/L, i.e., the US Clinical Laboratory Standards Institute’s breakpoint) and treated with piperacillin–tazobactam versus carbapenems [57]. The marked inoculum effect observed with tazobactam in ESBL-PE may contribute to explain why in vitro susceptibility does not systematically translate to clinical efficacy [58]. As a whole, the efficacy of BLBLI associations remains scarcely described in severe ESBL-PE infections [59], and has not been specifically investigated in ICU patients (Table 2). In this population, the pharmacokinetic properties of beta-lactams are dramatically modified [60–62], a pivotal point that may lead to sub-optimal BLBLI dosing even for ESBL-PE strains with relatively low MICs. Likewise, and according to the EUCAST guidelines [53], the use of cefepime might be discussed for infections due to ESBL-PE with MIC ≤1 mg/L [63, 64], but high-dose regimen should be used to overstep the risk of sub-optimal concentrations (Table 3) [18].
In parallel to ESBL, plasmid-borne cephalosporinases have gained increasing prominence in Enterobacteriaceae, including in community-acquired strains [21]. These beta-lactamases are actually encoded by chromosomal bla AmpC genes of Enterobacteriaceae that have been captured on MGE. CMY-2 from Citrobacter freundii is the most frequently encountered type [65]. Most of plasmid-borne cephalosporinases confer a similar pattern of resistance to that of derepressed AmpC.
As the prevalence of ESBL and plasmid-borne cephalosporinases rose, so did the consumption of carbapenems, which promoted the emergence of CRE through the diffusion of plasmid-borne carbapenemases. Unfortunately, their story shall be similar to that of ESBL-PE, namely a first step in K. pneumoniae (more rarely in Enterobacter sp.) affecting hospital settings with local outbreaks (e.g., VIM and KPC) [66, 67], and then emerging in the community in E. coli (NDM and OXA-48 variants) [68, 69] (Fig. 1). This new wave after the CTX-M pandemic raises high concerns as CRE are a step ahead of ESBL-PE in terms of multidrug resistance: for most, only colistin, tigecycline and gentamicin (for some KPC-producing strains) still have an activity (Table 3). OXA-48 is an exception as it hydrolyzes penicillins (with or without inhibitor) and carbapenems (low level of resistance), but not 3GC [70]. Yet, many OXA-48-producing Enterobacteriaceae coproduce an ESBL, jeopardizing all regular beta-lactam antibiotics [69]. While originally restricted to certain geographic areas (USA and Israel for KPC, Greece and Italy for VIM, India and Pakistan for NDM and the Eastern and Southern Mediterranean area for OXA-48) [71], CRE are currently spreading worldwide through travelers and repatriated patients [72–74], and are now isolated in subjects with no previous stay in endemic areas [75, 76]. Lastly, it should be underlined that carbapenemase production is not the sole mechanism of carbapenem resistance in Enterobacteriaceae, since this phenotype may also emerge under therapy in ESBL-PE or AmpC hyperproducers with acquired impermeability to carbapenems due to mutation-derived loss of outer membrane porins [77–79].
Resistance to other antimicrobials
Aminoglycosides resistance in Enterobacteriaceae mainly relies on AMEs that hamper antibiotic activity by engrafting various radicals (aminoglycoside phosphotransferase, APH, aminoglycoside nucleotidyltransferase, ANT and aminoglycoside acetyltransferase, AAC, see Additional file 1: Table S2). An intrinsic AME production is met in Providencia stuartii (AAC(2′), resistance to gentamicin and tobramycin) and Serratia marcescens (AAC(6′)-I, low-level resistance to tobramycin and amikacin). Other species are intrinsically susceptible but can acquire AME-encoding genes on plasmids that often carry multiple resistance determinants, including ESBL [80]. Current rates of co-resistance in hospital-acquired ESBL-PE are 50–60% for gentamicin and 10–20% to amikacin [81, 82], although local variations are observed. Methylases of the 16S ribosomal subunit (i.e., the target of aminoglycosides) have been more recently described, notably in NDM-producing strains [83]: these enzymes, named ArmA and Rmt, confer resistance to all aminoglycosides except neomycin.
All Enterobacteriaceae are naturally susceptible to quinolones and fluoroquinolones. High-level resistance emerges after successive chromosomal mutations in the DNA gyrase- and topoisomerase IV-encoding genes (gyrA and parC, respectively), each mutation causing a rise in the MICs [84]. Thus, strains with a single mutation can appear susceptible to fluoroquinolones but highly resistant to quinolones [53]. This phenotype may ease the emergence of mutants with high-level fluoroquinolone resistance under fluoroquinolone monotherapy, especially when the bacterial inoculum is high [85]. Chromosomal mutations may also lead to decreased permeability or overexpression of efflux pumps, resulting in reduced susceptibility. Besides mutations, plasmid-encoded resistance has emerged in the 2000s with Qnr (A, B, C, D and S subtypes), a small DNA-mimicking protein that confers low-level fluoroquinolone resistance [86], AAC(6′)-Ib-cr, an AME for which two mutations extend the resistance spectrum to ciprofloxacin and norfloxacin [87], and the QepA efflux pump [88]. It is noteworthy that these plasmid-borne determinants of fluoroquinolone resistance are frequently associated with ESBL [89].
Resistance to colistin, the last-resort antibiotic for CRE infections, is now under scrutiny. Proteus sp., Providencia sp., Serratia sp. and Morganella sp. are intrinsically resistant to colistin, and the acquisition of carbapenemase-encoding genes by these species is of major concern [71]. To date, no transferable resistance determinant has been described, and colistin resistance mainly rests on mutations in genes involved in the outer membrane polarity [90]. The spread of colistin-resistant Enterobacteriaceae, most notably K. pneumoniae, is alarming in environments with high prevalence of CRE, that is, in ICU with high volume of colistin consumption [91].
Antimicrobial resistance in non-fermenting GNB
Pseudomonas aeruginosa
Similarly to AmpC-producing Enterobacteriaceae, P. aeruginosa harbors an inducible AmpC-type cephalosporinase that can be derepressed following mutations in the regulation system [92]. Wild-type strains of P. aeruginosa are resistant to amoxicillin (with or without clavulanate), 1GC, 2GC, cefotaxime, ceftriaxone and ertapenem, while they remain susceptible to ticarcillin, piperacillin, ceftazidime, cefepime, imipenem, meropenem and doripenem. Aztreonam activity is variable. Unlike tazobactam, clavulanate is a strong inducer of AmpC in P. aeruginosa, and experimental data suggest a risk of clinical failure with the ticarcillin–clavulanate association [93]. AmpC-hyperproducing strains remain susceptible to carbapenems only.
P. aeruginosa has several three-component efflux systems, some of which confer resistance to beta-lactams when strongly expressed after mutations in their promoter regions (Table 4) [94]. The most frequently involved system is MexAB-OprM, whose overexpression confers resistance to ticarcillin, aztreonam, cefepime and meropenem. Efflux pumps are major determinants of the multidrug resistance phenotypes that are increasingly observed in P. aeruginosa. A key feature is that different antimicrobial classes may be substrates of a single pump: exposure to a given class (e.g., beta-lactams) may thereby select mutants with resistance to other classes (e.g., beta-lactams plus fluoroquinolones or aminoglycosides) [95].
Imipenem resistance in otherwise beta-lactam-susceptible strains of P. aeruginosa indicates the functional loss of OprD, a porin which manages the passage of imipenem through the outer membrane [95, 96]. The emergence of imipenem resistance under therapy results almost exclusively from the selection of OprD mutants, either from a previously imipenem-susceptible inoculum or, more occasionally, after cross-transmission of another clone [97]. The risk appears notably high in clinical practice. Indeed, in four randomized controlled trials (RCTs) including patients with hospital-acquired P. aeruginosa pneumonia, the average rate of resistance emergence under therapy was 30% (range, 6–53%) for imipenem, while only 15% (range, 6–36%) for other beta-lactams [98].
P. aeruginosa has the ability to develop resistance to all beta-lactams as the sole result of chromosomal mutations. Nonetheless, the species can acquire MGE-encoded beta-lactamases, including ESBL and carbapenemases (Table 4) [96]. Major hospital outbreaks have notably been observed with VIM or IMP carbapenemase-producing clones [99, 100].
Resistance to tobramycin mostly occurs through the acquisition of AMEs, while resistance to amikacin mostly depends on the over-expression of efflux pumps [101]. MGE-borne 16S rRNA methylases such as ArmA, RmtA and RmtD are also reported as an emerging mechanism of aminoglycoside resistance in P. aeruginosa [102]. Fluoroquinolone resistance results from mutations in the topoisomerase-encoding genes and/or the hyper-expression of efflux systems [95]. Lastly, and as for Enterobacteriaceae, colistin-resistant mutants of P. aeruginosa may emerge in settings with high frequency of colistin use [90].
Acinetobacter baumannii
Acinetobacter baumannii naturally produces a non-inducible AmpC-type cephalosporinase (ACE-1 or ACE-2) and an OXA-51-like oxacillinase which confer, at basal levels of expression, intrinsic resistance to aminopenicillins, 1GC, 2GC and aztreonam [103]. Ertapenem naturally lacks activity against A. baumannii. Together with a marked impermeability and the expression of multiple efflux systems, the plasticity of its genome enables the species to gather many resistance mechanisms, leading easily to multidrug resistance (Table 5). Most of the time, acquired resistance to carboxypenicillins, ureidopenicillins and 3GC rests on the overproduction of the AmpC-type cephalosporinase. However, in addition to plasmidic narrow-spectrum beta-lactamases, several ESBLs have also been acquired by A. baumannii: PER and VEB are the most frequently encountered types, particularly within pandemic clones [104]. In both cases, imipenem and meropenem remain the drugs of choice. More worrying are the emergence and dissemination of carbapenem-resistant clones since the end of the 1980s. Although carbapenem resistance can result from the over-expression of the chromosomal OXA-51-like enzyme [105], this phenotype is mostly due to the acquisition of plasmid-borne OXA-23-like, IMP, VIM, SIM or, more recently, NDM-type carbapenemases [102]. Of note, the prevalence of such carbapenemase-producing strains increases steadily from Northern to Southern European countries [104]. Acquired resistances to fluoroquinolones (mutations in gyrA and/or parC) and aminoglycosides (plasmid-borne AMEs—particularly AAC(3), AAC(6′) and APH(3′)—and 16S rRNA methylases) are commonly observed in ESBL- as well as carbapenemase-producing A. baumannii strains.
Colistin stands as the main therapeutic option for ICU-acquired infections due to extensively drug-resistant A. baumannii, and should be considered as part of the empirical antibiotic regimen in settings with high densities of carbapenem-resistant strains [106]. Nevertheless, colistin-resistant isolates are now increasingly reported worldwide, especially in patients previously exposed to this drug [107]. This phenotype mainly depends on the loss of lipopolysaccharide (LPS) production secondary to the insertion of the ISAba11 sequence in genes encoding the lipid A biosynthesis [108]. Increased expression of the PmrAB two-component regulatory system is another mechanism of LPS alteration resulting in colistin resistance [109]. Interestingly, the reduction of the negative charge of the lipid A, which lowers the affinity for colistin (positively charged), may also induce cross-resistance to host cationic antimicrobials such as lysozyme [110]. Furthermore, colistin exposure may select for a resistant fraction among an otherwise colistin-susceptible A. baumannii population [111, 112]. The prevalence of this mechanism of resistance—referred as heteroresistance—is poorly documented due to missed detection by conventional microbiological methods but could have significant clinical consequences [113].
For infection due to colistin-susceptible A. baumannii strains, the benefit of combination with rifampin has not been confirmed by a recent RCT [114]. Sulbactam, a BLI with intrinsic activity against A. baumannii, may be useful alone or in combination [103], although clinical data are still scarce. Clinical experience is also limited for minocycline, despite of a high in vitro activity against multidrug-resistant isolates [115]. The use of tigecycline may be discussed in the absence of other option (i.e., colistin resistance or toxicity) [104]: double-dose regimens appear well tolerated and could be more active than standard dosing owing to pharmacokinetic considerations, notably in patients with VAP [116].
Stenotrophomonas maltophilia
S. maltophilia is an emerging pathogen responsible for hospital-acquired infections in patients previously exposed to carbapenems or other broad-spectrum antibiotics [117, 118]. Its intrinsic multidrug resistance phenotype involves several chromosomal determinants. First, the species expresses various efflux systems and most notably the SmeDEF pump, which takes part in the extrusion of certain beta-lactams, quinolones and aminoglycosides [118]. Also, this GNB should be considered as naturally resistant to aminoglycosides, owing to the presence of a chromosomal AAC(6′)-Iz and the thermo-dependent permeability of its outer membrane to this antimicrobial class [119, 120]. Next, S. maltophilia produces two chromosomal beta-lactamases, namely, the inducible L1 carbapenemase (conferring an intrinsic resistance to all carbapenems) and the inducible, inhibitor-susceptible L2 cephalosporinase. Together, these enzymes may confer various resistance phenotypes, according to their respective degrees of expression and the concomitant levels of impermeability and efflux [118, 121]. The ticarcillin–clavulanate association remains usually the most effective beta-lactam regimen, while cephalosporins are almost constantly inactive.
S. maltophilia is highly susceptible to the trimethoprim–sulfamethoxazole combination, which is traditionally seen as the cornerstone of therapy [121]. Acquired resistance is however reported with various frequencies and rests on dihydropteroate synthases encoded by the MGE-borne sul genes. Fluoroquinolones, particularly ciprofloxacin, levofloxacin and moxifloxacin, are active despite the low-level expression of a Qnr protein encoded by the chromosomal SmQnr gene [122]. High-level resistance to fluoroquinolones may emerge through the selection of mutants with increased expression of SmQnr proteins or efflux pumps (SmeDEF or SmeVWX) [123].
The association of trimethoprim–sulfamethoxazole (high-dosing regimen) with ticarcillin–clavulanate or fluoroquinolones is generally advocated as a first-line regimen for serious infections [121]. Indeed, synergy with these combinations is observed in vitro for more than half of isolates [124]. Alternatives include monotherapy with trimethoprim–sulfamethoxazole, fluoroquinolones, or tigecycline [125, 126], with a possible synergic effect when the latter is associated to colistin [127, 128].
Is administration of combination therapy needed to prevent resistance?
To increase the likelihood of adequate coverage, the empirical antimicrobial regimen for VAP or other ICU-acquired infections in patients at risk for multidrug-resistant GNB usually combines a broad-spectrum beta-lactam with anti-pseudomonal activity and either an aminoglycoside or an anti-pseudomonal fluoroquinolone [29]. However, when both agents are active, the benefit of combination therapy over adequate monotherapy has not been proven in terms of clinical cure or microbiological eradication [129–131]. Convincing evidence is similarly lacking to support the routine use of antimicrobial combinations (including a beta-lactam) as definite regimen in an attempt to prevent the emergence of resistance under therapy [129, 132–134]. In P. aeruginosa infections, adding an aminoglycoside to an effective beta-lactam does not prevent from the emergence of beta-lactam resistance [133, 135, 136], including in patients treated with imipenem [137]. In Enterobacteriaceae, the main mechanism of acquired beta-lactam resistance under therapy is chromosomal AmpC derepression. In a prospective cohort of 218 patients infected with natural AmpC producers and receiving 3GC, the emergence of 3GC resistance was observed in 11 cases (5%): combining 3GC with an aminoglycoside or a fluoroquinolone did not significantly reduce the rate of mutant selection [138]. Therefore, once susceptibility testing results are known, monotherapy with the most active beta-lactam could be considered, with high-dosing regimen and optimized administration (Table 3). Clinical data remain scarce for infections due to multidrug-resistant A. baumannii [139], although in vitro studies indicate that combining colistin with rifampicine, carbapenems or tigecycline may be effective to prevent the emergence of colistin-resistant mutants [109].
A pivotal role for the gut microbiota
The intestinal microbiota forms the main reservoir of multidrug-resistant GNB in critically ill patients [30, 31, 140]. While similar data are currently not available for ICU-acquired infections, some studies have showed that high intestinal densities of resistant bacteria increase the risk of intestinal translocation [141], urinary tract infections [142] and cross-transmission [143]. Antibiotics that reach this microbiome promote the growth of resistant bacteria over the susceptible ones, and each daily dose may exert a significant impact in terms of selective pressure [144]. This appears notably relevant for carbapenems [26], fluoroquinolones [145] or cephalosporins with biliary excretion such as ceftriaxone [146]. Extended treatment with colistin has also been shown to increase the likelihood of colonization with colistin-resistant GNB, including both mutants from otherwise colistin-susceptible species, and intrinsically colistin-resistant Enterobacteriaceae [147].
The spectrum, duration of exposure and fecal concentration of the antibiotic may all play a role. Therefore, and although the ecological benefit of such an approach remains to be formally demonstrated [148], de-escalation to the antimicrobial regimen with the narrower spectrum and the lower intestinal excretion should be logically discussed when culture and susceptibility testing results become available. In this respect, new phenotypic and molecular diagnostic tools may fasten the detection of multidrug-resistant GNB—or rule them out precociously—thereby assisting ICU physicians for earlier adjustments of broad-spectrum empirical regimen [149–151].
Another unresolved issue is whether selective oral or digestive decontamination (SOD/SDD) with colistin and/or aminoglycosides compromises the efficacy of these agents by selecting resistant GNB in the ICU. SOD and SDD are infection prevention measures with proven efficacy in reducing the incidence of ICU-acquired bacteremia [152], the all-cause mortality rate at day 28 [153] and, for SDD combined with systemic antibiotic administration, the mortality attributable to VAP [154]. In a meta-analysis published in 2013, SOD and SDD were not associated with an increased risk of acquisition of aminoglycoside-resistant GNB, and were even protective against the acquisition of polymyxin-resistant GNB when compared to standard care [155]. However, the number of included studies was relatively low, most of them were conducted in the 1990s—that is, one decade before the pandemic of carbapenemase-producing Enterobacteriaceae—and carriage samples were pooled with clinical samples to assess the acquisition rates of resistant GNB, making the authors conclude that the impact of SOD/SDD on ICU-level antimicrobial resistance rates was understudied. In a recent RCT conducted in Dutch ICUs with low levels of resistance, the use of a tobramycin-based SDD regimen was associated with a gradual increase in the prevalence of aminoglycoside-resistant GNB when compared to SOD [156]. More strikingly, a worrisome rise in the rates of colistin and aminoglycoside resistance has been observed following the implementation of SOD/SDD policies with these antibiotics in ICUs facing outbreaks of ESBL- or carbapenemase-producing K. pneumoniae [157–159]. Conversely, two prospective trials reported that colistin-based SDD regimen might help eradicating CRE carriage [160, 161]. Overall, pending further longitudinal studies, SOD/SDD should probably be used with caution in environments with high prevalence of multidrug-resistant GNB to preserve the efficacy of polymyxins and aminoglycosides as last-resort agents [162, 163].
Concluding remarks and perspectives
The spread of multidrug-resistant GNB in the hospital setting is now seen as a globalized threat [15], and ICU patients are especially exposed to the risk. The number of potential novel agents in the pipeline is low; nevertheless, the development of new BLBLI combinations may raise significant hopes [164]. Avibactam (NXL104) is a synthetic BLI with activity on Ambler’s class A (including ESBL and KPC-type carbapenemases), class C (derepressed chromosomal AmpC or plasmid-borne AmpC) and some class D (oxacillinases) beta-lactamases [165]. In vitro, ceftazidime–avibactam, ceftaroline–avibactam and aztreonam–avibactam associations have shown promising results against 3GC-resistant and even KPC-producing Enterobacteriaceae [166, 167]. Avibactam also lessens the MICs of ceftazidime in AmpC-hyperproducing P. aeruginosa [168]. Other cephalosporin-BLI associations such as cefepime–tazobactam, ceftriaxone–sulbactam and ceftolozane–tazobactam are under evaluation [59, 169]. Clinical works addressing the yield of these new combinations in the ICU are highly warranted. Besides, improvements in the use of already available drugs are still possible (e.g., piperacillin–tazobactam for ESBL-PE with low MICs, or colistin for carbapenem-resistant GNB), both when indications and modalities of administration (including therapeutic drug monitoring) are considered. Unfortunately, the emergence of bacterial resistance following the introduction of new drugs appears as an unavoidable and endless process and every initiative aiming at limiting the selective pressure of antibiotics on the intestinal flora is more than ever justified.
Abbreviations
- AAC:
-
aminoglycoside acetyltransferase
- AME:
-
aminoglycoside-modifying enzyme
- ANT:
-
aminoglycoside nucleotidyltransferase
- APH:
-
aminoglycoside phosphotransferase
- CRE:
-
carbapenem-resistant Enterobacteriaceae
- BLBLI:
-
beta-lactam/beta-lactamase inhibitor
- ESBL:
-
extended-spectrum beta-lactamase
- ESBL-PE:
-
extended-spectrum beta-lactamase-producing Enterobacteriaceae
- 1GC:
-
first-generation cephalosporin
- 2GC:
-
second-generation cephalosporin
- 3GC:
-
third-generation cephalosporin
- GNB:
-
Gram-negative bacilli
- ICU:
-
intensive care unit
- MGE:
-
mobile genetic element
- MIC:
-
minimal inhibitory concentration
- LPS:
-
lipopolysaccharide
- RCT:
-
randomized controlled trial
- RNA:
-
ribonucleic acid
- SOD:
-
selective oral decontamination
- SDD:
-
selective digestive decontamination
- UTI:
-
urinary tract infection
- VAP:
-
ventilator-associated pneumonia
References
Barbier F, Andremont A, Wolff M, Bouadma L (2013) Hospital-acquired pneumonia and ventilator-associated pneumonia: recent advances in epidemiology and managment. Curr Opin Pulm Med 19(3):216–228
REA-Raisin Network (2012) Surveillance of nosocomial infections in critically ill adult patients, France, 2012. http://www.cclinparisnord.org. Accessed 26 Dec 2014
Rosenthal VD, Bijie H, Maki DG, Mehta Y, Apisarnthanarak A, Medeiros EA et al (2012) International Nosocomial Infection Control Consortium (INICC) report, data summary of 36 countries, for 2004–2009. Am J Infect Control 40(5):396–407
Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM et al (2013) Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 39(2):165–228
European Center for Disease Control and Prevention (2013) European Antimicrobial Resistance Surveillance (EARS-Net) 2013. http://www.ecdc.europa.eu/en/healthtopics/antimicrobial_resistance. Accessed 29 Dec 2014
Jones RN (2010) Microbial etiologies of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin Infect Dis 51(Suppl 1):S81–S87
Sader HS, Farrell DJ, Flamm RK, Jones RN (2014) Antimicrobial susceptibility of Gram-negative organisms isolated from patients hospitalised with pneumonia in US and European hospitals: results from the SENTRY Antimicrobial Surveillance Program, 2009–2012. Int J Antimicrob Agents 43(4):328–334
Chung DR, Song JH, Kim SH, Thamlikitkul V, Huang SG, Wang H et al (2011) High prevalence of multidrug-resistant nonfermenters in hospital-acquired pneumonia in Asia. Am J Respir Crit Care Med 184(12):1409–1417
Sader HS, Farrell DJ, Flamm RK, Jones RN (2014) Antimicrobial susceptibility of Gram-negative organisms isolated from patients hospitalized in intensive care units in United States and European hospitals (2009–2011). Diagn Microbiol Infect Dis 78(4):443–448
Shindo Y, Ito R, Kobayashi D, Ando M, Ichikawa M, Shiraki A et al (2013) Risk factors for drug-resistant pathogens in community-acquired and healthcare-associated pneumonia. Am J Respir Crit Care Med 188(8):985–995
Aliberti S, Cilloniz C, Chalmers JD, Zanaboni AM, Cosentini R, Tarsia P et al (2013) Multidrug-resistant pathogens in hospitalised patients coming from the community with pneumonia: an European perspective. Thorax 68:997–999
Razazi K, Derde LP, Verachten M, Legrand P, Lesprit P, Brun-Buisson C (2012) Clinical impact and risk factors for colonization with extended-spectrum beta-lactamase-producing bacteria in the intensive care unit. Intensive Care Med 38(11):1769–1778
Hsueh P-R, Badal RE, Hawser SP, Hoban DJ, Bouchillon SK, Ni Y et al (2010) Epidemiology and antimicrobial susceptibility profiles of aerobic and facultative Gram-negative bacilli isolated from patients with intra-abdominal infections in the Asia-Pacific region: 2008 results from SMART (Study for Monitoring Antimicrobial Resistance Trends). Int J Antimicrob Agents 36:408–414
Torres A, Cilloniz C, Ferrer M, Gabarrus A, Polverino E, Villegas S et al (2015) Bacteraemia and antibiotic-resistant pathogens in community acquired pneumonia: risk and prognosis. Eur Respir J 45(5):1353–1363
Center for Diseases Control and Prevention (2013) Threat report 2013. http://www.cdc.gov. Accessed 13 Mar 2015
Alekshun MN, Levy SB (2007) Molecular mechanisms of antibacterial multidrug resistance. Cell 128(6):1037–1050
Luyt CE, Brechot N, Trouillet JL, Chastre J (2014) Antibiotic stewardship in the intensive care unit. Crit Care 18(5):480
Bassetti M, De Waele JJ, Eggimann P, Garnacho-Montero J, Kahlmeter G, Menichetti F et al (2015) Preventive and therapeutic strategies in critically ill patients with highly resistant bacteria. Intensive Care Med 41(5):776–795
Meyer E, Schwab F, Schroeren-Boersch B, Gastmeier P (2010) Dramatic increase of third-generation cephalosporin-resistant E. coli in German intensive care units: secular trends in antibiotic drug use and bacterial resistance, 2001 to 2008. Crit Care 14(3):R113
Ambler RP (1980) The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci 289(1036):321–331
Jacoby GA (2009) AmpC beta-lactamases. Clin Microbiol Rev 22(1):161–182
Leclercq R, Canton R, Brown DF, Giske CG, Heisig P, MacGowan AP et al (2013) EUCAST expert rules in antimicrobial susceptibility testing. Clin Microbiol Infect 19(2):141–160
Livermore DM, Brown DF, Quinn JP, Carmeli Y, Paterson DL, Yu VL (2004) Should third-generation cephalosporins be avoided against AmpC-inducible Enterobacteriaceae? Clin Microbiol Infect 10(1):84–85
Harris PN, Ferguson JK (2012) Antibiotic therapy for inducible AmpC beta-lactamase-producing Gram-negative bacilli: what are the alternatives to carbapenems, quinolones and aminoglycosides? Int J Antimicrob Agents 40(4):297–305
Goldstein FW (2002) Cephalosporinase induction and cephalosporin resistance: a longstanding misinterpretation. Clin Microbiol Infect 8(12):823–825
Armand-Lefevre L, Angebault C, Barbier F, Hamelet E, Defrance G, Ruppé E et al (2013) Emergence of imipenem-resistant Gram-negative bacilli in intestinal flora of intensive care patients. Antimicrob Agents Chemother 57(3):1488–1495
Tamma PD, Girdwood SC, Gopaul R, Tekle T, Roberts AA, Harris AD et al (2013) The use of cefepime for treating AmpC beta-lactamase-producing Enterobacteriaceae. Clin Infect Dis 57(6):781–788
Hilty M, Sendi P, Seiffert SN, Droz S, Perreten V, Hujer AM et al (2013) Characterisation and clinical features of Enterobacter cloacae bloodstream infections occurring at a tertiary care university hospital in Switzerland: is cefepime adequate therapy? Int J Antimicrob Agents 41(3):236–249
American Thoracic Society (2005) Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 171(4):388–416
Thiebaut AC, Arlet G, Andremont A, Papy E, Sollet JP, Bernede-Bauduin C et al (2012) Variability of intestinal colonization with third-generation cephalosporin-resistant Enterobacteriaceae and antibiotic use in intensive care units. J Antimicrob Chemother 67(6):1525–1536
Grohs P, Podglajen I, Guerot E, Bellenfant F, Caumont-Prim A, Kac G et al (2014) Assessment of five screening strategies for optimal detection of carriers of third-generation cephalosporin-resistant Enterobacteriaceae in intensive care units using daily sampling. Clin Microbiol Infect 20(11):O879–O886
Livermore DM, Hope R, Reynolds R, Blackburn R, Johnson AP, Woodford N (2013) Declining cephalosporin and fluoroquinolone non-susceptibility among bloodstream Enterobacteriaceae from the UK: links to prescribing change? J Antimicrob Chemother 68(11):2667–2674
Beta-lactamase classification and amino-acid sequences for TEM, SHV and OXA extended-spectrum and inhibitor-resistant enzymes. http://www.lahey.org/studies. Accessed 23 Mar 2015
Sirot J, Chanal C, Petit A, Sirot D, Labia R, Gerbaud G (1988) Klebsiella pneumoniae and other Enterobacteriaceae producing novel plasmid-mediated beta-lactamases markedly active against third-generation cephalosporins: epidemiologic studies. Rev Infect Dis 10(4):850–859
Quale JM, Landman D, Bradford PA, Visalli M, Ravishankar J, Flores C et al (2002) Molecular epidemiology of a citywide outbreak of extended-spectrum beta-lactamase-producing Klebsiella pneumoniae infection. Clin Infect Dis 35(7):834–841
Mammeri H, Laurans G, Eveillard M, Castelain S, Eb F (2001) Coexistence of SHV-4- and TEM-24-producing Enterobacter aerogenes strains before a large outbreak of TEM-24-producing strains in a French hospital. J Clin Microbiol 39(6):2184–2190
Wiener J, Quinn JP, Bradford PA, Goering RV, Nathan C, Bush K et al (1999) Multiple antibiotic-resistant Klebsiella pneumoniae and Escherichia coli in nursing homes. JAMA 281(6):517–523
Bauernfeind A, Stemplinger I, Jungwirth R, Ernst S, Casellas JM (1996) Sequences of beta-lactamase genes encoding CTX-M-1 (MEN-1) and CTX-M-2 and relationship of their amino acid sequences with those of other beta-lactamases. Antimicrob Agents Chemother 40(2):509–513
Matsumoto Y, Ikeda F, Kamimura T, Yokota Y, Mine Y (1998) Novel plasmid-mediated beta-lactamase from Escherichia coli that inactivates oxyimino-cephalosporins. Antimicrob Agents Chemother 32:1243–1246
Barthelemy M, Peduzzi J, Bernard H, Tancrède C, Labia R (1992) Close amino acid sequence relationship between the new plasmid-mediated extended-spectrum beta-lactamase MEN-1 and chromosomally encoded enzymes of Klebsiella oxytoca. Biochim Biophys Acta 1122(1):15–22
D’Andrea MM, Arena F, Pallecchi L, Rossolini GM (2013) CTX-M-type beta-lactamases: a successful story of antibiotic resistance. Int J Med Microbiol 303(6–7):305–317
Woerther PL, Burdet C, Chachaty E, Andremont A (2013) Trends in human fecal carriage of extended-spectrum beta-lactamases in the community: toward the globalization of CTX-M. Clin Microbiol Rev 26(4):744–758
Ben-Ami R, Schwaber MJ, Navon-Venezia S, Schwartz D, Giladi M, Chmelnitsky I et al (2006) Influx of extended-spectrum beta-lactamase-producing Enterobacteriaceae into the hospital. Clin Infect Dis 42(7):925–934
Ruppé E, Pitsch A, Tubach F, de Lastours V, Chau F, Pasquet B et al (2012) Clinical predictive values of extended-spectrum beta-lactamase carriage in patients admitted to medical wards. Eur J Clin Microbiol Infect Dis 31(3):319–325
Kantele A, Laaveri T, Mero S, Vilkman K, Pakkanen SH, Ollgren J et al (2015) Antimicrobials increase travelers’ risk of colonization by extended-spectrum betalactamase-producing Enterobacteriaceae. Clin Infect Dis 60(6):837–846
Ruppé E, Armand-Lefevre L, Estellat C, Consigny PH, El Mniai A, Boussadia Y et al (2015) High rate of acquisition but short duration of carriage of multidrug-resistant Enterobacteriaceae after travel to the tropics. Clin Infect Dis. doi:10.1093/cid/civ333
Doi Y, Park YS, Rivera JI, Adams-Haduch JM, Hingwe A, Sordillo EM et al (2013) Community-associated extended-spectrum beta-lactamase-producing Escherichia coli infection in the United States. Clin Infect Dis 56(5):641–648
Derde LP, Cooper BS, Goossens H, Malhotra-Kumar S, Willems RJ, Gniadkowski M et al (2014) Interventions to reduce colonisation and transmission of antimicrobial-resistant bacteria in intensive care units: an interrupted time series study and cluster randomised trial. Lancet Infect Dis 14(1):31–39
Livermore DM, Hope R, Fagan EJ, Warner M, Woodford N, Potz N (2006) Activity of temocillin against prevalent ESBL- and AmpC-producing Enterobacteriaceae from south-east England. J Antimicrob Chemother 57(5):1012–1014
Soubirou JF, Rossi B, Couffignal C, Ruppe E, Chau F, Massias L et al (2015) Activity of temocillin in a murine model of urinary tract infection due to Escherichia coli producing or not producing the ESBL CTX-M-15. J Antimicrob Chemother 70(5):1466–1472
Lepeule R, Ruppe E, Le P, Massias L, Chau F, Nucci A et al (2012) Cefoxitin as an alternative to carbapenems in a murine model of urinary tract infection due to Escherichia coli harboring CTX-M-15-type extended-spectrum beta-lactamase. Antimicrob Agents Chemother 56(3):1376–1381
Balakrishnan I, Awad-El-Kariem FM, Aali A, Kumari P, Mulla R, Tan B et al (2011) Temocillin use in England: clinical and microbiological efficacies in infections caused by extended-spectrum and/or derepressed AmpC beta-lactamase-producing Enterobacteriaceae. J Antimicrob Chemother 66(11):2628–2631
European Committe on Antimicrobial Susceptibility Testing—Comité de l’antibiogramme de la Société Française de Microbiologie (2015) Guidelines 2015. http://www.eucast.org. Accessed 3 Mar 2015
Vardakas KZ, Tansarli GS, Rafailidis PI, Falagas ME (2012) Carbapenems versus alternative antibiotics for the treatment of bacteraemia due to Enterobacteriaceae producing extended-spectrum beta-lactamases: a systematic review and meta-analysis. J Antimicrob Chemother 67(12):2793–2803
Retamar P, Lopez-Cerero L, Muniain MA, Pascual A, Rodriguez-Bano J (2013) Impact of the MIC of piperacillin-tazobactam on the outcome of patients with bacteremia due to extended-spectrum-beta-lactamase-producing Escherichia coli. Antimicrob Agents Chemother 57(7):3402–3404
Rodriguez-Bano J, Navarro MD, Retamar P, Picon E, Pascual A (2012) beta-Lactam/beta-lactam inhibitor combinations for the treatment of bacteremia due to extended-spectrum beta-lactamase-producing Escherichia coli: a post hoc analysis of prospective cohorts. Clin Infect Dis 54(2):167–174
Tamma PD, Han JH, Rock C, Harris AD, Lautenbach E, Hsu AJ et al (2015) Carbapenem therapy is associated with improved survival compared with piperacillin-tazobactam for patients with extended-spectrum beta-lactamase bacteremia. Clin Infect Dis. doi:10.1093/cid/civ003
Lopez-Cerero L, Picon E, Morillo C, Hernandez JR, Docobo F, Pachon J et al (2010) Comparative assessment of inoculum effects on the antimicrobial activity of amoxycillin-clavulanate and piperacillin-tazobactam with extended-spectrum beta-lactamase-producing and extended-spectrum beta-lactamase-non-producing Escherichia coli isolates. Clin Microbiol Infect 16(2):132–136
Harris PN, Tambyah PA, Paterson DL (2015) Beta-lactam and beta-lactamase inhibitor combinations in the treatment of extended-spectrum beta-lactamase producing Enterobacteriaceae: time for a reappraisal in the era of few antibiotic options? Lancet Infect Dis 15(4):475–485
Roberts JA, Paul SK, Akova M, Bassetti M, De Waele JJ, Dimopoulos G et al (2014) DALI: defining antibiotic levels in intensive care unit patients: are current beta-lactam antibiotic doses sufficient for critically ill patients? Clin Infect Dis 58(8):1072–1083
De Waele JJ, Carrette S, Carlier M, Stove V, Boelens J, Claeys G et al (2014) Therapeutic drug monitoring-based dose optimisation of piperacillin and meropenem: a randomised controlled trial. Intensive Care Med 40(3):380–387
De Waele JJ, Lipman J, Akova M, Bassetti M, Dimopoulos G, Kaukonen M et al (2014) Risk factors for target non-attainment during empirical treatment with beta-lactam antibiotics in critically ill patients. Intensive Care Med 40(9):1340–1351
Lee NY, Lee CC, Huang WH, Tsui KC, Hsueh PR, Ko WC (2013) Cefepime therapy for monomicrobial bacteremia caused by cefepime-susceptible extended-spectrum beta-lactamase-producing Enterobacteriaceae: MIC matters. Clin Infect Dis 56(4):488–495
Goethaert K, Van Looveren M, Lammens C, Jansens H, Baraniak A, Gniadkowski M et al (2006) High-dose cefepime as an alternative treatment for infections caused by TEM-24 ESBL-producing Enterobacter aerogenes in severely-ill patients. Clin Microbiol Infect 12(1):56–62
Pascual V, Ortiz G, Simo M, Alonso N, Garcia MC, Xercavins M et al (2015) Epidemiology and risk factors for infections due to AmpC beta-lactamase-producing Escherichia coli. J Antimicrob Chemother 70(3):899–904
Bratu S, Landman D, Haag R, Recco R, Eramo A, Alam M et al (2005) Rapid spread of carbapenem-resistant Klebsiella pneumoniae in New York City: a new threat to our antibiotic armamentarium. Arch Intern Med 165(12):1430–1435
Souli M, Galani I, Antoniadou A, Papadomichelakis E, Poulakou G, Panagea T et al (2010) An outbreak of infection due to beta-Lactamase Klebsiella pneumoniae Carbapenemase 2-producing K. pneumoniae in a Greek University Hospital: molecular characterization, epidemiology, and outcomes. Clin Infect Dis 50(3):364–373
Walsh TR, Weeks J, Livermore DM, Toleman MA (2011) Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis 11(5):355–362
Canton R, Akova M, Carmeli Y, Giske CG, Glupczynski Y, Gniadkowski M et al (2012) Rapid evolution and spread of carbapenemases among Enterobacteriaceae in Europe. Clin Microbiol Infect 18(5):413–431
Evans BA, Amves SG (2014) OXA beta-lactamases. Clin Microbiol Rev 27(2):241–263
Nordmann P, Naas T, Poirel L (2011) Global spread of carbapenemase-producing Enterobacteriaceae. Emerg Infect Dis 17(10):1791–1798
Ruppé E, Armand-Lefevre L, Estellat C, El-Mniai A, Boussadia Y, Consigny PH et al (2014) Acquisition of carbapenemase-producing Enterobacteriaceae by healthy travellers to India, France, February 2012 to March 2013. Euro Surveill 19(14)
Birgand G, Armand-Lefevre L, Lepainteur M, Lolom I, Neulier C, Reibel F et al (2014) Introduction of highly resistant bacteria into a hospital via patients repatriated or recently hospitalized in a foreign country. Clin Microbiol Infect 20(11):O887–O890
Barbier F, Ruppé E, Giakkoupi P, Wildenberg L, Lucet J-C, Vatopoulos A et al (2010) Genesis of a KPC-producing Klebsiella pneumoniae after in vivo transfer from an imported Greek strain. Euro Surveill 15(1):19457
Institut National de Veille Sanitaire (2013) Episodes impliquant des entérobactéries productrices de carbapénèmases en France: situation épidémiologique du 1er avril 2013. http://www.invs.sante.fr. Accessed 29 Dec 2014
Poirel L, Potron A, Nordmann P (2012) OXA-48-like carbapenemases: the phantom menace. J Antimicrob Chemother 67:1597–1606
Skurnik D, Nucci A, Ruimy R, Lasocki S, Muller-Serieys C, Montravers P et al (2010) Emergence of carbapenem-resistant Hafnia: the fall of the last soldier. Clin Infect Dis 50(10):1429–1431
Findlay J, Hamouda A, Dancer SJ, Amyes SG (2012) Rapid acquisition of decreased carbapenem susceptibility in a strain of Klebsiella pneumoniae arising during meropenem therapy. Clin Microbiol Infect 18(2):140–146
Goessens WH, van der Bij AK, van Boxtel R, Pitout JD, van Ulsen P, Melles DC et al (2013) Antibiotic trapping by plasmid-encoded CMY-2 beta-lactamase combined with reduced outer membrane permeability as a mechanism of carbapenem resistance in Escherichia coli. Antimicrob Agents Chemother 57(8):3941–3949
Billard-Pomares T, Tenaillon O, Le Nagard H, Rouy Z, Cruveiller S, Medigue C et al (2011) Complete nucleotide sequence of plasmid pTN48, encoding the CTX-M-14 extended-spectrum beta-lactamase from an Escherichia coli O102-ST405 strain. Antimicrob Agents Chemother 55(3):1270–1273
Lowe CF, McGeer A, Muller MP, Katz K (2012) Decreased susceptibility to noncarbapenem antimicrobials in extended-spectrum-beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae isolates in Toronto. Canada. Antimicrob Agents Chemother 56(7):3977–3980
French National Observatory for Bacterial Resistance (ONERBA). 2009–10 Report. http://www.onerba.org. Accessed 26 Dec 2014
Galimand M, Courvalin P, Lambert T (2003) Plasmid-mediated high-level resistance to aminoglycosides in Enterobacteriaceae due to 16S rRNA methylation. Antimicrob Agents Chemother 47(8):2565–2571
Deguchi T, Yasuda M, Nakano M, Ozeki S, Kanematsu E, Nishino Y et al (1997) Detection of mutations in the gyrA and parC genes in quinolone-resistant clinical isolates of Enterobacter cloacae. J Antimicrob Chemother 40(4):543–549
Jacoby GA (2005) Mechanisms of resistance to quinolones. Clin Infect Dis 41(Suppl 2):S120–S126
Allou N, Cambau E, Massias L, Chau F, Fantin B (2009) Impact of low-level resistance to fluoroquinolones due to qnrA1 and qnrS1 genes or a gyrA mutation on ciprofloxacin bactericidal activity in a murine model of Escherichia coli urinary tract infection. Antimicrob Agents Chemother 53(10):4292–4297
Strahilevitz J, Jacoby GA, Hooper DC, Robicsek A (2009) Plasmid-mediated quinolone resistance: a multifaceted threat. Clin Microbiol Rev 22(4):664–689
Périchon B, Courvalin P, Galimand M (2007) Transferable resistance to aminoglycosides by methylation of G1405 in 16S rRNA and to hydrophilic fluoroquinolones by QepA-mediated efflux in Escherichia coli. Antimicrob Agents Chemother 51(7):2464–2469
Filippa N, Carricajo A, Grattard F, Fascia P, El Sayed F, Defilippis JP et al (2013) Outbreak of multidrug-resistant Klebsiella pneumoniae carrying qnrB1 and blaCTX-M15 in a French intensive care unit. Ann Intensive Care 3(1):18
Olaitan AO, Morand S, Rolain JM (2014) Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol 5:643
Zagorianou A, Sianou E, Iosifidis E, Dimou V, Protonotariou E, Miyakis S et al (2012) Microbiological and molecular characteristics of carbapenemase-producing Klebsiella pneumoniae endemic in a tertiary Greek hospital during 2004-2010. Euro Surveill 17(7)
Juan C, Macia MD, Gutierrez O, Vidal C, Perez JL, Oliver A (2005) Molecular mechanisms of beta-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother 49(11):4733–4738
Lister PD, Gardner VM, Sanders CC (1999) Clavulanate induces expression of the Pseudomonas aeruginosa AmpC cephalosporinase at physiologically relevant concentrations and antagonizes the antibacterial activity of ticarcillin. Antimicrob Agents Chemother 43(4):882–889
Mesaros N, Nordmann P, Plesiat P, Roussel-Delvallez M, Van Eldere J, Glupczynski Y et al (2007) Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin Microbiol Infect 13(6):560–578
Lister PD, Wolter DJ, Hanson ND (2009) Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22(4):582–610
Mesaros N, Glupczynski Y, Avrain L, Caceres NE, Tulkens PM, Van Bambeke F (2007) A combined phenotypic and genotypic method for the detection of Mex efflux pumps in Pseudomonas aeruginosa. J Antimicrob Chemother 59(3):378–386
Tsai MH, Wu TL, Su LH, Lo WL, Chen CL, Liang YH et al (2014) Carbapenem-resistant-only Pseudomonas aeruginosa infection in patients formerly infected by carbapenem-susceptible strains. Int J Antimicrob Agents 44(6):541–545
Zilberberg MD, Chen J, Mody SH, Ramsey AM, Shorr AF (2010) Imipenem resistance of Pseudomonas in pneumonia: a systematic literature review. BMC Pulm Med 10:45
Koutsogiannou M, Drougka E, Liakopoulos A, Jelastopulu E, Petinaki E, Anastassiou ED et al (2013) Spread of multidrug-resistant Pseudomonas aeruginosa clones in a university hospital. J Clin Microbiol 51(2):665–668
Willmann M, Bezdan D, Zapata L, Susak H, Vogel W, Schroppel K et al (2015) Analysis of a long-term outbreak of XDR Pseudomonas aeruginosa: a molecular epidemiological study. J Antimicrob Chemother 70(5):1322–1330
Kos VN, Deraspe M, McLaughlin RE, Whiteaker JD, Roy PH, Alm RA et al (2015) The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother 59(1):427–436
Potron A, Poirel L, Nordmann P (2015) Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int J Antimicrob Agents. doi:10.1016/j.ijantimicag.2015.03.001
Munoz-Price LS, Weinstein RA (2008) Acinetobacter infection. N Engl J Med 358(12):1271–1281
Kempf M, Rolain JM (2012) Emergence of resistance to carbapenems in Acinetobacter baumannii in Europe: clinical impact and therapeutic options. Int J Antimicrob Agents 39(2):105–114
Turton JF, Ward ME, Woodford N, Kaufmann ME, Pike R, Livermore DM et al (2006) The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS Microbiol Lett 258(1):72–77
Falagas ME, Rafailidis PI (2007) When to include polymyxins in the empirical antibiotic regimen in critically ill patients with fever? A decision analysis approach. Shock 27(6):605–609
Qureshi ZA, Hittle LE, O’Hara JA, Rivera JI, Syed A, Shields RK et al (2015) Colistin-resistant Acinetobacter baumannii: beyond carbapenemresistance. Clin Infect Dis 60(9):1295–1303
Pogue JM, Cohen DA, Marchaim D (2015) Polymyxin-resistant Acinetobacter baumannii: urgent action needed. Clin Infect Dis 60(9):1304–1307
Cai Y, Chai D, Wang R, Liang B, Bai N (2012) Colistin resistance of Acinetobacter baumannii: clinical reports, mechanisms and antimicrobial strategies. J Antimicrob Chemother 67(7):1607–1615
Napier BA, Burd EM, Satola SW, Cagle SM, Ray SM, McGann P et al (2013) Clinical use of colistin induces cross-resistance to host antimicrobials in Acinetobacter baumannii. MBio 4(3):e00013–e00021
Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE et al (2006) Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 50(9):2946–2950
Hawley JS, Murray CK, Jorgensen JH (2008) Colistin heteroresistance in acinetobacter and its association with previous colistin therapy. Antimicrob Agents Chemother 52(1):351–352
Rodriguez CH, Bombicino K, Granados G, Nastro M, Vay C, Famiglietti A (2009) Selection of colistin-resistant Acinetobacter baumannii isolates in postneurosurgical meningitis in an intensive care unit with high presence of heteroresistance to colistin. Diagn Microbiol Infect Dis 65(2):188–191
Durante-Mangoni E, Signoriello G, Andini R, Mattei A, De Cristoforo M, Murino P et al (2013) Colistin and rifampicin compared with colistin alone for the treatment of serious infections due to extensively drug-resistant Acinetobacter baumannii: a multicenter, randomized clinical trial. Clin Infect Dis 57(3):349–358
Ritchie DJ, Garavaglia-Wilson A (2014) A review of intravenous minocycline for treatment of multidrug-resistant Acinetobacter infections. Clin Infect Dis 59(Suppl 6):S374–S380
De Pascale G, Montini L, Pennisi M, Bernini V, Maviglia R, Bello G et al (2014) High dose tigecycline in critically ill patients with severe infections due to multidrug-resistant bacteria. Crit Care 18(3):R90
Soubirou JF, Gault N, Alfaiate T, Lolom I, Tubach F, Andremont A et al (2014) Ventilator-associated pneumonia due to carbapenem-resistant Gram-negative bacilli in an intensive care unit without carbapenemase-producing Enterobacteriaceae or epidemic Acinetobacter baumannii. Scand J Infect Dis 46(3):215–220
Brooke JS (2012) Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin Microbiol Rev 25:2–41
Lambert T, Ploy MC, Denis F, Courvalin P (1999) Characterization of the chromosomal aac(6’)-Iz gene of Stenotrophomonas maltophilia. Antimicrob Agents Chemother 43(10):2366–2371
Rahmati-Bahram A, Magee JT, Jackson SK (1996) Temperature-dependent aminoglycoside resistance in Stenotrophomonas (Xanthomonas) maltophilia: alterations in protein and lipopolysaccharide with growth temperature. J Antimicrob Chemother 37(4):665–676
Looney WJ, Narita M, Mühlemann K (2009) Stenotrophomonas maltophilia: an emerging opportunist human pathogen. Lancet Infect Dis 9(5):312–323
Sanchez MB, Martinez JL (2010) SmQnr contributes to intrinsic resistance to quinolones in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 54(1):580–581
Garcia-Leon G, Ruiz de Alegria Puiq C, Garcia de la Fuente C, Matinez-Martinez L, Martinez JL, Sanchez MB (2015) High-level quinolone resistance is associated with the overexpression of SmeVWX in Stenotrophomonas maltophilia clinical isolates. Clin Microbiol Infect. doi:10.1016/j.cmi.2015.01.007
San Gabriel P, Zhou J, Tabibi S, Chen Y, Trauzzi M, Saiman L (2004) Antimicrobial susceptibility and synergy studies of Stenotrophomonas maltophilia isolates from patients with cystic fibrosis. Antimicrob Agents Chemother 48(1):168–171
Wang YL, Scipione MR, Dubrovskaya Y, Papadopoulos J (2014) Monotherapy with fluoroquinolone or trimethoprim-sulfamethoxazole for treatment of Stenotrophomonas maltophilia infections. Antimicrob Agents Chemother 58(1):176–182
Tekce YT, Erbay A, Cabadak H, Sen S (2012) Tigecycline as a therapeutic option in Stenotrophomonas maltophilia infections. J Chemother 24(3):150–154
Betts JW, Phee LM, Woodford N, Wareham DW (2014) Activity of colistin in combination with tigecycline or rifampicin against multidrug-resistant Stenotrophomonas maltophilia. Eur J Clin Microbiol Infect Dis 33(9):1565–1572
Church D, Lloyd T, Peirano G, Pitout J (2013) Antimicrobial susceptibility and combination testing of invasive Stenotrophomonas maltophilia isolates. Scand J Infect Dis 45(4):265–270
Paul M, Lador A, Grozinsky-Glasberg S, Leibovici L (2014) Beta lactam antibiotic monotherapy versus beta lactam-aminoglycoside antibiotic combination therapy for sepsis. Cochrane Database Syst Rev 1:CD003344
Paul M, Silbiger I, Grozinsky S, Soares-Weiser K, Leibovici L (2006) Beta lactam antibiotic monotherapy versus beta lactam-aminoglycoside antibiotic combination therapy for sepsis. Cochrane Database Syst Rev 1:CD003344
Pena C, Suarez C, Ocampo-Sosa A, Murillas J, Almirante B, Pomar V et al (2013) Effect of adequate single-drug vs combination antimicrobial therapy on mortality in Pseudomonas aeruginosa bloodstream infections: a post-hoc analysis of a prospective cohort. Clin Infect Dis 57(2):208–216
Tamma PD, Cosgrove SE, Maragakis LL (2012) Combination therapy for treatment of infections with gram-negative bacteria. Clin Microbiol Rev 25(3):450–470
Bliziotis IA, Samonis G, Vardakas KZ, Chrysanthopoulou S, Falagas ME (2005) Effect of aminoglycoside and beta-lactam combination therapy versus beta-lactam monotherapy on the emergence of antimicrobial resistance: a meta-analysis of randomized, controlled trials. Clin Infect Dis 41(2):149–158
Lopez-Cortes LE, Cisneros JM, Fernandez-Cuenca F, Bou G, Tomas M, Garnacho-Montero J et al (2014) Monotherapy versus combination therapy for sepsis due to multidrug-resistant Acinetobacter baumannii: analysis of a multicentre prospective cohort. J Antimicrob Chemother 69(11):3119–3126
Carmeli Y, Troillet N, Eliopoulos GM, Samore MH (1999) Emergence of antibiotic-resistant Pseudomonas aeruginosa: comparison of risks associated with different antipseudomonal agents. Antimicrob Agents Chemother 43(6):1379–1382
Manian FA, Meyer L, Jenne J, Owen A, Taff T (1996) Loss of antimicrobial susceptibility in aerobic gram-negative bacilli repeatedly isolated from patients in intensive-care units. Infect Control Hosp Epidemiol 17(4):222–226
Cometta A, Baumgartner JD, Lew D, Zimmerli W, Pittet D, Chopart P et al (1994) Prospective randomized comparison of imipenem monotherapy with imipenem plus netilmicin for treatment of severe infections in nonneutropenic patients. Antimicrob Agents Chemother 38(6):1309–1313
Choi SH, Lee JE, Park SJ, Lee SO, Jeong JY, Kim MN et al (2008) Emergence of antibiotic resistance during therapy for infections caused by Enterobacteriaceae producing AmpC beta-lactamase: implications for antibiotic use. Antimicrob Agents Chemother 52(3):995–1000
Poulikakos P, Tansarli GS, Falagas ME (2014) Combination antibiotic treatment versus monotherapy for multidrug-resistant, extensively drug-resistant, and pandrug-resistant Acinetobacter infections: a systematic review. Eur J Clin Microbiol Infect Dis 33(10):1675–1685
Latibeaudiere R, Rosa R, Laowansiri P, Arheart K, Namias N, Munoz-Price LS (2015) Surveillance cultures growing carbapenem-Resistant Acinetobacter baumannii predict the development of clinical infections: a retrospective cohort study. Clin Infect Dis 60(3):415–422
Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A et al (2012) Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 55(7):905–914
Ruppé E, Lixandru B, Cojocaru R, Buke C, Paramythiotou E, Angebault C et al (2013) Relative fecal abundance of extended-spectrum-beta-lactamase-producing Escherichia coli strains and their occurrence in urinary tract infections in women. Antimicrob Agents Chemother 57(9):4512–4517
Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM et al (2000) Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 343(26):1925–1932
Ruppé E, Andremont A (2013) Causes, consequences, and perspectives in the variations of intestinal density of colonization of multidrug-resistant enterobacteria. Front Microbiol 4:129
Nseir S, Di Pompeo C, Soubrier S, Delour P, Lenci H, Roussel-Delvallez M et al (2005) First-generation fluoroquinolone use and subsequent emergence of multiple drug-resistant bacteria in the intensive care unit. Crit Care Med 33(2):283–289
Grohs P, Kerneis S, Sabatier B, Lavollay M, Carbonnelle E, Rostane H et al (2014) Fighting the spread of AmpC-hyperproducing Enterobacteriaceae: beneficial effect of replacing ceftriaxone with cefotaxime. J Antimicrob Chemother 69(3):786–789
Kontopidou F, Plachouras D, Papadomichelakis E, Koukos G, Galani I, Poulakou G et al (2011) Colonization and infection by colistin-resistant Gram-negative bacteria in a cohort of critically ill patients. Clin Microbiol Infect 17(11):E9–E11
Weiss E, Zahar JR, Lesprit P, Ruppé E, Leone M, Chastre J et al (2015) Elaboration of a consensual definition of de-escalation allowing a ranking of beta-lactams. Clin Microbiol Infect. doi:10.1016/j.cmi.2015.03.013
Le Dorze M, Gault N, Foucrier A, Ruppé E, Mourvillier B, Woerther PL et al (2014) Perfomance and impact of a rapid method combining mass spectrometry and direct antimicrobial susceptibility testing on treatment adequacy of patients with ventilator-associated pneumonia. Clin Microbiol Infect. doi:10.1016/j.cmi.2014.12.007
Morosini MI, Garcia-Castillo M, Tato M, Gijon D, Valverde A, Ruiz-Garbajosa P et al (2014) Rapid detection of beta-lactamase-hydrolyzing extended-spectrum cephalosporins in Enterobacteriaceae by use of the new chromogenic Betalacta-test. J Clin Microbiol 52(5):1741–1744
Jamal W (2014) Al Roomi E, Abdulaziz LR, Rotimi VO. Evaluation of Curetis Unyvero, a multiplex PCR-based testing system, for rapid detection of bacteria and antibiotic resistance and impact of the assay on management of severe nosocomial pneumonia. J Clin Microbiol 52(7):2487–2492
de Smet AM, Kluytmans JA, Blok HE, Mascini EM, Benus RF, Bernards AT et al (2011) Selective digestive tract decontamination and selective oropharyngeal decontamination and antibiotic resistance in patients in intensive-care units: an open-label, clustered group-randomised, crossover study. Lancet Infect Dis 11(5):372–380
de Smet AM, Kluytmans JA, Cooper BS, Mascini EM, Benus RF, van der Werf TS et al (2009) Decontamination of the digestive tract and oropharynx in ICU patients. N Engl J Med 360(1):20–31
Roquilly A, Marret E, Abraham E, Asehnoune K (2015) Pneumonia prevention to decrease mortality in intensive care unit: a systematic review and meta-analysis. Clin Infect Dis 60(1):64–75
Daneman N, Sarwar S, Fowler RA, Cuthbertson BH (2013) Effect of selective decontamination on antimicrobial resistance in intensive care units: a systematic review and meta-analysis. Lancet Infect Dis 13(4):328–341
Oostdijk EA, Kesecioglu J, Schultz MJ, Visser CE, de Jonge E, van Essen EH et al (2014) Effects of decontamination of the oropharynx and intestinal tract on antibiotic resistance in ICUs: a randomized clinical trial. JAMA 312(14):1429–1437
Halaby T, Al Naiemi N, Kluytmans J, van der Palen J, Vandenbroucke-Grauls CM (2013) Emergence of colistin resistance in Enterobacteriaceae after the introduction of selective digestive tract decontamination in an intensive care unit. Antimicrob Agents Chemother 57(7):3224–3229
Lubbert C, Faucheux S, Becker-Rux D, Laudi S, Durrbeck A, Busch T et al (2013) Rapid emergence of secondary resistance to gentamicin and colistin following selective digestive decontamination in patients with KPC-2-producing Klebsiella pneumoniae: a single-centre experience. Int J Antimicrob Agents 42(6):565–570
Brink AJ, Coetzee J, Corcoran C, Clay CG, Hari-Makkan D, Jacobson RK et al (2013) Emergence of OXA-48 and OXA-181 carbapenemases among Enterobacteriaceae in South Africa and evidence of in vivo selection of colistin resistance as a consequence of selective decontamination of the gastrointestinal tract. J Clin Microbiol 51(1):369–372
Saidel-Odes L, Polachek H, Peled N, Riesenberg K, Schlaeffer F, Trabelsi Y et al (2012) A randomized, double-blind, placebo-controlled trial of selective digestive decontamination using oral gentamicin and oral polymyxin E for eradication of carbapenem-resistant Klebsiella pneumoniae carriage. Infect Control Hosp Epidemiol 33(1):14–19
Oren I, Sprecher H, Finkelstein R, Hadad S, Neuberger A, Hussein K et al (2013) Eradication of carbapenem-resistant Enterobacteriaceae gastrointestinal colonization with nonabsorbable oral antibiotic treatment: A prospective controlled trial. Am J Infect Control 41(12):1167–1172
Bassetti M, Righi E (2014) SDD and colistin resistance: end of a dream? Intensive Care Med 40(7):1066–1067
Kollef MH, Micek ST (2014) Rational use of antibiotics in the ICU: balancing stewardship and clinical outcomes. JAMA 312(14):1403–1404
Drawz SM, Papp-Wallance KM, Bonomo RA (2014) New beta-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob Agents Chemother 58(4):1835–1846
Zhanel GG, Lawson CD, Adam H, Schweizer F, Zelenitsky S, Lagacé-Wiens PR et al (2013) Ceftazidime-avibactam: a novel cephalosporin/beta-lactamase inhibitor combination. Drugs 73(2):159–177
Testa R, Canton R, Giani T, Morosini MI, Nichols WW, Seifert H et al (2015) In vitro activity of ceftazidime, ceftaroline and aztreonam alone and in combination with avibactam against European Gram-negative and Gram-positive clinical isolates. Int J Antimicrob Agents. doi:10.1016/j.ijantimicag.2014.12.033
Li H, Estabrook M, Jacoby GA, Nichols WW, Testa R, Bush K (2015) In vitro susceptibility of characterized beta-lactamase-producing strains tested with avibactam combinations. Antimicrob Agents Chemother 59(3):1789–1793
Sader HS, Castanheira M, Flamm RK, Farrell DJ, Jones RN (2014) Antimicrobial activity of ceftazidime-avibactam against Gram-negative organisms collected from U.S. medical centers in 2012. Antimicrob Agents Chemother 58(3):1684–1692
Solomkin J, Hershberger E, Miller B, Popejoy M, Friedland I, Steenbergen J et al (2015) Ceftolozane/tazobactam plus metronidazole for complicated intra-abdominal infections in an era of multidrug resistance: results from a randomized, double-blind, phase 3 trial (ASPECT-cIAI). Clin Infect Dis. doi:10.1093/cid/civ097
Falagas ME, Tansarli GS, Ikawa K, Vardakas KZ (2013) Clinical outcomes with extended or continuous versus short-term intravenous infusion of carbapenems and piperacillin/tazobactam: a systematic review and meta-analysis. Clin Infect Dis 56(2):272–282
Dalfino L, Puntillo F, Mosca A, Monno R, Spada ML, Coppolecchia S et al (2012) High-dose, extended-interval colistin administration in critically ill patients: is this the right dosing strategy? A preliminary study. Clin Infect Dis 54(12):1720–1726
Lu Q, Luo R, Bodin L, Yang J, Zahr N, Aubry A et al (2012) Efficacy of high-dose nebulized colistin in ventilator-associated pneumonia caused by multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Anesthesiology 117(6):1335–1347
Authors’ contribution
ER, P-LW and FB contributed equally to the literature search and writing of the review. All authors read and approved the final manuscript.
Compliance with ethical guidelines
Competing interests FB received lecture fees from Novartis, conference invitation from Pfizer, and consulting fees from MSD, ER and P-LW have no potential conflict of interest reledonevant to the present work.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
Table S1. Intrinsic beta-lactam resistance in clinically relevant Enterobacteriaceae species. Table S2. Aminoglycoside-modifying enzymes in Gram-negative bacilli: main clinically relevant types and corresponding resistance profiles.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ruppé, É., Woerther, PL. & Barbier, F. Mechanisms of antimicrobial resistance in Gram-negative bacilli. Ann. Intensive Care 5, 21 (2015). https://doi.org/10.1186/s13613-015-0061-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13613-015-0061-0