
Abstract
Post-traumatic stress disorder (PTSD) is accompanied by disturbed sleep and an impaired ability to learn and remember extinction of conditioned fear. Following a traumatic event, the full spectrum of PTSD symptoms typically requires several months to develop. During this time, sleep disturbances such as insomnia, nightmares, and fragmented rapid eye movement sleep predict later development of PTSD symptoms. Only a minority of individuals exposed to trauma go on to develop PTSD. We hypothesize that sleep disturbance resulting from an acute trauma, or predating the traumatic experience, may contribute to the etiology of PTSD. Because symptoms can worsen over time, we suggest that continued sleep disturbances can also maintain and exacerbate PTSD. Sleep disturbance may result in failure of extinction memory to persist and generalize, and we suggest that this constitutes one, non-exclusive mechanism by which poor sleep contributes to the development and perpetuation of PTSD. Also reviewed are neuroendocrine systems that show abnormalities in PTSD, and in which stress responses and sleep disturbance potentially produce synergistic effects that interfere with extinction learning and memory. Preliminary evidence that insomnia alone can disrupt sleep-dependent emotional processes including consolidation of extinction memory is also discussed. We suggest that optimizing sleep quality following trauma, and even strategically timing sleep to strengthen extinction memories therapeutically instantiated during exposure therapy, may allow sleep itself to be recruited in the treatment of PTSD and other trauma and stress-related disorders.
Similar content being viewed by others

Review
Introduction
This review explores the possibility that disruption of sleep by acute or chronic stress may lead to alterations in emotional memory processing and, thereby, contribute to psychiatric illnesses such as post-traumatic stress disorder (PTSD) [1]. Here, one particular form of emotional memory, extinction of a conditioned fear response (i.e., learning that something that once signaled danger no longer does so) is emphasized. Extinction is a form of emotional memory that is important to normal emotion regulation [2], influenced by normal sleep and its disturbance [3–5], impaired in anxiety disorders [6], and exploited in their treatment [7]. Recent experimental findings, which are reviewed in reference [8], suggest that sleep may play key roles in the consolidation, integration, and balance of fear and extinction memory. The current review focuses on clinical issues and puts forward the hypothesis that one mechanism leading from psychological trauma to PTSD is stress-related sleep disturbances that interfere with sleep-dependent consolidation of emotion-regulatory neuroplasticity such as fear extinction and habituation.
Disturbances of sleep and of emotions are reciprocally related
Healthy sleep is associated with normal emotion regulation [9, 10]. Conversely, sleep disturbance is both a common behavioral sequela of acute and chronic stress [11, 12] and a prominent symptom of anxiety and mood disorders [13, 14]. Specifically, sleep disturbance is a characteristic sequela of psychological trauma although subjective reports often indicate far greater severity than objective measurements in the immediately post-trauma period [15, 16]. While daytime affective symptoms and associated neural, physiological, and endocrine disturbances can adversely affect sleep, there is growing evidence that sleep disturbances (e.g., insomnia) can reciprocally impact daytime symptoms. For instance, epidemiological and prospective studies show that sleep disturbances that are present prior to trauma exposure, or that occur soon after trauma exposure, are a robust risk factor of poor psychiatric outcomes including PTSD, anxiety disorders, mood disorders, suicidality, and alcohol/substance use disorders [17–20]. Similarly, pre-existing insomnia has been shown to be a risk factor for incident depression [21–23]. The presence of untreated sleep disturbances comorbid with psychiatric disorders tends to attenuate treatment response and increase the risk of relapse [24–27]. Conversely, the persistence of consolidated sleep following stress or trauma exposure, as well as sleep improvements over the course of treatment for affective disorders, are associated with better mental health outcomes [28, 17].
As a result of such observations, it has been widely hypothesized that sleep disturbance is crucially involved in the etiology of PTSD rather than being solely a symptom arising secondarily from this disorder [16, 29–36]. In a comprehensive review on the temporal sequence of sleep disruption following traumatic events and the subsequent emergence of PTSD, Babson and Feldner [16] have shown that, in many cases following psychological trauma, subjective and, to a lesser extent, objective sleep disturbances can precede PTSD diagnosis thus providing clear evidence that such an etiological role of sleep is a distinct possibility. They note, however, the study of potential mechanisms for such a role is only in its infancy. The current review begins to explore evidence of one such factor, impaired fear extinction.
Involvement of sleep disturbance in the pathophysiology of PTSD does not, of course, exclude the more traditional view that psychiatric illness produces unique sleep disturbances or exacerbates pre-existing ones. Moreover, it is likely that a third vulnerability factor, such as individual variability in the degree to which psychological stress provokes enduring arousal in central limbic and autonomic circuits, can contribute to both poor sleep and increased risk of psychopathology. For example, waking hypervigilance and sleep disturbance could both arise from excess sympathetic activation without a direct interaction between the waking and sleep effects of such hyperarousal. As discussed below, chronic hyperarousal is increasingly implicated in the development of insomnia [37–42]. Similarly, repetitive nightmares and daytime traumatic memory intrusions may reflect a similar priming or disinhibition of retrieval for stored representations of the traumatic event, again without direct interaction between these two phenomena. Moreover, it has been suggested that sleep loss may secondarily diminish daytime coping strategies increasing the likelihood of developing psychopathology. Similarly, nightmares may sensitize individuals to waking trauma cues, or sleep disruption may directly exacerbate anxiety (reviewed in [16]). As in other disorders of biological systems, it is likely that pathogenic factors interact and that impaired negative feedback, escalating positive feedback, or compensatory allostatic mechanisms allow abnormalities in one domain to exacerbate those in others [43]. Therefore, we suggest that sleep disturbance and its negative effect on extinction memory is one of a number of neurocognitive and physiological pathways that could exacerbate risk of developing PTSD following a traumatic experience. For example, other neurocognitive factors potentially escalating risk of PTSD following initial trauma might include persistent threat of re-traumatization (enhanced conditioning), whereas physiological factors might include poor nutritional status (impairment of memory processing).
The temporal development of PTSD following psychological trauma
Before proceeding to consider how sleep disturbance following trauma could contribute to the development of PTSD, it must first be established that PTSD is a disorder that can, in fact, develop over time following trauma rather than simply being an acute stress disorder (ASD) [1] that persists beyond an arbitrary 1-month threshold [1, 44]. What is the evidence that this is the case?
First, in a systematic review of prospective studies, among 19 studies of adults, following a median 6-month follow-up, a median of only 50 % of those with ASD subsequently met criteria for PTSD, whereas a median of only 47 % of those with PTSD previously met criteria for ASD [45]. Second, in a study of over 1000 traumatic injury survivors, only about a third of persons who developed PTSD by 1 year following a traumatic event showed ASD immediately following the trauma [46]. Importantly, this percentage increased by only about 9 % when a more liberal (subsyndromal) definition of ASD, not requiring dissociative symptoms, was used [46]. A similar percentage of persons with ASD (36 %) or subsyndromal ASD (30 %) went on to develop PTSD, although 65 % did eventually develop some psychiatric disorder.
Third, among post-combat military populations, it is not uncommon for diagnosed PTSD to emerge only after a delay of several months post-deployment [47]. For example, 88,235 Army soldiers were evaluated immediately upon returning from the Iraq war with a self-administered Post-Deployment Health Assessment that included specific screening questions for PTSD [48]. These same individuals were then re-evaluated with an assessment that contained the same PTSD screening questions at a median of 6 months after finishing the first evaluation [47]. In this re-assessment, report of psychological distress was markedly higher and reports of PTSD symptoms increased from 11.8 to 16.7 % in active-service Army and from 12.7 to 24.5 % in Reserve and National Guard [47]. Notably, among those who had reported PTSD symptoms on the immediately post-deployment assessment, approximately half reported improvement of these symptoms at re-assessment [47]. Therefore, the increased proportion of individuals reporting PTSD symptoms at re-assessment must have included individuals in whom symptoms appeared following their first assessment.
Fourth, although delayed-onset PTSD, defined most strictly as onset of any PTSD symptoms only after 6 months or more following trauma, is controversial and rare [49, 50], exacerbation of existing symptoms is common [49]. For example, a comprehensive review reported that, over the 6 months following a traumatic experience, worsening of existing PTSD symptoms or re-emergence of previously experienced symptoms was reported by 15.3 % of civilians and 38.2 % of military personnel [49]. Therefore, sleep disturbance may directly worsen existing symptoms, or, as here suggested, result in a failure to ameliorate such symptoms through consolidation of naturalistic or therapeutic extinction learning. In either case, the characteristic PTSD symptoms of intrusions (including nightmares), avoidance, negative affect, and hyperarousal [1] clearly may emerge or worsen over the initial months following a traumatic event.
Fear conditioning and extinction
Fear conditioning occurs when an emotionally neutral stimulus is associated with an inherently aversive experience (unconditioned stimulus or US). The neutral stimulus thereby becomes a conditioned stimulus (CS) with the capability, on its own, to evoke a fearful conditioned response (CR). When the CS is subsequently presented repeatedly without the US, extinction (reduction) of the CR typically takes place. However, rather than erasing the CS-US association, extinction represents formation of a new memory, an “extinction memory,” signifying “CS-no US,” that competitively inhibits the memory of the CS-US contingency and expression of its associated CR when the CS is again encountered [51–59]. Neuroimaging research using de novo fear-conditioning and extinction paradigms has revealed areas in the brain associated with experiencing conditioned fear (a “fear expression network”) in the amygdala and dorsal anterior cingulate cortex (dACC) and other areas associated with memory for the extinction (inhibition) of this fear (an “extinction memory network”) that includes the hippocampus and ventromedial prefrontal cortex (vmPFC) [8, 59–61] (Fig. 1).

The “anterior paralimbic REM activation area” overlaps with fear and extinction circuits. 18Fluoro-deoxyglucose PET image of areas that reactivate during REM sleep following relative quiescence during NREM sleep. Dashed lines surround approximate cortical regions commonly activated in experimental protocols during fear conditioning (yellow lines) and during recall of extinguished conditioned fear (white) based upon Milad and Rauch [61], Fig. 3. Solid lines encircle approximate anatomic loci of subcortical structures similarly activated during fear conditioning (yellow) and extinction recall (white). The anterior paralimbic REM activation area includes the amygdala (A), and regions of dorsal anterior cingulate (dACC) and insular (not shown) cortices linked to a putative fear expression network. Additionally, this region includes the ventromedial prefrontal (vmPFC) and hippocampal (H) areas [127–129] linked to a putative extinction memory network
Extinction learning, viz. learning the “CS-no US” contingency, is the neurobehavioral basis for the efficacy of exposure therapy [7, 62]. Means by which memory for this therapeutic learning may be strengthened, and relapse to dominance of the fear (CS-US) memory prevented, are currently the subject of extensive clinical research [62–64]. It is also, however, important to recognize that extinction is a process that is ongoing in the course of everyday life. For example, individuals who display resilience and recovery, without any therapeutic intervention, following a psychologically traumatic event, presumably acquire extinction memories based upon spontaneous encounters with reminders of the trauma. And these extinction memories, in turn, prevent subsequent trauma cues from triggering fearful responses. And, as is the case for other forms of emotional memory [10], healthy sleep may be of ongoing, and cumulative importance in the consolidation of memory for both therapeutically induced and naturally learned extinction.
Sleep-dependent memory consolidation
Extinction memory must be encoded, consolidated, and then retrieved in order to oppose conditioned fear. For declarative and procedural forms of memory, sleep has been widely demonstrated to promote the consolidation stage of memory formation [65–68], including processes related to prioritization and integration of newly acquired memories with existing stores [69–72]. Moreover, prior sleep can facilitate memory retrieval via such functions as protection from retroactive interference [73] and enhanced updating during reconsolidation [74]. Rapid eye movement (REM) sleep is associated with consolidation of emotional memory (reviewed in [8–10]), and REM sleep has been suggested to be the stage of sleep during which emotion is regulated [75]. For example, in the “Sleep to Remember, Sleep to Forget” model, Walker and colleagues suggest that REM sleep serves the dual purpose of consolidating the content of emotional memory and diminishing the memory’s emotional charge [75, 10]. Similarly, regulation of mood and working through of emotional responses to intra- and interpersonal stressors have been linked with REM sleep and associated dreaming [76–78]. Significantly, a broad anterior midline area of cortex and subcortex (the “anterior paramedian REM sleep activation area” detailed below) selectively activates during REM sleep following relative deactivation during non-REM (NREM) sleep [79], and this region encompasses both the fear expression and extinction memory networks (Fig. 1).
Physiological processes underlying sleep’s effects on memory consolidation have been demonstrated in animals and include replay, during sleep, of patterns of hippocampal place-cell firing that accompanied learning [80, 81]. Similar encoding-induced changes in subsequent sleep physiology are reported human polysomnographic and neuroimaging studies (reviewed in [67]). Post-learning sleep may facilitate synaptic, second messenger, gene transcription, and protein synthesis steps required for memory consolidation [82] such as N-methyl-D-aspartate (NMDA) receptor-dependent hippocampal long-term potentiation [83, 84]. Critical periods requiring sleep, including specifically REM sleep, for memory consolidation following encoding have been demonstrated in animals and humans [85], and such a period for extinction memory has recently been demonstrated for REM sleep [86].
Extinction and disorders with abnormal levels of anxiety
Abnormal levels of anxiety seen in Diagnostic and Statistical Manual of Mental Disorders—5th ed. (DSM-5) Anxiety Disorders and Trauma- and Stressor-Related Disorders suggest a deficiency in emotion-regulatory mechanisms. A deficit in the ability to encode, consolidate, or retrieve extinction memory is believed to play a role in the development and perpetuation of such disorders [6, 59, 61, 87].
Having hypothesized deficient extinction in PTSD, at what point in the processes of fear acquisition, extinction learning, and extinction memory does this problem arise? Deficient memory for extinction has been shown to differentiate individuals with PTSD from trauma-exposed controls at both the behavioral and neural levels [88–90]. Notably, in these particular studies, acquisition of fear conditioning and extinction learning did not differ between these groups [88–90]. Other studies have specifically implicated a deficiency in PTSD to use contextual information to disambiguate danger versus safety [91]. Such findings would suggest that a deficiency in emotional memory systems might be of primary etiological importance in PTSD as might be expected given the above-cited abundant evidence for sleep effects on memory consolidation. Nonetheless, other studies do suggest greater de novo fear conditioning in PTSD [92] and deficient acquisition of extinction [93–95]. In addition, the degree of recall of de novo fear conditioning may predict later development of PTSD symptoms [96, 97]. Moreover, enhanced physiological reactivity to acoustic startle stimuli compared to controls is also commonly noted in this disorder [98–100]. Therefore, lesser capacity to acquire extinction, possibly due, in part, to enhanced ability to acquire a conditioned fear that is itself related to augmented autonomic and limbic reactivity may also play a role, particularly in the hyperarousal symptoms of PTSD. Interestingly, such hyperarousal also produces sleep disturbance that, in turn, may further disrupt sleep-dependent memory processes as detailed below.
The gold standard treatment for certain disorders with abnormal levels of anxiety involves formation of therapeutic extinction using exposure therapy [7, 62]. In such treatment, the patient is exposed to imaginal, pictorial, video, virtual reality, or in vivo representations of feared stimuli for a sufficient duration that anxiety is experienced and withstood and the patient thereby develops a new, inhibitory memory that opposes subsequent fear responses [7, 62, 101, 102]. Exposure is especially effective when fearful symptoms are associated with specific stimuli as in the case of PTSD [103], social anxiety disorder [104], obsessive-compulsive disorder [105], and specific phobia [106]. It is especially important to promote the generalization of extinction memories acquired during exposure sessions to prevent the return of fear outside of the safe, therapeutic context [3, 62, 64, 107–110].
An important distinction made during exposure therapy is between within-session learning by which extinction/habituation is initially acquired and between-session extinction/habituation, or the persistence of such learning across time from one exposure session to another [62]. (The combined term “extinction/habituation” is used because habituation is difficult to differentiate from extinction in clinical practice [111].) Note, however, that typically in exposure therapy, within-session extinction/habituation is continued following each session in the form of exposure homework (e.g., [103, 112]); therefore, the encoding and consolidation of extinction/habituation is, in reality, an iterative process. Between-session extinction/habituation corresponds to memory for what was learned within session and thus requires consolidation to persist over time. In the case of between-session extinction, this involves consolidation of an associative memory (e.g., of the CS-no US contingency) and, in the case of between-session habituation, the consolidation is of neural changes corresponding to a non-associative learning process [8]. Current animal research suggests that within-session and between-session extinction are dissociable processes [113], and studies of exposure therapy also show that the degree of within-session extinction does not predict the extent of the between-session extinction that, cumulatively, leads to clinical improvement [62]. Consequently, much research has gone into ways to strengthen this new learning via the timing, spacing, and gradation of intensity of exposures, manipulations of aspects of the environments or stimuli in which it is carried out, pharmacological interventions to potentiate the encoding and consolidation of inhibitory memory, and prevention of spontaneous recovery, renewal, or reinstatement of fear responses [62–64, 101, 114]. Sleep strategically timed so as to promote consolidation of extinction memory constitutes a potential new technique directed toward this same goal [3, 115]. Memory consolidation processes also provide opportunity for extinction/habituation to generalize, and sleep appears to augment this process as well [3, 107]. Specific clinical implications regarding the use of sleep as a means to enhance extinction/habituation are discussed in the “Sleep and exposure therapy” section below.
Brain bases of deficient extinction in PTSD
PTSD patients show structural abnormalities in limbic regions associated with extinction recall including the perigenual anterior cingulate, amygdala, and hippocampus [6, 116, 117]. These are accompanied by greater functional activation of the fear expression network (amygdala and dACC) and lesser activation of the extinction network (hippocampus and vmPFC) during de novo fear-conditioning and extinction experiments [59, 60, 118–120]. Compared with trauma-exposed controls, those with PTSD show greater amygdala activation during extinction learning, and, during extinction recall, lesser activation of the vmPFC and hippocampus but greater activation of the dACC [88]. Therefore, in PTSD, there is both hyperactivation of the fear expression and hypoactivation of the extinction memory networks [59, 60]. However, not all neuroimaging studies show functional differences between PTSD and trauma-exposed controls in all of these loci or at the same anatomic coordinates within them. While a complete review of this diverse literature is beyond the scope of this article, excellent reviews can be found in [116, 119–126].
Importantly, the midline limbic and paralimbic areas that selectively activate during REM sleep (Fig. 1) encompass these same networks that show structural and functional abnormalities in PTSD. For example, this “anterior paralimbic REM sleep activation area” [79] includes the amygdala, and regions of anterior cingulate and insular cortex [127–129] that are linked to a putative fear expression network [61]. Similarly, this region includes the ventromedial prefrontal and hippocampal areas [127–129] linked to a putative extinction memory network [61]. As noted, these fear-related structures are hyperactive and extinction-related areas hypoactive in PTSD [88, 130].
Sleep and the anxiety-related disorders
These common mechanisms in etiology, perpetuation, and treatment suggest that factors that strengthen or weaken extinction, such as good and poor sleep, respectively, may apply similarly across anxiety, traumatic stress, and obsessive-compulsive disorders. Sleep disruption is a DSM-5 [1] diagnostic criterion for generalized anxiety disorder and PTSD, is common in panic disorder [131, 132], and appears, more subtly, in obsessive-compulsive disorder [133]. Because both sleep and extinction appear to be degraded in PTSD, their interaction represents one putative mechanism contributing to the development and persistence of PTSD symptoms. And because treatment of PTSD with exposure-based therapies relies upon formation and strengthening of extinction memory, the memory-enhancing function of healthy sleep may play a role in recovery and disturbed sleep in treatment resistance.
Sleep disruption in PTSD
Degradation of subjective and/or objective sleep quality is commonly reported in studies of individuals with PTSD [13, 16, 31, 134–136]. Sleep disruption and repetitive nightmares meet DSM-5 PTSD criteria for “alterations in arousal and reactivity” and “intrusion symptoms,” respectively [1]. For example, in a self-report study, the severity of PTSD symptoms predicted sleep problems to a much greater degree than age, gender, psychiatric comorbidity, type of trauma, or chronicity of PTSD [137]. Persistent trauma-related nightmares of a replicative nature are a near-universal symptom of PTSD [29, 138].
For objective sleep measures, a recent meta-analysis [134] found that, among the highly variable alterations of sleep in PTSD compared to control groups, increased stage 1 NREM sleep, decreased slow wave sleep (SWS) (see also [139]), and increased average number of rapid eye movements per minute in REM sleep (REM sleep density) were the most consistent abnormalities across studies. Additional abnormalities expressed by subgroups of PTSD patients included shorter total sleep time (TST), increased sleep onset latency, reduced stage 2 NREM sleep, and increased REM sleep as percent of TST [134, 140]. Polysomnographic studies have also shown that EEG spectral power at delta frequencies is significantly decreased in PTSD patients [139, 141, 142]. These abnormalities are consistent with an underlying hyperarousal in PTSD that lightens sleep, prevents deeper, more restorative sleep stages, and alters the distinct physiology of REM sleep [20, 29, 36, 134, 143]. Evidence that PTSD may influence the quality versus absolute quantity of REM includes not only greater REM density [134], but the fact that some studies have shown greater percent REM in PTSD [140, 139].
Therefore, both objective and subjective sleep disturbances represent core features of PTSD [36, 134, 136]. As noted, however, specific polysomnographic sleep abnormalities reported in different studies of PTSD are highly variable in type and severity [134] and can vary with age, sex, comorbidities, and other factors (for reviews, see [13, 134, 139, 144, 145]).
Sleep abnormalities predict PTSD
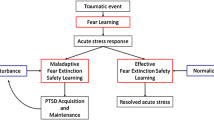
Objective and subjective sleep abnormalities, including insomnia complaints, that either precede or follow traumatic experiences, predict later development of PTSD (reviewed in [16, 33]). For example, motor vehicle accident survivors who later developed PTSD, unlike survivors who did not, had more severe immediately post-trauma sleep disturbances that failed to normalize over time [146]. Similarly, Mellman and colleagues showed that subjective insomnia, nightmare severity, and abnormalities of REM sleep, especially its fragmentation in the early aftermath of a traumatic injury, predicted later development of PTSD [20, 147–149]. In addition, higher sympathetic drive during REM sleep within 1 month of trauma was associated with development of PTSD symptoms at 2 months post-trauma [149]. Such sleep disruption might impede normal processing of emotional memories following trauma [20] including the ability to consolidate memory for the extinction of fear associated with traumatic memories (Fig. 2).

Possible pathway whereby sleep disruption accompanying acute response to trauma can lead to PTSD. In vulnerable individuals, acute post-traumatic insomnia can become chronic and disrupt processes of sleep-dependent emotional memory consolidation, thereby contributing to the etiology of PTSD. Chronic sleep disruption can subsequently perpetuate PTSD symptoms by continued interference with normal processing of emotional memories as well as impaired consolidation of therapeutic extinction memories if exposure therapy has been initiated. Stars indicate possible strategic points for sleep interventions to prevent PTSD onset or enhance exposure-based treatment
It is noteworthy that only a minority of individuals who have experienced a traumatic event develop PTSD. For example, among 99 studies on diverse disasters, the prevalence of PTSD at first assessment averaged 27 % [150]. Similarly, prevalence of PTSD in combat-exposed infantry is only around 20 % [151]. And, as noted above, only around 30 % of individuals with ASD go on to develop PTSD [46]. Therefore, factors other than trauma exposure alone or the acute reaction to trauma must contribute to the development of PTSD. In light of the preceding findings, we suggest that alterations in the emotion regulatory functions of sleep might be one such factor.
Extinction and sleep in PTSD
Because sleep deprivation reduces amygdala-vmPFC functional connectivity [152] as well as task-related activation of the ventral anterior cingulate cortex (part of the vmPFC) in a positron emission tomography (PET) study [153], trauma-induced sleep loss might specifically impair consolidation of extinction memory via interference with vmPFC-amygdala circuitry. A finding that longer sleep on the night preceding functional magnetic resonance imaging (fMRI) scans was positively associated with both resting-state amygdala-vmPFC functional connectivity and higher self-report indices of mental health indicates that even mild restriction of sleep can diminish vmPFC-amygdala connectivity [154]. Animal studies show that stress-associated cues continue to interfere with REM sleep long after the original stressful experience [155–157], raising the possibility that REM sleep alterations in humans may play a role in both the acquisition and perpetuation of PTSD (Fig. 2). Although, there are not yet published studies on both sleep and extinction in PTSD patients, circuits involved in fear and extinction learning and memory are implicated in sleep-related symptoms of PTSD such as nightmares [138]. For example, in combat-exposed veterans with versus without PTSD, REM sleep is characterized by increased metabolic activity in the amygdala and anterior paralimbic regions, and reduced metabolism in hippocampal regions [158]. It is important to note that, in addition to effects of sleep on amygdala-vmPFC connectivity, trauma itself may affect such circuits, as is suggested by reports of structural abnormalities in these areas in PTSD [116, 120].
Sleep-related neuroendocrine abnormalities in PTSD—relationship to emotional memory consolidation?
By what mechanisms might both sleep and extinction memory become progressively degraded following traumatic stress? One possibility is that physiological stress responses produce sleep disruption that, via positive feedback, perpetuates these stress responses. In the rat, following experimental stress induction paradigms, sleep shows a number of compelling parallels to changes in human sleep following traumatic stress and in PTSD. For example, in the rat, fear conditioning and other forms of inescapable stress lead to disruption of sleep and fragmentation of REM sleep, conditioned reminders produce similar sleep-disruptive effects for several weeks post conditioning, and extinction training reverses these sleep effects (reviewed in ref. [157]). Such sleep disturbances in the rat have been linked with the actions of central stress systems including the sympathetic response, the hypothalamic-pituitary-adrenal (HPA) axis, and the central extra-hypothalamic stress system (reviewed in refs. [155, 157]). Within and between these stress systems, there are positive feedback mechanisms whereby neuroendocrine responses lead to elevated arousal and sleep disturbance that can, in turn, further activate stress responses. Abnormal activation of these stress systems has also been reported in PTSD, and these systems may interact following traumatic stress in a manner analogous to findings in animal models of stress. As depicted in Fig. 3, following a traumatic stressor, such interactions may disrupt sleep as well as sleep-mediated processing of extinction memory producing an escalating abnormality potentially leading to PTSD. The following section first describes neuroendocrine abnormalities in these three stress systems reported in PTSD. We then examine their potential impact on fear and extinction memory and interactions with sleep.

Hypothetical interactions among activated stress systems and disrupted sleep in PTSD. Note that multiple positive feedback loops result in depicted effects at any one node further driving effects at other nodes. Experimental evidence for many specific interactions depicted is provided in text. For clarity, the following mechanisms mentioned in the text are not depicted: 1) interaction between sympathetic activation and the HPA axis, 2) possible negative feedback mechanisms involving hypothalamic corticotropin releasing factor (CRF) that may explain hypocortisolemia in PTSD, and 3) direct effects of stress systems on extinction memory and habituation. The dashed line depicts an additional positive feedback mechanism whereby poor extinction memory promotes continued activation of neuroendocrine stress systems by failing to inhibit expression of conditioned fears. HPA hypothalamic-pituitary-adrenal, CRF corticotropin releasing factor, NE norepinephrine
Noradrenergic abnormalities [“Sympathetic activation (NE)”]
PTSD is associated with elevated levels of central [159, 160] and urinary [161] norepinephrine, including measurements taken during sleep [161]. Normal NREM sleep is associated with a marked decrease in sympathetic and increase in parasympathetic drive [162–164]. Central secretion of norepinephrine (NE), the catecholamine responsible for the acute sympathetic stress response, acts to oppose REM sleep [165], and NE normally declines with sleep onset and deepening NREM sleep to reach its nadir in REM sleep [165]. Because nocturnal secretion of NE may remain relatively elevated in PTSD [159–161], this may be one factor serving to fragment REM sleep [29]. The success of the alpha-adrenergic antagonist prazosin in treating PTSD nightmares is strong evidence of NE involvement in the pathophysiology of this disorder [166, 167].
HPA-axis abnormalities [“HPA axis (cortisol)”]
Persons with PTSD frequently show abnormalities of the HPA axis [168]. The initiating factor of the HPA response is corticotropin-releasing factor (CRF), a polypeptide neurohormone whose secretion by the paraventricular nucleus (PVN) of the hypothalamus triggers release of adrenocorticotropic hormone (ACTH) from the anterior pituitary leading to the secretion of adrenal glucocorticoids [169]. Paradoxically, although CRF is elevated in the cerebrospinal fluid (CSF) of patients with PTSD [170–172], abnormally low baseline levels of plasma cortisol are typically observed in this disorder [173], possibly due to downregulation of pituitary CRF receptors resulting from elevated CRF [174, 175].
Central actions of CRF [“Central stress response (CRF)”]
Although CRF triggers release of cortisol via ACTH and PTSD may be characterized by low peripheral (plasma) levels of cortisol, variations in levels of central CRF and plasma cortisol are dissociable. This is illustrated, for example, by their circadian rhythms [164]. CRF sampled hourly in the cerebrospinal fluid of healthy volunteers showed an evening acrophase and morning nadir [176]—directly opposite to the pattern of plasma cortisol [177]. In addition to its effect on the HPA axis, CRF from the PVN as well as from the central nucleus of the amygdala (CeA) is a key neuromodulator activating the central extrahypothalamic stress system via CRF1 receptors in the CeA, basolateral nucleus of the amygdala (BLA), bed nucleus of the stria terminalis (BNST), and locus coerulus (LC) [178, 179]. Activation of CRF receptors in the BNST is associated with persistent (versus acute) threat responses in the rat [180, 181]. BNST activation also tracks sustained anxiety in humans [182], and sustained anxiety may better predict symptoms of PTSD than acute fear responses [183].
Positive feedback between central stress systems, disrupted sleep and REM sleep, and impaired extinction memory as a putative pathway for the escalation of post-traumatic psychopathology
Figure 3 suggests that activation of central stress systems not only mutually elevates activity among these systems themselves but also produces sleep disruption—itself a stressor that can further activate stress systems. Here, we present evidence for mutual activation between stress systems, followed by evidence for their reciprocal relationship with disrupted sleep and, finally, by suggestion of how impairment of extinction by poor sleep can further exacerbate stress responses.
CRF-ergic activation promotes secretion of NE by the LC [184, 185]. In turn, increased NE can stimulate the PVN resulting in further CRF release and activation of the HPA and central stress responses [184, 186]. Therefore, NE and CRF can reciprocally stimulate release of the other to escalate central stress responses [178] (Fig. 3).
Exogenous CRF disrupts sleep [187], endogenous CRF promotes waking [188], and sleep deprivation elevates endogenous CRF [189]. Recent studies in rodents suggest that the stress-induced reduction in REM sleep is attributable to the actions of CRF [190–192] as is the more general post-stress sleep fragmentation [193]. Chronically disturbed sleep can produce a persistent elevation of sympathetic activity and central NE [164, 194, 195]. Increased NE, in turn, activates other stress systems via its action on subcortical limbic structures such as the amygdala [178]. For example, based upon studies using a single-prolonged stress model of PTSD in the rat [196], a specific noradrenergic mechanism during sleep has been recently proposed to act on hippocampal-prefrontal systems and impair processing of traumatic memories in PTSD [197]. Therefore, sleep deprivation or restriction can generate a positive feedback cascade whereby central stress responses and sleep disruption mutually reinforce one another (Fig. 3). Thus, trauma exposure may precipitate a failure of sleep-dependent neuroendocrine processes that normally promote return to emotional homeostasis via nocturnal reductions in catecholamine levels, sympathetic drive, as well as HPA axis, and central CRF-ergic activity. Such changes may contribute to the development of PTSD in vulnerable individuals.
However, in addition to interactions taking place entirely within the interacting physiology of stress and sleep, failure to extinguish fear may also exacerbate stress and further drive the potentially pathogenic physiological interactions described above. Deprivation, curtailment, and fragmentation of sleep, and specifically REM sleep, can affect the processing of emotional memory including the consolidation and generalization of extinction (reviewed in [8]). Moreover, the direct effects of stress and stress hormones on memory are multifold (reviewed in [198]), and memory for extinction of fear conditioning may be especially susceptible to stress effects [199]. Therefore, the persistence of conditioned fear, in the face of its failed extinction (dashed line in Fig. 3), may continue to activate stress systems and further exacerbate positive feedback mechanisms that lead to further impairment of extinction and the persistence of pathological fear.
The exact ways in which REM sleep is altered in the period following a traumatic event, as well as after PTSD symptoms have developed, are not yet fully understood, and as noted above, a simple consistent quantitative change is not observed. Nonetheless, there is suggestive evidence in the fragmentation of REM following trauma [147, 148] or following inescapable stress in the rat [157] as well as in the increased REM density once PTSD has developed [134] that hyperarousal of limbic structures during REM may be one characteristic abnormality. The neurochemical changes in arousal systems observed in PTSD detailed above may underlie or contribute to such limbic hyperarousal during REM sleep, and recurrent REM sleep nightmares may be a subjective manifestation. The effects of limbic hyperarousal in REM sleep on consolidation of conditioned fear and its extinction may be to bias consolidation processes taking place via neuronal replay and other mechanisms during sleep (reviewed in reference [8]) toward the fear expression and away from the fear extinction networks described above. The underlying changes in REM sleep in PTSD thus remain areas in need of additional study.
Insomnia, emotional dysregulation, and PTSD
The preceding discussion reviewed evidence that sleep disturbance is a cardinal symptom of PTSD that can appear prior to and predict PTSD symptoms. We reviewed evidence that stress responses and sleep disturbance can mutually exacerbate each other via neuroendocrine systems that also show abnormalities in PTSD, and that such abnormalities could potentially interfere with extinction learning and memory. Evidence that experimental sleep manipulations can influence fear conditioning and extinction is reviewed separately in [8]. However, to what extent can sleep disturbance predating or acutely following trauma itself initiate these pathogenic events? Examining psychopathological correlates and consequences of insomnia may begin to address this question.
The ubiquity of chronic insomnia both as a primary disorder and comorbid with psychiatric [200, 201] and non-psychiatric [202] conditions suggests that it reflects a trait vulnerability that can be triggered by a variety of stressors. Stressful events are a significant predictor of insomnia with odds of incident insomnia increasing in a dose-response manner for each such event [203]. The following section considers insomnia as a potential contributor to PTSD.
Emotional dysregulation and hyperarousal in insomnia
Insomnia is associated with dysregulation of emotions pertaining to sleep itself [204, 205]. However, a more general emotional dysregulation is a characteristic of many individuals with insomnia [206] that can be reflected in personality variables [207] such as a tendency to internalize conflict [208] as well as by the high comorbidity of insomnia with mood and anxiety disorders [22, 32, 200, 209]. Such findings have led to the suggestion that emotional reactivity is both a risk and perpetuating factor for the development of chronic insomnia [206, 210].
Contributing to this emotional dysregulation is the now well-replicated evidence for chronic hyperarousal in insomnia [37–39]. Such hyperarousal is manifested in both peripheral [37] and central [38] physiology as well as in pre- and post-morbid cognitive style [40, 41] and sensitivity of sleep quality to acute stress [42]. Acute insomnia is ubiquitous following a wide variety of stressors [211], and insomnia following traumatic events [146] including combat [33] is predictive of later development of PTSD [16].
REM sleep disruption in insomnia
As noted above, there is strong evidence that REM sleep is important in the emotion-regulatory function of sleep. For example, REM sleep fragmentation following a traumatic event is predictive of later development of PTSD [147, 148].
Although early polysomnographic studies of insomnia reported little change or small reductions in REM sleep compared to good sleepers [212], there is now increasing evidence for both percentage reductions [204] and fragmentation [213–215] of REM sleep in insomnia. Being the stage of sleep with the highest level of forebrain arousal [38], REM sleep may also be the most vulnerable stage to disruption by awakenings due to chronic physiological and cognitive arousal. This is because, in this activated behavioral state, the brain is closer to its threshold for awakening [213, 214].
Neuroimaging studies of Insomnia
Evidence is accumulating that functional abnormalities in emotional regulatory networks that overlap with the fear expression and fear extinction networks also occur in insomnia. Patients with insomnia showed higher levels of arousal (greater glucose metabolism) during NREM sleep compared with good sleepers, and objective and subjective increases in sleep disruption were positively associated with metabolic activity in the anterior cingulate cortex [38, 216]. Poor quality of sleep may negatively impact the ability of the vmPFC to consolidate and later express extinction memory. During resting-state fMRI, functional connectivity between the amygdala and other brain areas was reduced in persons with insomnia compared with healthy controls [217]. Specifically, amygdala connectivity with the insula, striatum, and thalamus was reduced, again suggesting dysfunction in emotion regulatory circuits.
Our studies suggest that insomnia patients show hyperactivation of the dACC and hypoactivation of the vmPFC during REM sleep. Using 18FDG-PET, insomnia patients showed a greater increase in cerebral glucose metabolism from wakefulness to REM sleep compared to good sleepers in an anterior midline region (Fig. 4a) in close proximity to the region of the dACC that has been associated with fear expression (Fig. 1). In addition, insomnia patients showed a smaller increase in cerebral glucose metabolism in the vmPFC from wakefulness to REM sleep (Fig. 4b). As noted, this latter area is associated with the memory and expression of fear extinction (Fig. 1). Thus, a closer investigation of the effects of chronic insomnia on fear learning and memory may provide novel insights into psychophysiological and neural mechanisms underlying anxiety and mood disorders.

Comparison of REM activations in individuals with insomnia versus without insomnia. When comparing REM to wake, there is a greater increase in regional cerebral glucose metabolism (18fluoro-deoxyglucose PET) in an anterior midline region in close proximity to the region of the dACC that has been associated with fear expression (a). However, in a comparison of two different groups, the insomnia group showed lesser increase in the vmPFC, an area associated with the memory and expression of fear extinction (b)
Insomnia and PTSD
The normal, sleep-disrupting after-effects of a traumatic experience may develop into diagnosed chronic insomnia disorderFootnote 1 or may be expressed as a more short-term, reactive sleep disturbance that does not meet the duration criteria of a chronic disorder.Footnote 2 In either case, the likelihood that PTSD will later develop may increase due to the neurohormonal and mnemonic processes detailed above. Similarly, if an individual has poor sleep quality due to a pre-existing sleep disorder (such as obstructive sleep apnea) or is experiencing poor sleep due to limited sleep opportunity or sleep during an unfavorable circadian phase (as is common in the military), these same factors may increase vulnerability to PTSD irrespective of formal insomnia diagnoses. Indeed, among military service members, pre-deployment symptoms of insomnia have been shown to confer increased risk of post-deployment PTSD symptoms [17] and individuals with self-reported, pre-existing sleep problems had increased likelihood of developing PTSD following Hurricane Andrew [218]. Poor extinction memory may impair the ability to modulate arousal that results from stressors and thus could synergize with the physiological and cognitive hyperarousal of insomnia [37–40] to further elevate the risk of developing PTSD. Further evidence that insomnia can be primary is the fact that, whereas insomnia comorbid with anxiety disorders responds well to cognitive behavioral therapies developed for primary insomnia [209, 219], sleep disturbance often persists following successful treatment of PTSD [31]. Moreover, sleep-focused treatments can significantly improve both sleep and daytime symptoms of PTSD [220–222]. Therefore, insomnia may represent an emotionally dysregulated state that can contribute to the development of PTSD as well as exacerbate its symptoms and impede its treatment.
Sex differences in extinction memory, insomnia and PTSD
Prevalence is greater in females than in males for both insomnia [223] and PTSD [224]. Translational studies with both humans [225–229] and rodents [230, 227, 228] have shown that extinction memory is sexually dimorphic (better in males) and that it varies across the menstrual cycle in females [225, 226]. Gender differences in the relationship between PTSD and sleep are also now being reported [144, 231]. For example, following a traumatic event, females who progressed to PTSD showed greater wake time after sleep onset than males who similarly developed PTSD [231]. Sex differences in the sleep symptoms of existing PTSD are also noted. For example, a study comparing sleep in PTSD and healthy controls in both sexes reported a gender × diagnosis interaction whereby, among females, those with PTSD showed greater REM duration and percent than controls whereas, among males, this difference appeared (non-significantly) in the opposite direction [139]. Additionally, as in the case of extinction memory in the experimental setting [232], it has been suggested that sleep symptoms in women may vary with hormonal levels and phase of the menstrual cycle [144].
Sleep and exposure therapy
The ability to remember fear extinction is a key element of both normal recovery from trauma [118] and of psychotherapeutic treatment of PTSD using exposure therapy [7, 62, 101]. One mechanism by which sleep disturbance might precipitate or perpetuate PTSD is by preventing the consolidation and generalization of naturally occurring or therapeutically induced extinction memories during sleep [29]. The degree to which extinction learning can generalize from the specific stimuli extinguished in therapy to similar stimuli encountered outside the treatment setting will strongly impact the efficacy of such therapy [3, 62, 64, 108–110]. For example, fearful responding may re-emerge when the patient encounters an exemplar of a feared category of objects (e.g., spiders) that differs from the specific exemplar (e.g., species of spider) for which fear was extinguished in therapy [110, 233]. Similarly, gains achieved in exposure therapy may be compromised by fear renewal when the patient encounters a feared stimulus (e.g., a trauma reminder) outside of the therapeutic context in which it was extinguished [101, 108]. Such “return of fear” phenomena [234] may be conceptualized as re-emergence of conditioned fear due to failure of extinction memory to generalize from the treatment setting to diverse stimuli and settings that evoke such fears in the real world [63].
Extinction generalization may be particularly relevant to the treatment of PTSD, a disorder in which the opposing effect, generalization of fear responses, is ubiquitous [235]. Moreover, in PTSD, the same traumatic event can produce conditioned fear to multiple stimuli in multiple perceptual modalities each of which then becomes a warning signal of impending danger [236]. Generalization and multiplication of fear responses in PTSD may occur via processes such as second-order fear conditioning to primary trauma reminder cues [237]. Improved therapeutic extinction generalization thus might mitigate mechanisms by which fear generalization exacerbates the number and fear relevance of trauma reminders.
Clinical strategies to maximize extinction generalization include exposing patients to a variety of exemplars in a class of feared objects [101, 110], exposure of patients to feared stimuli in a variety of different contexts [101, 109] and in vivo exposure sessions [103]. A promising pharmacological approach for enhancing exposure therapy involves using D-cycloserine, an NMDA receptor partial agonist, that promotes NMDA-dependent memory consolidation of therapeutic extinction memory [238–240]. Some studies have suggested that outcomes from exposure therapy for PTSD can benefit from administration of D-cycloserine in temporal proximity to exposure sessions [241, 242]. Since, sleep [83] and, specifically, REM sleep [84] have also been shown to be important for NMDA-dependent long-term potentiation, sleep itself might be employed to help strengthen and generalize extinction [107].
In a preliminary application of this hypothesis to anxiety disorders [3], highly spider-fearful, young adult women were repeatedly exposed to a spider video after which half, who were exposed in the evening, had a normal night’s sleep and other half, exposed in the morning, had an equal (12-h) duration of continuous wakefulness. Following the delay, all groups viewed the same video and then videos of a new spider. Only in the wake group was there loss of psychophysiological and self-reported extinction and evidence of sensitization between sessions. Only the sleep group was there psychophysiological evidence of enhanced extinction retention and generalization between sessions. Because these effects did not differentiate control groups exposed and tested entirely in the morning or evening, a time-of-day explanation was ruled out. Thus, following exposure therapy, sleep may promote retention and generalization of extinction and prevent sensitization. These findings have been replicated in a recent study that used virtual-reality exposure therapy for DSM-IV diagnosed spider phobia [115]. Yet more recently, a large study of cognitive behavioral therapy in social anxiety disorder has shown that better self-reported baseline sleep was associated with better post-exposure treatment outcome on measures of anxiety [243].
Important caveats
Impaired consolidation of extinction is unlikely to be the only sleep-related factor contributing to PTSD. Sleep disruption may lead to fatigue [244, 245], executive deficits [246, 247], mood dysregulation [10], and psychosocial impairments [248], all of which may degrade psychological resilience and exacerbate symptoms. Moreover, post-trauma disturbed sleep is unlikely, by itself, sufficient to produce PTSD—a disorder that also shows the above neuroendocrine abnormalities [161, 172, 173] as well as neurocognitive changes [249, 250], emergent psychosocial stressors [251], and genetic predispositions [252].
Caveats to animal models of physiological sleep disturbance and PTSD need also be emphasized. First, the sleep- and REM sleep-disruptive effects of experimental stressors appear with inescapable forms of stress, of which Pavlovian cued and contextual fear conditioning are canonical examples [157, 253] as, of course, are most traumatic events that precipitate PTSD in the human. In contrast, escapable shock, such as occurs in active avoidance learning paradigms, can instead lead to enhanced total and REM sleep with robust rebound of any loss resulting from the stress manipulation [156, 157, 253]. Therefore, aspects of the stressor such as controllability, predictability, and even the specific form of stress (e.g., restraint versus footshock) may produce different and even opposing effects on sleep and sleep-dependent mnemonic processes [156, 157]
Therefore, although putative pathways from traumatic stress to sleep disturbance and thence to poor extinction memory are compelling, the current state of knowledge cannot attribute development of PTSD solely or in part to disturbed sleep-dependent extinction, nor, indeed, to disturbed sleep alone. Nonetheless, among sleep-mediated sources of waking symptoms in PTSD, impaired consolidation and/or generalization of extinction memory during sleep remains a hypothetical mechanism highly suited to future investigation.
Essential directions for future research
Despite the growing number of studies investigating sleep in PTSD, sleep and extinction memory, as well as extinction memory in PTSD and in anxiety disorders, there have been, to date, no studies specifically addressing sleep and extinction memory in patients with PTSD. Such studies, therefore, will be essential to test whether sleep-mediated effects on extinction memory seen in healthy subjects are altered in PTSD. Similarly, the interactions of extinction learning and memory with time-of-day [254] as well as sleep quality and chronotype [255, 256] described in healthy subjects (reviewed in ref. [8]) should also be examined in PTSD. Especially informative would be prospective longitudinal studies initiated following a traumatic event to monitor sleep physiology, circadian patterns of sleep-waking, HPA-axis function, mood, and nightmare frequency/content in order to examine potential linkages between these measures and emergent symptomatology in those individuals who progress to PTSD compared to those who prove resilient.
There have been some early attempts to examine sleep augmentation of pharmacological interventions that may be used to enhance exposure therapy. For example, in healthy volunteers following conditioning and extinction learning, valproic acid, a histone deacetylase inhibitor, enhanced delayed resistance to reinstatement following sleep but D-cycloserine enhanced such resistance following an awake delay [257]. Therefore, combining pharmacotherapy with strategically timed post-exposure sleep may further enhance exposure therapy [258].
Finally, future studies might also examine the effects of sleep on the newly described phenomenon of fear erasure using reconsolidation blockade following retrieval of traumatic memory [2, 57, 259–261]. Given findings that bidirectional plasticity, that includes depotentiation as well as long-term potentiation (LTP), may require REM sleep [197], it is possible that sleep following such reactivation would allow depotentiation to better compete with reconsolidation processes that require LTP. As in the pursuit of enhanced extinction, a sleep component might be added to pharmacological interventions designed to impede reconsolidation of aversive memory such as blockade of noradrenergic transmission [262, 263].
Conclusions
Sleep, acting as a modulator of physiological stress and emotional memory, is of crucial importance in maintaining day-to-day emotional homeostasis and long-term mental health. Sleep disturbance predating or acutely resulting from a traumatic event, particularly if it develops into chronic insomnia, may initiate positive feedback and allostatic mechanisms that impair emotional regulation and promote the pathophysiology of PTSD. The findings reviewed herein have compelling research and clinical implications. First, the effects of sleep deprivation and restriction on extinction learning and recall as well as their neural bases in healthy individuals (reviewed in ref. [8]) should be further investigated. Second, the interaction of sleep deficit, extinction recall, and clinical diagnosis will require studies in which extinction learning and recall are visualized in the brains of PTSD patients with greater and lesser sleep disturbance and these findings compared to trauma-exposed controls as well as patients with non-PTSD-related insomnia. Importantly, however, clinical applications of accruing knowledge need not await definitive findings but can be concurrently investigated to help address the urgent need for innovation in treatments for PTSD. For example, just as disrupted sleep may impair emotional recovery during the critical period following traumatic stress, healthy sleep may be protective at this same point in time. As has often been suggested, proactive treatment of any acute sleep disruption may be a crucial first intervention in the prevention or early treatment of PTSD symptoms [20, 29, 31, 36]. Although the evidence here reviewed indicates the specific importance of REM sleep, behavioral techniques to selectively enhance this sleep stage (e.g., prior REM sleep deprivation) involve further sleep disruption. Therefore, to preserve REM sleep following a trauma, optimizing overall sleep quality by treating comorbid insomnia or other sleep disorders and improving sleep hygiene is important. Another consideration that requires more research is whether REM-sleep suppressive agents, such as many aminergic antidepressants, should be avoided in the early aftermath of trauma. There is also preliminary evidence that the alpha-1 adrenoreceptor antagonist, prazosin, which is effective in relieving nightmares in PTSD, may also serve to normalize REM sleep [264]. Additionally, evaluation of pre-existing sleep disorders may serve as a screening criterion to identify individuals entering high-stress professions such as the military or first responders who are at greatest risk of developing PTSD [17, 265]. Lastly, the memory-enhancing function of sleep might be exploited to strengthen therapeutic extinction learned in exposure-based therapy using strategically timed sleep bouts [3, 115].
Notes
In various studies, diagnosed insomnia is typically DSM-IV [266] Primary Insomnia, DSM-5 [44] Insomnia Disorder, International Classification of Sleep Disorders Second Edition (ICSD-2) [267] Psychophysiological Insomnia, or ICSD-3 Chronic Insomnia Disorder [268] all of which have similar criteria including difficulty initiating or maintaining sleep, daytime fatigue, malaise or dissatisfaction with sleep, duration of at least 3 months (1 month in DSM-IV) and symptom occurrence on 3 or more days per week. Insomnia is frequently comorbid with mood or anxiety disorders in which case it is variously classified as Insomnia Due to Mental Disorder (ICSD-2, ICSD-3) Insomnia Related to another Mental Disorder (DSM-IV) or Insomnia Disorder with Non Sleep-Disorder Mental Comorbidity (DSM-5).
When sleep disturbance does not meet the duration criteria for the above definitions, it is variously classified as Situational/acute Insomnia (DSM-5), Adjustment Insomnia (ICSD-2), or Short-Term Insomnia Disorder (ICSD-3).
Abbreviations
- 1: BLA:
-
basolateral nuclei (amygdala)
- BNST:
-
bed nucleus of the stria terminalis
- CeA:
-
central nucleus (amygdala)
- CRF:
-
corticotropin-releasing factor
- CS:
-
conditioned stimulus
- CSF:
-
cerebrospinal fluid
- dACC:
-
dorsal anterior cingulate cortex
- DSM-5:
-
Diagnostic and Statistical Manual of Mental Disorders—5th ed.
- EEG:
-
electroencephalography
- fMRI:
-
functional magnetic resonance imaging
- HPA axis:
-
hypothalamic-pituitary-adrenal axis
- LC:
-
locus coeruleus
- LTP:
-
long-term potentiation
- NE:
-
norepinephrine
- NMDA:
-
N-methyl-D-aspartate receptor
- NREM:
-
non-REM sleep
- PSQI:
-
Pittsburgh Sleep Quality Index
- PTSD:
-
post-traumatic stress disorder
- PVN:
-
paraventricular nucleus (hypothalamus)
- REM:
-
rapid eye movement
- TST:
-
total sleep time
- US:
-
unconditioned stimulus
- vmPFC:
-
ventromedial prefrontal cortex
References
American_Psychiatric_Association. Diagnostic and statistical manual of mental disorders (Fifth ed.). Arlington, VA: American Psychiatric Publishing; 2013.
Schiller D, Delgado MR. Overlapping neural systems mediating extinction, reversal and regulation of fear. Trends Cogn Sci. 2010;14(6):268–76.
Pace-Schott EF, Verga PW, Bennett TS, Spencer RM. Sleep promotes consolidation and generalization of extinction learning in simulated exposure therapy for spider fear. J Psychiatr Res. 2012;46:1036–44. doi:10.1016/j.jpsychires.2012.04.015.
Spoormaker VI, Schroter MS, Andrade KC, Dresler M, Kiem SA, Goya-Maldonado R, et al. Effects of rapid eye movement sleep deprivation on fear extinction recall and prediction error signaling. Hum Brain Mapp. 2012;33(10):2362–76. doi:10.1002/hbm.21369.
Spoormaker VI, Sturm A, Andrade KC, Schroter MS, Goya-Maldonado R, et al. The neural correlates and temporal sequence of the relationship between shock exposure, disturbed sleep and impaired consolidation of fear extinction. J Psychiatr Res. 2010;44:1121–8.
Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73(1):61–71.
McNally RJ. Mechanisms of exposure therapy: how neuroscience can improve psychological treatments for anxiety disorders. Clin Psychol Rev. 2007;27(6):750–9.
Pace-Schott EF, Germain A, Milad MR. Effects of sleep on memory for conditioned fear and fear extinction. Psychol Bull. 2015;Apr 20. [Epub ahead of print].
Deliens G, Gilson M, Peigneux P. Sleep and the processing of emotions. Exp Brain Res. 2014;232(5):1403–14. doi:10.1007/s00221-014-3832-1.
Goldstein AN, Walker MP. The role of sleep in emotional brain function. Annual Reviews of Clinical Psychology. 2014;10:679–708.
Kim EJ, Dimsdale JE. The effect of psychosocial stress on sleep: a review of polysomnographic evidence. Behav Sleep Med. 2007;5(4):256–78.
Vandekerckhove M, Cluydts R. The emotional brain and sleep: an intimate relationship. Sleep Med Rev. 2010;14(4):219–26. doi:10.1016/j.smrv.2010.01.002.
Mellman TA. Sleep and anxiety disorders. Sleep Med Clinics. 2008;3:261–8.
Peterson MJ, Benca RM. Sleep in mood disorders. Psychiatr Clin North Am. 2006;29(4):1009–32. abstract ix.
Lavie P. Sleep disturbances in the wake of traumatic events. N Engl J Med. 2001;345(25):1825–32. doi:10.1056/NEJMra012893.
Babson KA, Feldner MT. Temporal relations between sleep problems and both traumatic event exposure and PTSD: a critical review of the empirical literature. J Anxiety Disord. 2010;24(1):1–15. doi:10.1016/j.janxdis.2009.08.002.
Gehrman P, Seelig AD, Jacobson IG, Boyko EJ, Hooper TI, Gackstetter GD, et al. Predeployment sleep duration and insomnia symptoms as risk factors for new-onset mental health disorders following military deployment. Sleep. 2013;36(7):1009–18.
Bryant RA, Creamer M, O’Donnell M, Silove D, McFarlane AC. Sleep disturbance immediately prior to trauma predicts subsequent psychiatric disorder. Sleep. 2010;33(1):69–74.
Breslau N, Roth T, Rosenthal L, Andreski P. Sleep disturbance and psychiatric disorders: a longitudinal epidemiological study of young adults. Biol Psychiatry. 1996;39(6):411–8.
Mellman TA, Hipolito MM. Sleep disturbances in the aftermath of trauma and posttraumatic stress disorder. CNS Spectr. 2006;11(8):611–5.
Ford DE, Kamerow DB. Epidemiologic study of sleep disturbances and psychiatric disorders. An opportunity for prevention? JAMA. 1989;262(11):1479–84.
Baglioni C, Battagliese G, Feige B, Spiegelhalder K, Nissen C, Voderholzer U, et al. Insomnia as a predictor of depression: a meta-analytic evaluation of longitudinal epidemiological studies. J Affect Disord. 2011;135(1-3):10–9. doi:10.1016/j.jad.2011.01.011.
Ford DE, Cooper-Patrick L. Sleep disturbances and mood disorders: an epidemiologic perspective. Depress Anxiety. 2001;14(1):3–6.
Marcks BA, Weisberg RB, Edelen MO, Keller MB. The relationship between sleep disturbance and the course of anxiety disorders in primary care patients. Psychiatry Res. 2010;178(3):487–92. doi:10.1016/j.psychres.2009.07.004.
Hasler BP, Germain A, Nofzinger EA, Kupfer DJ, Krafty RT, Rothenberger SD, et al. Chronotype and diurnal patterns of positive affect and affective neural circuitry in primary insomnia. J Sleep Res. 2012. doi:10.1111/j.1365-2869.2012.01002.x.
Buysse DJ, Hall M, Begley A, Cherry CR, Houck PR, Land S, et al. Sleep and treatment response in depression: new findings using power spectral analysis. Psychiatry Res. 2001;103(1):51–67.
Buysse DJ, Tu XM, Cherry CR, Begley AE, Kowalski J, Kupfer DJ, et al. Pretreatment REM sleep and subjective sleep quality distinguish depressed psychotherapy remitters and nonremitters. Biol Psychiatry. 1999;45(2):205–13.
Harvey AG, Talbot LS, Gershon A. Sleep disturbance in bipolar disorder across the lifespan. Clin Psychol Publication Division Clinical Psychol Am Psychol Assoc. 2009;16(2):256–77. doi:10.1111/j.1468-2850.2009.01164.x.
Germain A, Buysse DJ, Nofzinger E. Sleep-specific mechanisms underlying posttraumatic stress disorder: integrative review and neurobiological hypotheses. Sleep Med Rev. 2008;12(3):185–95.
Mellman TA. Sleep and post-traumatic stress disorder: a roadmap for clinicians and researchers. Sleep Med Rev. 2008;12(3):165–7.
Spoormaker VI, Montgomery P. Disturbed sleep in post-traumatic stress disorder: secondary symptom or core feature? Sleep Med Rev. 2008;12(3):169–84.
Alvaro PK, Roberts RM, Harris JK. A systematic review assessing bidirectionality between sleep disturbances, anxiety, and depression. Sleep. 2013;36(7):1059–68. doi:10.5665/sleep.2810.
Wright KM, Britt TW, Bliese PD, Adler AB, Picchioni D, Moore D. Insomnia as predictor versus outcome of PTSD and depression among Iraq combat veterans. J Clin Psychol. 2011;67(12):1240–58. doi:10.1002/jclp.20845.
Babson KA, Badour CL, Feldner MT, Bunaciu L. The relationship of sleep quality and PTSD to anxious reactivity from idiographic traumatic event script-driven imagery. J Trauma Stress. 2012;25(5):503–10. doi:10.1002/jts.21739.
Babson KA, Blonigen DM, Boden MT, Drescher KD, Bonn-Miller MO. Sleep quality among U.S. military veterans with PTSD: a factor analysis and structural model of symptoms. J Trauma Stress. 2012;25(6):665–74. doi:10.1002/jts.21757.
Germain A. Sleep disturbances as the hallmark of PTSD: where are we now? Am J Psychiatry. 2013;170(4):372–82. doi:10.1176/appi.ajp.2012.12040432.
Bonnet MH, Arand DL. Hyperarousal and insomnia: state of the science. Sleep Med Rev. 2010;14(1):9–15.
Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, Kupfer DJ. Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry. 2004;161(11):2126–8.
Riemann D, Spiegelhalder K, Feige B, Voderholzer U, Berger M, Perlis M, et al. The hyperarousal model of insomnia: a review of the concept and its evidence. Sleep Med Rev. 2010;14(1):19–31. doi:10.1016/j.smrv.2009.04.002.
Fernandez-Mendoza J, Vela-Bueno A, Vgontzas AN, Ramos-Platon MJ, Olavarrieta-Bernardino S, Bixler EO, et al. Cognitive-emotional hyperarousal as a premorbid characteristic of individuals vulnerable to insomnia. Psychosom Med. 2010;72(4):397–403. doi:10.1097/PSY.0b013e3181d75319.
Harvey AG. A cognitive model of insomnia. Behav Res Ther. 2002;40(8):869–93.
Drake CL, Friedman NP, Wright Jr KP, Roth T. Sleep reactivity and insomnia: genetic and environmental influences. Sleep. 2011;34(9):1179–88. doi:10.5665/SLEEP.1234.
McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873–904. doi:10.1152/physrev.00041.2006.
Harvey AG, Bryant RA. Acute stress disorder: a synthesis and critique. Psychol Bull. 2002;128:886–902.
Bryant RA. Acute stress disorder as a predictor of posttraumatic stress disorder: a systematic review. J Clin Psychiatry. 2011;72(2):233–9. doi:10.4088/JCP.09r05072blu.
Bryant RA, Creamer M, O’Donnell M, Silove D, McFarlane AC. The capacity of acute stress disorder to predict posttraumatic psychiatric disorders. J Psychiatr Res. 2012;46(2):168–73. doi:10.1016/j.jpsychires.2011.10.007.
Milliken CS, Auchterlonie JL, Hoge CW. Longitudinal assessment of mental health problems among active and reserve component soldiers returning from the Iraq war. JAMA. 2007;298(18):2141–8. doi:10.1001/jama.298.18.2141.
Hoge CW, Auchterlonie JL, Milliken CS. Mental health problems, use of mental health services, and attrition from military service after returning from deployment to Iraq or Afghanistan. JAMA. 2006;295(9):1023–32. doi:10.1001/jama.295.9.1023.
Andrews B, Brewin CR, Philpott R, Stewart L. Delayed-onset posttraumatic stress disorder: a systematic review of the evidence. Am J Psychiatry. 2007;164(9):1319–26. doi:10.1176/appi.ajp.2007.06091491.
Fikretoglu D, Liu A. Prevalence, correlates, and clinical features of delayed-onset posttraumatic stress disorder in a nationally representative military sample. Soc Psychiatry Psychiatr Epidemiol. 2012;47(8):1359–66. doi:10.1007/s00127-011-0444-y.
Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36(4):567–84.
Bouton ME, Westbrook RF, Corcoran KA, Maren S. Contextual and temporal modulation of extinction: behavioral and biological mechanisms. Biol Psychiatry. 2006;60(4):352–60.
Konorski J. Integrative activity of the brain. Chicago: University of Chicago Press; 1967.
Pavlov I. Conditioned reflexes. London: Oxford University Press; 1927.
Ji J, Maren S. Hippocampal involvement in contextual modulation of fear extinction. Hippocampus. 2007;17(9):749–58.
Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33(1):56–72.
Maren S. Seeking a spotless mind: extinction, deconsolidation, and erasure of fear memory. Neuron. 2011;70(5):830–45. doi:10.1016/j.neuron.2011.04.023.
Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Luthi A. Neuronal circuits of fear extinction. Eur J Neurosci. 2010;31(4):599–612.
Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–51. doi:10.1146/annurev.psych.121208.131631.
Graham BM, Milad MR. The study of fear extinction: implications for anxiety disorders. Am J Psychiatry. 2011;168(12):1255–65. doi:10.1176/appi.ajp.2011.11040557.
Milad MR, Rauch SL. Obsessive-compulsive disorder: beyond segregated cortico-striatal pathways. Trends Cogn Sci. 2012;16(1):43–51. doi:10.1016/j.tics.2011.11.003.
Craske MG, Kircanski K, Zelikowsky M, Mystkowski J, Chowdhury N, Baker A. Optimizing inhibitory learning during exposure therapy. Behav Res Ther. 2008;46(1):5–27.
Vervliet B, Baeyens F, Van den Bergh O, Hermans D. Extinction, generalization, and return of fear: a critical review of renewal research in humans. Biol Psychol. 2013;92(1):51–8. doi:10.1016/j.biopsycho.2012.01.006.
Vervliet B, Craske MG, Hermans D. Fear extinction and relapse: state of the art. Annu Rev Clin Psychol. 2013;9:215–48. doi:10.1146/annurev-clinpsy-050212-185542.
Diekelmann S, Wilhelm I, Born J. The whats and whens of sleep-dependent memory consolidation. Sleep Med Rev. 2009;13(5):309–21. doi:10.1016/j.smrv.2008.08.002.
Diekelmann S, Born J. The memory function of sleep. Nature Rev. 2010;11(2):114–26.
Rasch B, Born J. About sleep’s role in memory. Physiol Rev. 2013;93(2):681–766. doi:10.1152/physrev.00032.2012.
Stickgold R. Sleep-dependent memory consolidation. Nature. 2005;437(7063):1272–8.
Landmann N, Kuhn M, Piosczyk H, Feige B, Baglioni C, Spiegelhalder K, et al. The reorganisation of memory during sleep. Sleep Med Rev. 2014. doi:10.1016/j.smrv.2014.03.005.
Payne JD, Chambers AM, Kensinger EA. Sleep promotes lasting changes in selective memory for emotional scenes. Front Integrative Neurosci. 2012;6:108. doi:10.3389/fnint.2012.00108.
Pace-Schott EF, Nave G, Morgan A, Spencer RM. Sleep-dependent modulation of affectively guided decision-making. J Sleep Res. 2012;21(1):30–9. doi:10.1111/j.1365-2869.2011.00921.x.
Stickgold R, Walker MP. Sleep-dependent memory triage: evolving generalization through selective processing. Nat Neurosci. 2013;16(2):139–45. doi:10.1038/nn.3303.
Ellenbogen JM, Hulbert JC, Stickgold R, Dinges DF, Thompson-Schill SL. Interfering with theories of sleep and memory: sleep, declarative memory, and associative interference. Curr Biol. 2006;16(13):1290–4.
Deliens G, Schmitz R, Caudron I, Mary A, Leproult R, Peigneux P. Does recall after sleep-dependent memory consolidation reinstate sensitivity to retroactive interference? PLoS One. 2013;8(7):e68727. doi:10.1371/journal.pone.0068727.
Walker MP, van der Helm E. Overnight therapy? The role of sleep in emotional brain processing. Psychol Bull. 2009;135(5):731–48.
Cartwright R, Luten A, Young M, Mercer P, Bears M. Role of REM sleep and dream affect in overnight mood regulation: a study of normal volunteers. Psychiatry Res. 1998;81(1):1–8.
Cartwright R, Young MA, Mercer P, Bears M. Role of REM sleep and dream variables in the prediction of remission from depression. Psychiatry Res. 1998;80(3):249–55.
Cartwright RD, Kravitz HM, Eastman CI, Wood E. REM latency and the recovery from depression: getting over divorce. Am J Psychiatry. 1991;148(11):1530–5.
Nofzinger EA, Buysse DJ, Germain A, Carter C, Luna B, Price JC, et al. Increased activation of anterior paralimbic and executive cortex from waking to rapid eye movement sleep in depression. Arch Gen Psychiatry. 2004;61(7):695–702.
Skaggs WE, McNaughton BL. Replay of neuronal firing sequences in rat hippocampus during sleep following spatial experience. Science. 1996;271(5257):1870–3.
Louie K, Wilson MA. Temporally structured replay of awake hippocampal ensemble activity during rapid eye movement sleep. Neuron. 2001;29(1):145–56.
Graves L, Pack A, Abel T. Sleep and memory: a molecular perspective. Trends Neurosci. 2001;24(4):237–43.
Kopp C, Longordo F, Nicholson JR, Luthi A. Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J Neurosci. 2006;26(48):12456–65.
Ishikawa A, Kanayama Y, Matsumura H, Tsuchimochi H, Ishida Y, Nakamura S. Selective rapid eye movement sleep deprivation impairs the maintenance of long-term potentiation in the rat hippocampus. Eur J Neurosci. 2006;24(1):243–8.
Smith C. Sleep states and memory processes in humans: procedural versus declarative memory systems. Sleep Med Rev. 2001;5(6):491–506.
Fu J, Li P, Ouyang X, Gu C, Song Z, Gao J, et al. Rapid eye movement sleep deprivation selectively impairs recall of fear extinction in hippocampus-independent tasks in rats. Neuroscience. 2007;144(4):1186–92.
Milad MR, Furtak SC, Greenberg JL, Keshaviah A, Im JJ, Falkenstein MJ, et al. Deficits in conditioned fear extinction in obsessive-compulsive disorder and neurobiological changes in the fear circuit. JAMA Psychiatry. 2013;70(6):608–18. doi:10.1001/jamapsychiatry.2013.914. quiz 554.
Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66(12):1075–82. doi:10.1016/j.biopsych.2009.06.026.
Milad MR, Orr SP, Lasko NB, Chang Y, Rauch SL, Pitman RK. Presence and acquired origin of reduced recall for fear extinction in PTSD: results of a twin study. J Psychiatr Res. 2008;42(7):515–20.
Shvil E, Sullivan GM, Schafer S, Markowitz JC, Campeas M, Wager TD, et al. Sex differences in extinction recall in posttraumatic stress disorder: a pilot fMRI study. Neurobiol Learn Mem. 2014;113:101–8. doi:10.1016/j.nlm.2014.02.003.
Garfinkel SN, Abelson JL, King AP, Sripada RK, Wang X, Gaines LM, et al. Impaired contextual modulation of memories in PTSD: an fMRI and psychophysiological study of extinction retention and fear renewal. J Neurosci. 2014;34(40):13435–43. doi:10.1523/JNEUROSCI.4287-13.2014.
Orr SP, Metzger LJ, Lasko NB, Macklin ML, Peri T, Pitman RK. De novo conditioning in trauma-exposed individuals with and without posttraumatic stress disorder. J Abnorm Psychol. 2000;109(2):290–8.
Blechert J, Michael T, Vriends N, Margraf J, Wilhelm FH. Fear conditioning in posttraumatic stress disorder: evidence for delayed extinction of autonomic, experiential, and behavioural responses. Behav Res Ther. 2007;45(9):2019–33. doi:10.1016/j.brat.2007.02.012.
Lommen MJ, Engelhard IM, Sijbrandij M, van den Hout MA, Hermans D. Pre-trauma individual differences in extinction learning predict posttraumatic stress. Behav Res Ther. 2013;51(2):63–7. doi:10.1016/j.brat.2012.11.004.
Jovanovic T, Norrholm SD, Fennell JE, Keyes M, Fiallos AM, Myers KM, et al. Posttraumatic stress disorder may be associated with impaired fear inhibition: relation to symptom severity. Psychiatry Res. 2009;167(1-2):151–60. doi:10.1016/j.psychres.2007.12.014.
Guthrie RM, Bryant RA. Extinction learning before trauma and subsequent posttraumatic stress. Psychosom Med. 2006;68(2):307–11.
Orr SP, Lasko NB, Macklin ML, Pineles SL, Chang Y, Pitman RK. Predicting post-trauma stress symptoms from pre-trauma psychophysiologic reactivity, personality traits and measures of psychopathology. Biology Mood Anxiety Disorders. 2012;2(1):8. doi:10.1186/2045-5380-2-8.
Glover EM, Phifer JE, Crain DF, Norrholm SD, Davis M, Bradley B, et al. Tools for translational neuroscience: PTSD is associated with heightened fear responses using acoustic startle but not skin conductance measures. Depress Anxiety. 2011;28(12):1058–66. doi:10.1002/da.20880.
Fani N, Tone EB, Phifer J, Norrholm SD, Bradley B, et al. Attention bias toward threat is associated with exaggerated fear expression and impaired extinction in PTSD. Psychol Med. 2012;42:533–43.
Carson MA, Paulus LA, Lasko NB, Metzger LJ, Wolfe J, Orr SP, et al. Psychophysiologic assessment of posttraumatic stress disorder in Vietnam nurse veterans who witnessed injury or death. J Consult Clin Psychol. 2000;68(5):890–7.
Hermans D, Craske MG, Mineka S, Lovibond PF. Extinction in human fear conditioning. Biol Psychiatry. 2006;60(4):361–8.
Hofmann SG. Enhancing exposure-based therapy from a translational research perspective. Behav Res Ther. 2007;45(9):1987–2001.
Foa E, Hembree E, Rothbaum B. Prolonged exposure therapy for PTSD. Emotional processing of traumatic experiences. Therapist Guide: Oxford University Press; 2007.
Hofmann SG. Exposure therapy for social anxiety disorder. (unpublished treatment manual). 2004.
Williams MT, Mugno B, Franklin M, Faber S. Symptom dimensions in obsessive-compulsive disorder: phenomenology and treatment outcomes with exposure and ritual prevention. Psychopathology. 2013;46(6):365–76. doi:10.1159/000348582.
Choy Y, Fyer AJ, Lipsitz JD. Treatment of specific phobia in adults. Clin Psychol Rev. 2007;27(3):266–86. doi:10.1016/j.cpr.2006.10.002.
Pace-Schott EF, Milad MR, Orr SP, Rauch SL, Stickgold R, Pitman RK. Sleep promotes generalization of extinction of conditioned fear. Sleep. 2009;32(1):19–26.
Vansteenwegen D, Hermans D, Vervliet B, Francken G, Beckers T, Baeyens F, et al. Return of fear in a human differential conditioning paradigm caused by a return to the original acquisition context. Behav Res Ther. 2005;43(3):323–36.
Vansteenwegen D, Vervliet B, Iberico C, Baeyens F, Van den Bergh O, Hermans D. The repeated confrontation with videotapes of spiders in multiple contexts attenuates renewal of fear in spider-anxious students. Behav Res Ther. 2007;45(6):1169–79.
Rowe MK, Craske MG. Effects of varied-stimulus exposure training on fear reduction and return of fear. Behav Res Ther. 1998;36(7-8):719–34.
McSweeney FK, Swindell S. Common processes may contribute to extinction and habituation. J Gen Psychol. 2002;129(4):364–400. doi:10.1080/00221300209602103.
Hofmann SG, Otto MW. Cognitive behavioral therapy for social anxiety disorder: evidence-based and disorder-specific treatment techniques. New York, NY: Routledge; 2008.
Plendl W, Wotjak CT. Dissociation of within- and between-session extinction of conditioned fear. J Neurosci. 2010;30(14):4990–8. doi:10.1523/JNEUROSCI.6038-09.2010.
Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118–26. doi:10.1016/j.biopsych.2008.01.012.
Kleim B, Wilhelm FH, Temp L, Margraf J, Wiederhold BK, Rasch B. Sleep enhances exposure therapy. Psychol Med. 2013:1-9. doi:10.1017/S0033291713001748.
Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research—past, present, and future. Biol Psychiatry. 2006;60(4):376–82.
Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79.
Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, et al. Biological studies of post-traumatic stress disorder. Nature Rev. 2012;13(11):769–87. doi:10.1038/nrn3339.
Liberzon I, Sripada CS. The functional neuroanatomy of PTSD: a critical review. Prog Brain Res. 2008;167:151–69. doi:10.1016/S0079-6123(07)67011-3.
Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35(1):169–91. doi:10.1038/npp.2009.83.
Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164(10):1476–88. doi:10.1176/appi.ajp.2007.07030504.
Liberzon I, Martis B. Neuroimaging studies of emotional responses in PTSD. Ann N Y Acad Sci. 2006;1071:87–109. doi:10.1196/annals.1364.009.
Sartory G, Cwik J, Knuppertz H, Schurholt B, Lebens M, Seitz RJ, et al. In search of the trauma memory: a meta-analysis of functional neuroimaging studies of symptom provocation in posttraumatic stress disorder (PTSD). PLoS One. 2013;8(3), e58150. doi:10.1371/journal.pone.0058150.
Daniels JK, Lamke JP, Gaebler M, Walter H, Scheel M. White matter integrity and its relationship to PTSD and childhood trauma—a systematic review and meta-analysis. Depress Anxiety. 2013;30(3):207–16. doi:10.1002/da.22044.
Simmons AN, Matthews SC. Neural circuitry of PTSD with or without mild traumatic brain injury: a meta-analysis. Neuropharmacology. 2012;62(2):598–606. doi:10.1016/j.neuropharm.2011.03.016.
Karl A, Schaefer M, Malta LS, Dorfel D, Rohleder N, Werner A. A meta-analysis of structural brain abnormalities in PTSD. Neurosci Biobehav Rev. 2006;30(7):1004–31. doi:10.1016/j.neubiorev.2006.03.004.
Nofzinger EA, Mintun MA, Wiseman M, Kupfer DJ, Moore RY. Forebrain activation in REM sleep: an FDG PET study. Brain Res. 1997;770(1-2):192–201.
Braun AR, Balkin TJ, Wesenten NJ, Carson RE, Varga M, Baldwin P, et al. Regional cerebral blood flow throughout the sleep-wake cycle. An H2(15)O PET study. Brain. 1997;120(Pt 7):1173–97.
Maquet P, Ruby P, Maudoux A, Albouy G, Sterpenich V, Dang-Vu T, et al. Human cognition during REM sleep and the activity profile within frontal and parietal cortices: a reappraisal of functional neuroimaging data. Prog Brain Res. 2005;150:219–27.
Bremner JD, Elzinga B, Schmahl C, Vermetten E. Structural and functional plasticity of the human brain in posttraumatic stress disorder. Prog Brain Res. 2008;167:171–86.
Sheikh JI, Woodward SH, Leskin GA. Sleep in post-traumatic stress disorder and panic: convergence and divergence. Depress Anxiety. 2003;18(4):187–97.
Lepola U, Koponen H, Leinonen E. Sleep in panic disorders. J Psychosom Res. 1994;38 Suppl 1:105–11.
Paterson JL, Reynolds AC, Ferguson SA, Dawson D. Sleep and obsessive-compulsive disorder (OCD). Sleep Med Rev. 2013. doi:10.1016/j.smrv.2012.12.002.
Kobayashi I, Boarts JM, Delahanty DL. Polysomnographically measured sleep abnormalities in PTSD: a meta-analytic review. Psychophysiology. 2007;44(4):660–9.
Mellman TA. Sleep and anxiety disorders. Psychiatr Clin North Am. 2006;29(4):1047–58.
Ross RJ, Ball WA, Sullivan KA, Caroff SN. Sleep disturbance as the hallmark of posttraumatic stress disorder. Am J Psychiatry. 1989;146(6):697–707.
Germain A, Buysse DJ, Shear MK, Fayyad R, Austin C. Clinical correlates of poor sleep quality in posttraumatic stress disorder. J Trauma Stress. 2004;17(6):477–84.
Levin R, Nielsen TA. Disturbed dreaming, posttraumatic stress disorder, and affect distress: a review and neurocognitive model. Psychol Bull. 2007;133(3):482–528.
Richards A, Metzler TJ, Ruoff LM, Inslicht SS, Rao M, Talbot LS, et al. Sex differences in objective measures of sleep in post-traumatic stress disorder and healthy control subjects. J Sleep Res. 2013;22(6):679–87. doi:10.1111/jsr.12064.
Engdahl BE, Eberly RE, Hurwitz TD, Mahowald MW, Blake J. Sleep in a community sample of elderly war veterans with and without posttraumatic stress disorder. Biol Psychiatry. 2000;47(6):520–5.
Neylan TC, Lenoci M, Maglione ML, Rosenlicht NZ, Metzler TJ, Otte C, et al. Delta sleep response to metyrapone in post-traumatic stress disorder. Neuropsychopharmacology. 2003;28(9):1666–76.
Woodward SH, Murburg MM, Bliwise DL. PTSD-related hyperarousal assessed during sleep. Physiol Behav. 2000;70(1-2):197–203.
Ebdlahad S, Nofzinger EA, James JA, Buysse DJ, Price JC, Germain A. Comparing neural correlates of REM sleep in posttraumatic stress disorder and depression: a neuroimaging study. Psychiatry Res. 2013;214(3):422–8. doi:10.1016/j.pscychresns.2013.09.007.
Kobayashi I, Cowdin N, Mellman TA. One’s sex, sleep, and posttraumatic stress disorder. Biology Sex Differences. 2012;3(1):29. doi:10.1186/2042-6410-3-29.
Harvey AG, Jones C, Schmidt DA. Sleep and posttraumatic stress disorder: a review. Clin Psychol Rev. 2003;23(3):377–407.
Koren D, Arnon I, Lavie P, Klein E. Sleep complaints as early predictors of posttraumatic stress disorder: a 1-year prospective study of injured survivors of motor vehicle accidents. Am J Psychiatry. 2002;159(5):855–7.
Mellman TA, Bustamante V, Fins AI, Pigeon WR, Nolan B. REM sleep and the early development of posttraumatic stress disorder. Am J Psychiatry. 2002;159(10):1696–701.
Mellman TA, Pigeon WR, Nowell PD, Nolan B. Relationships between REM sleep findings and PTSD symptoms during the early aftermath of trauma. J Trauma Stress. 2007;20(5):893–901.
Mellman TA, Knorr BR, Pigeon WR, Leiter JC, Akay M. Heart rate variability during sleep and the early development of posttraumatic stress disorder. Biol Psychiatry. 2004;55(9):953–6.
Galea S, Nandi A, Vlahov D. The epidemiology of post-traumatic stress disorder after disasters. Epidemiol Rev. 2005;27:78–91. doi:10.1093/epirev/mxi003.
Kok BC, Herrell RK, Thomas JL, Hoge CW. Posttraumatic stress disorder associated with combat service in Iraq or Afghanistan: reconciling prevalence differences between studies. J Nerv Ment Dis. 2012;200(5):444–50. doi:10.1097/NMD.0b013e3182532312.
Yoo SS, Gujar N, Hu P, Jolesz FA, Walker MP. The human emotional brain without sleep—a prefrontal amygdala disconnect. Curr Biol. 2007;17(20):R877–8.
Thomas M, Sing H, Belenky G, Holcomb H, Mayberg H, Dannals R, et al. Neural basis of alertness and cognitive performance impairments during sleepiness. I. Effects of 24 h of sleep deprivation on waking human regional brain activity. J Sleep Res. 2000;9(4):335–52.
Killgore WD. Self-reported sleep correlates with prefrontal-amygdala functional connectivity and emotional functioning. Sleep. 2013;36(11):1597–608. doi:10.5665/sleep.3106.
Pawlyk AC, Morrison AR, Ross RJ, Brennan FX. Stress-induced changes in sleep in rodents: models and mechanisms. Neurosci Biobehav Rev. 2008;32(1):99–117.
Suchecki D, Tiba PA, Machado RB. REM sleep rebound as an adaptive response to stressful situations. Frontiers Neurol. 2012;3:41. doi:10.3389/fneur.2012.00041.
Sanford LD, Suchecki D, Meerlo P. Stress, arousal, and sleep. Curr Top Behav Neurosci. 2014. doi:10.1007/7854_2014_314.
Germain A, James J, Insana S, Herringa RJ, Mammen O, Price J, et al. A window into the invisible wound of war: functional neuroimaging of REM sleep in returning combat veterans with PTSD. Psychiatry Res. 2013;211(2):176–9. doi:10.1016/j.pscychresns.2012.05.007.
Southwick SM, Bremner JD, Rasmusson A, Morgan 3rd CA, Arnsten A, Charney DS. Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry. 1999;46(9):1192–204.
Geracioti Jr TD, Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB, et al. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry. 2001;158(8):1227–30.
Mellman TA, Kumar A, Kulick-Bell R, Kumar M, Nolan B. Nocturnal/daytime urine noradrenergic measures and sleep in combat-related PTSD. Biol Psychiatry. 1995;38(3):174–9. doi:10.1016/0006-3223(94)00238-X.
Trinder J, Kleiman J, Carrington M, Smith S, Breen S, Tan N, et al. Autonomic activity during human sleep as a function of time and sleep stage. J Sleep Res. 2001;10(4):253–64.
Brandenberger G, Ehrhart J, Piquard F, Simon C. Inverse coupling between ultradian oscillations in delta wave activity and heart rate variability during sleep. Clin Neurophysiol. 2001;112(6):992–6.
Meerlo P, Sgoifo A, Suchecki D. Restricted and disrupted sleep: effects on autonomic function, neuroendocrine stress systems and stress responsivity. Sleep Med Rev. 2008;12(3):197–210.
Pace-Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nature Rev. 2002;3(8):591–605.
Kung S, Espinel Z, Lapid MI. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87(9):890–900. doi:10.1016/j.mayocp.2012.05.015.
Raskind MA, Peterson K, Williams T, Hoff DJ, Hart K, Holmes H, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013. doi:10.1176/appi.ajp.2013.12081133.
Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann N Y Acad Sci. 2009;1179:56–69. doi:10.1111/j.1749-6632.2009.04979.x.
Aguilera G. HPA axis responsiveness to stress: implications for healthy aging. Exp Gerontol. 2011;46(2-3):90–5. doi:10.1016/j.exger.2010.08.023.
Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, et al. Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry. 1997;154(5):624–9.
Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, et al. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry. 1999;156(4):585–8.
Sautter FJ, Bissette G, Wiley J, Manguno-Mire G, Schoenbachler B, Myers L, et al. Corticotropin-releasing factor in posttraumatic stress disorder (PTSD) with secondary psychotic symptoms, nonpsychotic PTSD, and healthy control subjects. Biol Psychiatry. 2003;54(12):1382–8.
Morris MC, Compas BE, Garber J. Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: a systematic review and meta-analysis. Clin Psychol Rev. 2012;32(4):301–15. doi:10.1016/j.cpr.2012.02.002.
Heim C, Nemeroff CB. Neurobiology of posttraumatic stress disorder. CNS Spectr. 2009;14(1 Suppl 1):13–24.
Yehuda R. Advances in understanding neuroendocrine alterations in PTSD and their therapeutic implications. Ann N Y Acad Sci. 2006;1071:137–66. doi:10.1196/annals.1364.012.
Kling MA, DeBellis MD, O’Rourke DK, Listwak SJ, Geracioti Jr TD, McCutcheon IE, et al. Diurnal variation of cerebrospinal fluid immunoreactive corticotropin-releasing hormone levels in healthy volunteers. J Clin Endocrinol Metab. 1994;79(1):233–9. doi:10.1210/jcem.79.1.8027234.
Kalsbeek A, van der Spek R, Lei J, Endert E, Buijs RM, Fliers E. Circadian rhythms in the hypothalamo-pituitary-adrenal (HPA) axis. Mol Cell Endocrinol. 2012;349(1):20–9. doi:10.1016/j.mce.2011.06.042.
Koob GF. Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry. 1999;46(9):1167–80.
Heinrichs SC, Koob GF. Corticotropin-releasing factor in brain: a role in activation, arousal, and affect regulation. J Pharmacol Exp Ther. 2004;311(2):427–40.
Davis M, Walker DL, Miles L, Grillon C. Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology. 2010;35(1):105–35. doi:10.1038/npp.2009.109.
Walker DL, Miles LA, Davis M. Selective participation of the bed nucleus of the stria terminalis and CRF in sustained anxiety-like versus phasic fear-like responses. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(8):1291–308. doi:10.1016/j.pnpbp.2009.06.022.
Somerville LH, Whalen PJ, Kelley WM. Human bed nucleus of the stria terminalis indexes hypervigilant threat monitoring. Biol Psychiatry. 2010;68(5):416–24. doi:10.1016/j.biopsych.2010.04.002.
Pole N, Neylan TC, Otte C, Henn-Hasse C, Metzler TJ, Marmar CR. Prospective prediction of posttraumatic stress disorder symptoms using fear potentiated auditory startle responses. Biol Psychiatry. 2009;65(3):235–40. doi:10.1016/j.biopsych.2008.07.015.
Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. 2001;49(12):1023–39.
Krohg K, Hageman I, Jorgensen MB. Corticotropin-releasing factor (CRF) in stress and disease: a review of literature and treatment perspectives with special emphasis on psychiatric disorders. Nord J Psychiatry. 2008;62(1):8–16. doi:10.1080/08039480801983588.
Dunn AJ, Swiergiel AH, Palamarchouk V. Brain circuits involved in corticotropin-releasing factor-norepinephrine interactions during stress. Ann N Y Acad Sci. 2004;1018:25–34. doi:10.1196/annals.1296.003.
Sanford LD, Yang L, Wellman LL, Dong E, Tang X. Mouse strain differences in the effects of corticotropin releasing hormone (CRH) on sleep and wakefulness. Brain Res. 2008;1190:94–104.
Chang FC, Opp MR. Corticotropin-releasing hormone (CRH) as a regulator of waking. Neurosci Biobehav Rev. 2001;25(5):445–53.
Fadda P, Fratta W. Stress-induced sleep deprivation modifies corticotropin releasing factor (CRF) levels and CRF binding in rat brain and pituitary. Pharmacol Res. 1997;35(5):443–6.
Yang L, Tang X, Wellman LL, Liu X, Sanford LD. Corticotropin releasing factor (CRF) modulates fear-induced alterations in sleep in mice. Brain Res. 2009;1276:112–22.
Wellman LL, Yang L, Ambrozewicz MA, Machida M, Sanford LD. Basolateral amygdala and the regulation of fear-conditioned changes in sleep: role of corticotropin-releasing factor. Sleep. 2013;36(4):471–80. doi:10.5665/sleep.2526.
Liu X, Wellman LL, Yang L, Ambrozewicz MA, Tang X, Sanford LD. Antagonizing corticotropin-releasing factor in the central nucleus of the amygdala attenuates fear-induced reductions in sleep but not freezing. Sleep. 2011;34(11):1539–49. doi:10.5665/sleep.1394.
Philbert J, Beeske S, Belzung C, Griebel G. The CRF1 receptor antagonist SSR125543 prevents stress-induced long-lasting sleep disturbances in a mouse model of PTSD: comparison with paroxetine and d-cycloserine. Behav Brain Res. 2015;279:41–6. doi:10.1016/j.bbr.2014.11.006.
Kim Y, Chen L, McCarley RW, Strecker RE. Sleep allostasis in chronic sleep restriction: the role of the norepinephrine system. Brain Res. 2013;1531:9–16. doi:10.1016/j.brainres.2013.07.048.
McEwen BS. Sleep deprivation as a neurobiologic and physiologic stressor: allostasis and allostatic load. Metabolism. 2006;55 Suppl 2:S20–3.
Liberzon I, Krstov M, Young EA. Stress-restress: effects on ACTH and fast feedback. Psychoneuroendocrinology. 1997;22(6):443–53.
Vanderheyden WM, Poe GR, Liberzon I. Trauma exposure and sleep: using a rodent model to understand sleep function in PTSD. Exp Brain Res. 2014;232(5):1575–84. doi:10.1007/s00221-014-3890-4.
Schwabe L, Wolf OT. Stress and multiple memory systems: from ‘thinking’ to ‘doing’. Trends Cogn Sci. 2013;17(2):60–8. doi:10.1016/j.tics.2012.12.001.
Parsons RG, Ressler KJ. Implications of memory modulation for post-traumatic stress and fear disorders. Nat Neurosci. 2013;16(2):146–53. doi:10.1038/nn.3296.
Riemann D. Insomnia and comorbid psychiatric disorders. Sleep Med. 2007;8(4):S15–20. doi:10.1016/S1389-9457(08)70004-2.
Papadimitriou GN, Linkowski P. Sleep disturbance in anxiety disorders. Int Rev Psychiatry (Abingdon, England). 2005;17(4):229–36. doi:10.1080/09540260500104524.
Budhiraja R, Roth T, Hudgel DW, Budhiraja P, Drake CL. Prevalence and polysomnographic correlates of insomnia comorbid with medical disorders. Sleep. 2011;34(7):859–67. doi:10.5665/SLEEP.1114.
Pillai V, Steenburg LA, Ciesla JA, Roth T, Drake CL. A seven day actigraphy-based study of rumination and sleep disturbance among young adults with depressive symptoms. J Psychosom Res. 2014;77(1):70–5. doi:10.1016/j.jpsychores.2014.05.004.
Baglioni C, Regen W, Teghen A, Spiegelhalder K, Feige B, Nissen C, et al. Sleep changes in the disorder of insomnia: a meta-analysis of polysomnographic studies. Sleep Med Rev. 2013. doi:10.1016/j.smrv.2013.04.001.
Baglioni C, Lombardo C, Bux E, Hansen S, Salveta C, Biello S, et al. Psychophysiological reactivity to sleep-related emotional stimuli in primary insomnia. Behav Res Ther. 2010;48(6):467–75. doi:10.1016/j.brat.2010.01.008.
Baglioni C, Spiegelhalder K, Lombardo C, Riemann D. Sleep and emotions: a focus on insomnia. Sleep Med Rev. 2010;14(4):227–38. doi:10.1016/j.smrv.2009.10.007.
van de Laar M, Verbeek I, Pevernagie D, Aldenkamp A, Overeem S. The role of personality traits in insomnia. Sleep Med Rev. 2010;14(1):61–8. doi:10.1016/j.smrv.2009.07.007.
Kales A, Caldwell AB, Soldatos CR, Bixler EO, Kales JD. Biopsychobehavioral correlates of insomnia. II. Pattern specificity and consistency with the Minnesota Multiphasic Personality Inventory. Psychosom Med. 1983;45(4):341–56.
Stepanski EJ, Rybarczyk B. Emerging research on the treatment and etiology of secondary or comorbid insomnia. Sleep Med Rev. 2006;10(1):7–18. doi:10.1016/j.smrv.2005.08.002.
Espie CA. Insomnia: conceptual issues in the development, persistence, and treatment of sleep disorder in adults. Annu Rev Psychol. 2002;53:215–43. doi:10.1146/annurev.psych.53.100901.135243.
Ellis JG, Gehrman P, Espie CA, Riemann D, Perlis ML. Acute insomnia: current conceptualizations and future directions. Sleep Med Rev. 2012;16(1):5–14. doi:10.1016/j.smrv.2011.02.002.
Reite M, Buysse D, Reynolds C, Mendelson W. The use of polysomnography in the evaluation of insomnia. Sleep. 1995;18(1):58–70.
Riemann D, Spiegelhalder K, Nissen C, Hirscher V, Baglioni C, Feige B. REM sleep instability—a new pathway for insomnia? Pharmacopsychiatry. 2012;45(5):167–76. doi:10.1055/s-0031-1299721.
Feige B, Al-Shajlawi A, Nissen C, Voderholzer U, Hornyak M, Spiegelhalder K, et al. Does REM sleep contribute to subjective wake time in primary insomnia? A comparison of polysomnographic and subjective sleep in 100 patients. J Sleep Res. 2008;17(2):180–90.
Perusse AD, Pedneault-Drolet M, Rancourt C, Turcotte I, St-Jean G, Bastien CH. REM sleep as a potential indicator of hyperarousal in psychophysiological and paradoxical insomnia sufferers. Int J Psychophysiol. 2015;95(3):372–8. doi:10.1016/j.ijpsycho.2015.01.005.
Nofzinger EA, Nissen C, Germain A, Moul D, Hall M, Price JC, et al. Regional cerebral metabolic correlates of WASO during NREM sleep in insomnia. J Clin Sleep Med. 2006;2(3):316–22.
Huang Z, Liang P, Jia X, Zhan S, Li N, Ding Y, et al. Abnormal amygdala connectivity in patients with primary insomnia: evidence from resting state fMRI. Eur J Radiol. 2012;81(6):1288–95. doi:10.1016/j.ejrad.2011.03.029.
Mellman TA, David D, Kulick-Bell R, Hebding J, Nolan B. Sleep disturbance and its relationship to psychiatric morbidity after Hurricane Andrew. Am J Psychiatry. 1995;152(11):1659–63.
Morin CM, Benca R. Chronic insomnia. Lancet. 2012;379(9821):1129–41. doi:10.1016/S0140-6736(11)60750-2.
Davidson JR, Rothbaum BO, van der Kolk BA, Sikes CR, Farfel GM. Multicenter, double-blind comparison of sertraline and placebo in the treatment of posttraumatic stress disorder. Arch Gen Psychiatry. 2001;58(5):485–92.
Galovski TE, Monson C, Bruce SE, Resick PA. Does cognitive-behavioral therapy for PTSD improve perceived health and sleep impairment? J Trauma Stress. 2009;22(3):197–204. doi:10.1002/jts.20418.
Talbot LS, Maguen S, Metzler TJ, Schmitz M, McCaslin SE, Richards A, et al. Cognitive behavioral therapy for insomnia in posttraumatic stress disorder: a randomized controlled trial. Sleep. 2014;37(2):327–41.
Zhang B, Wing YK. Sex differences in insomnia: a meta-analysis. Sleep. 2006;29(1):85–93.
Ditlevsen DN, Elklit A. Gender, trauma type, and PTSD prevalence: a re-analysis of 18 nordic convenience samples. Annals General Psychiatry. 2012;11(1):26. doi:10.1186/1744-859X-11-26.
Milad MR, Goldstein JM, Orr SP, Wedig MM, Klibanski A, Pitman RK, et al. Fear conditioning and extinction: influence of sex and menstrual cycle in healthy humans. Behav Neurosci. 2006;120(6):1196–203.
Milad MR, Zeidan MA, Contero A, Pitman RK, Klibanski A, Rauch SL, et al. The influence of gonadal hormones on conditioned fear extinction in healthy humans. Neuroscience. 2010;168(3):652–8.
Graham BM, Milad MR. Blockade of estrogen by hormonal contraceptives impairs fear extinction in female rats and women. Biol Psychiatry. 2013;73(4):371–8. doi:10.1016/j.biopsych.2012.09.018.
Zeidan MA, Igoe SA, Linnman C, Vitalo A, Levine JB, Klibanski A, et al. Estradiol modulates medial prefrontal cortex and amygdala activity during fear extinction in women and female rats. Biol Psychiatry. 2011;70(10):920–7. doi:10.1016/j.biopsych.2011.05.016.
Lebron-Milad K, Abbs B, Milad MR, Linnman C, Rougemount-Bucking A, Zeidan MA, et al. Sex differences in the neurobiology of fear conditioning and extinction: a preliminary fMRI study of shared sex differences with stress-arousal circuitry. Biology Mood Anxiety Disorders. 2012;2(1):7. doi:10.1186/2045-5380-2-7.
Milad MR, Igoe SA, Lebron-Milad K, Novales JE. Estrous cycle phase and gonadal hormones influence conditioned fear extinction. Neuroscience. 2009;164(3):887–95.
Kobayashi I, Mellman TA. Gender differences in sleep during the aftermath of trauma and the development of posttraumatic stress disorder. Behav Sleep Med. 2012;10(3):180–90. doi:10.1080/15402002.2011.654296.
Lebron-Milad K, Milad MR. Sex differences, gonadal hormones and the fear extinction network: implications for anxiety disorders. Biology Mood Anxiety Disorders. 2012;2(1):3. doi:10.1186/2045-5380-2-3.
Vervliet B, Vansteenwegen D, Baeyens F, Hermans D, Eelen P. Return of fear in a human differential conditioning paradigm caused by a stimulus change after extinction. Behav Res Ther. 2005;43(3):357–71.
Rachman S. The return of fear: review and prospect. Clin Psychol Rev. 1989;9:147–68.
Sauerhofer E, Pamplona FA, Bedenk B, Moll GH, Dawirs RR, von Horsten S, et al. Generalization of contextual fear depends on associative rather than non-associative memory components. Behav Brain Res. 2012;233(2):483–93. doi:10.1016/j.bbr.2012.05.016.
Ehlers A, Hackmann A, Steil R, Clohessy S, Wenninger K, Winter H. The nature of intrusive memories after trauma: the warning signal hypothesis. Behav Res Ther. 2002;40(9):995–1002.
Wessa M, Flor H. Failure of extinction of fear responses in posttraumatic stress disorder: evidence from second-order conditioning. Am J Psychiatry. 2007;164(11):1684–92.
Hofmann SG. D-cycloserine for treating anxiety disorders: making good exposures better and bad exposures worse. Depress Anxiety. 2014;31:175–7.
Bontempo A, Panza KE, Bloch MH. D-cycloserine augmentation of behavioral therapy for the treatment of anxiety disorders: a meta-analysis. J Clin Psychiatry. 2012;73(4):533–7. doi:10.4088/JCP.11r07356.
Grillon C. D-cycloserine facilitation of fear extinction and exposure-based therapy might rely on lower-level, automatic mechanisms. Biol Psychiatry. 2009;66(7):636–41.
Litz BT, Salters-Pedneault K, Steenkamp MM, Hermos JA, Bryant RA, Otto MW, et al. A randomized placebo-controlled trial of D-cycloserine and exposure therapy for posttraumatic stress disorder. J Psychiatr Res. 2012;46(9):1184–90. doi:10.1016/j.jpsychires.2012.05.006.
de Kleine RA, Hendriks GJ, Kusters WJ, Broekman TG, van Minnen A. A randomized placebo-controlled trial of D-cycloserine to enhance exposure therapy for posttraumatic stress disorder. Biol Psychiatry. 2012;71(11):962–8. doi:10.1016/j.biopsych.2012.02.033.
Zalta AK, Dowd S, Rosenfield D, Smits JA, Otto MW, Simon NM et al. Sleep quality predicts treatment outcome in CBT for social anxiety disorder. Depress Anxiety. 2013. doi:10.1002/da.22170.
Eglinton R, Chung MC. The relationship between posttraumatic stress disorder, illness cognitions, defence styles, fatigue severity and psychological well-being in chronic fatigue syndrome. Psychiatry Res. 2011;188(2):245–52. doi:10.1016/j.psychres.2011.04.012.
Lerdal A, Lee KA, Rokne B, Knudsen Jr O, Wahl AK, Dahl AA. A population-based study of associations between current posttraumatic stress symptoms and current fatigue. J Trauma Stress. 2010;23(5):606–14. doi:10.1002/jts.20562.
Jones K, Harrison Y. Frontal lobe function, sleep loss and fragmented sleep. Sleep Med Rev. 2001;5(6):463–75.
McKenna BS, Eyler LT. Overlapping prefrontal systems involved in cognitive and emotional processing in euthymic bipolar disorder and following sleep deprivation: a review of functional neuroimaging studies. Clin Psychol Rev. 2012;32(7):650–63. doi:10.1016/j.cpr.2012.07.003.
Tavernier R, Willoughby T. Bidirectional associations between sleep (quality and duration) and psychosocial functioning across the university years. Dev Psychol. 2014;50(3):674–82. doi:10.1037/a0034258.
Dolan S, Martindale S, Robinson J, Kimbrel NA, Meyer EC, Kruse MI, et al. Neuropsychological sequelae of PTSD and TBI following war deployment among OEF/OIF veterans. Neuropsychol Rev. 2012;22(1):21–34. doi:10.1007/s11065-012-9190-5.
Qureshi SU, Long ME, Bradshaw MR, Pyne JM, Magruder KM, Kimbrell T, et al. Does PTSD impair cognition beyond the effect of trauma? J Neuropsychiatry Clin Neurosci. 2011;23(1):16–28. doi:10.1176/appi.neuropsych.23.1.16.
Charuvastra A, Cloitre M. Social bonds and posttraumatic stress disorder. Annu Rev Psychol. 2008;59:301–28. doi:10.1146/annurev.psych.58.110405.085650.
Broekman BF, Olff M, Boer F. The genetic background to PTSD. Neurosci Biobehav Rev. 2007;31(3):348–62. doi:10.1016/j.neubiorev.2006.10.001.
Sanford LD, Yang L, Wellman LL, Liu X, Tang X. Differential effects of controllable and uncontrollable footshock stress on sleep in mice. Sleep. 2010;33(5):621–30.
Pace-Schott EF, Spencer RM, Vijayakumar S, Ahmed NA, Verga PW, Orr SP, et al. Extinction of conditioned fear is better learned and recalled in the morning than in the evening. J Psychiatr Res. 2013;47(11):1776–84. doi:10.1016/j.jpsychires.2013.07.027.
Pace-Schott EF, Rubin Z, Verga PW, Spencer RMC, Orr SP, Milad MR. Chronotype, sleep quality and extinction memory, an actigraphic study. Sleep 37. 2014;(Abstract Supplement)(in press).
Pace-Schott EF, Rubin Z, Tracy LE, Spencer RMC, Orr SP, Verga PW. Emotional trait and memory associates of sleep timing and quality. Psychiatry Res. 2015. (in press).
Kuriyama K, Honma M, Soshi T, Fujii T, Kim Y. Effect of D-cycloserine and valproic acid on the extinction of reinstated fear-conditioned responses and habituation of fear conditioning in healthy humans: a randomized controlled trial. Psychopharmacology (Berl). 2011;218(3):589–97. doi:10.1007/s00213-011-2353-x.
Kuriyama K, Honma M, Yoshiike T, Kim Y. Valproic acid but not D-cycloserine facilitates sleep-dependent offline learning of extinction and habituation of conditioned fear in humans. Neuropharmacology. 2013;64:424–31. doi:10.1016/j.neuropharm.2012.07.045.
Schiller D, Monfils MH, Raio CM, Johnson DC, Ledoux JE, Phelps EA. Preventing the return of fear in humans using reconsolidation update mechanisms. Nature. 2010;463(7277):49–53. doi:10.1038/nature08637.
Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324(5929):951–5. doi:10.1126/science.1167975.
Quirk GJ, Pare D, Richardson R, Herry C, Monfils MH, Schiller D, et al. Erasing fear memories with extinction training. J Neurosci. 2010;30(45):14993–7.
Hoge EA, Worthington JJ, Nagurney JT, Chang Y, Kay EB, Feterowski CM, et al. Effect of acute posttrauma propranolol on PTSD outcome and physiological responses during script-driven imagery. CNS Neurosci Ther. 2012;18(1):21–7. doi:10.1111/j.1755-5949.2010.00227.x.
Brunet A, Poundja J, Tremblay J, Bui E, Thomas E, Orr SP, et al. Trauma reactivation under the influence of propranolol decreases posttraumatic stress symptoms and disorder: 3 open-label trials. J Clin Psychopharmacol. 2011;31(4):547–50. doi:10.1097/JCP.0b013e318222f360.
Taylor FB, Martin P, Thompson C, Williams J, Mellman TA, Gross C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry. 2008;63(6):629–32. doi:10.1016/j.biopsych.2007.07.001.
van Liempt S, van Zuiden M, Westenberg H, Super A, Vermetten E. Impact of impaired sleep on the development of PTSD symptoms in combat veterans: a prospective longitudinal cohort study. Depress Anxiety. 2013. doi:10.1002/da.22054.
APA. Diagnostic and statistical manual of mental disorders DSM-IV-TR Fourth Edition (Text Revision). Washington, DC: American Psychiatric Association; 2000.
AASM. International classification of sleep disorders—second edition (ICSD-2). American Academy of Sleep Medicine: Darien, IL; 2005.
AASM. International classification of sleep disorders—third edition (ICSD-3). American Academy of Sleep Medicine: Darien, IL; 2013.
Acknowledgements
The research was supported by MH101567, MH090357, USAMRAA Log11293006, and MH097965. The authors would like to thank Marie-France Marin, Ph.D., Lisa Maeng, Ph.D., and Kara Cover for helpful comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EFP-S was the lead writer of this review. AG and MRM were involved in critically revising the manuscript for important intellectual content. All authors read and approved the final manuscript.
Authors’ information
Edward F. Pace-Schott, Ph.D., is Assistant Professor of Psychiatry at Massachusetts General Hospital and Harvard Medical School. Anne Germain, Ph.D., is Associate Professor of Psychiatry and Psychology at the University of Pittsburgh School of Medicine and is Director of the Veterans Sleep and PTSD Research Program. Mohammed R. Milad, Ph.D., is Associate Professor of Psychiatry at Massachusetts General Hospital and Harvard Medical School and Director of the Massachusetts General Hospital Behavioral Neuroscience Program.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Pace-Schott, E.F., Germain, A. & Milad, M.R. Sleep and REM sleep disturbance in the pathophysiology of PTSD: the role of extinction memory. Biol Mood Anxiety Disord 5, 3 (2015). https://doi.org/10.1186/s13587-015-0018-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13587-015-0018-9