
Abstract
Immune checkpoint inhibitors (ICIs) targeting CTLA-4 and PD-1/PD-L1 to boost tumor-specific T lymphocyte immunity have opened up new avenues for the treatment of various histological types of malignancies, with the possibility of durable responses and improved survival. However, the development of acquired resistance to ICI therapy over time after an initial response remains a major obstacle in cancer therapeutics. The potential mechanisms of acquired resistance to ICI therapy are still ambiguous. In this review, we focused on the current understanding of the mechanisms of acquired resistance to ICIs, including the lack of neoantigens and effective antigen presentation, mutations of IFN‐γ/JAK signaling, and activation of alternate inhibitory immune checkpoints, immunosuppressive tumor microenvironment, epigenetic modification, and dysbiosis of the gut microbiome. Further, based on these mechanisms, potential therapeutic strategies to reverse the resistance to ICIs, which could provide clinical benefits to cancer patients, are also briefly discussed.
Similar content being viewed by others


Introduction
Over the last decade, the emergence of anticancer immunotherapies, especially immune checkpoint inhibitors (ICIs) targeting cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed death 1 (PD-1), or PD-ligand 1(PD-L1) to boost tumor-specific T lymphocyte immunity, has opened a brand new chapter for treatment of multiple histological types of malignancies with durable responses and unsurpassed clinical efficacy [1]. Compared to chemotherapeutic agents or targeted therapies, ICIs are characterized by a persistent response that can be translated into long-term survival benefits. A pooled analysis from clinical trials revealed that advanced melanoma patients treated with ICIs, such as ipilimumab and pembrolizumab, exhibited durable clinical responses, as evidenced by a three-year survival rate of about 70% and an overall survival (OS) exceeding 10 years in over 21% patients [2, 3].
Despite the unprecedented durable responses and survival benefits that have been observed, the majority of patients are less sensitive to ICI monotherapy, demonstrating primary resistance. The objective response rates of ICI monotherapy seldom exceed 40% for most tumor types (and are generally much beneath this figure), with a large proportion of patients having a partial response [4,5,6]. And more worryingly, an encouraging initial decrease in overall tumor burden observed in a number of patients can be offset by the evolution of acquired resistance to ICI therapy, which eventually leads to radiological and/or clinical disease progression [7, 8]. A study showed that approximately 25% of melanoma patients developed acquired resistance following an initial response to pembrolizumab, with disease progression evident during the median follow-up of 21 months [9]. Another study also suggested that after a five-year follow-up, 39% of patients with melanoma who initially responded to nivolumab had disease progression [10]. Similarly, more than 30% non-small cell lung cancer (NSCLC) patients were found to have relapsed after an objective initial response to nivolumab at the 2-year follow-up [11]. Accordingly, the pooled analysis of four clinical trials of nivolumab in NSCLC patients revealed that up to 65% of responders had progressed at 4 years follow-up [12]. Several studies attempted to infer the incidence of acquired resistance by analyzing durable response data, ranging from 11 to 77% across different tumor types [13]. However, up to now, no uniform definition has been established for acquired resistance to ICI therapy. Although the immunotherapy resistance taskforce of the Society for Immunotherapy of Cancer (SITC) considered acquired resistance as the occurrence of disease progression in the setting of ongoing treatment in the patient who had previously achieved a documented, confirmed objective response or stable disease lasting for more than 6 months following antineoplastic therapy, the response evaluation criteria remain divided [8] As in contrast to primary resistance various mechanisms of which have been identified during the past decade, until recently, mechanisms of acquired resistance remain ambiguous, thereby few reliable predictive biomarkers and effective treatment options can be used in such patients [14]. In this review, we outlined the present understanding of mechanisms underlying acquired resistance to ICI therapy and briefly explored potential strategies to counter the resistance and improve overall outcomes from the perspective of resistant mechanisms.
The cancer immunity cycle and acquired resistance to immune checkpoint inhibitors
The process of anti-tumor immunity is a complex sequence of multiple steps, which can be defined as the “Cancer-Immunity Cycle” (Fig. 1). First, the released tumor-associated neoantigens are captured by the antigen-presenting cells (APCs), subsequently migrating to secondary lymphoid organs as well as to tumor-related tertiary lymphoid structures [15]. Second, the APCs process the captured neoantigens into immunogenic polypeptides and present them to CD8+ T cells via the binding of the antigen peptide-major histocompatibility complex class I (MHC-I) molecule complex on the APC surface and the T cell receptor (TCR) on the surface of native CD8+ T cells, which leads to the activation and proliferation of CD8+ T cells [16]. The engagement of costimulatory molecules, CD28 and B7, is a necessary condition for the complete activation of naive T cells, and is strictly regulated by inhibitory immune checkpoints like CTLA-4 or PD-1 and their ligands. Third, the activated CD8+ T‐cells home-in to the tumor by extravasating via the endothelium and infiltrating through the surrounding stromal tissue [17]. Lastly, the TCRs on the surface of infiltrating CD8+ T‐cells need to establish contact with the peptide MHC-I complexes on the surface of APCs to release perforin and lytic granules in the immune synapse, thereby mediating tumor destruction [18]. After tumor cells are eradicated, memory T cells are formed which keep quiescent until a re-exposure to the neoantigens [19]. Under natural conditions, immune checkpoints play a negative feedback role in regulating immune responses after T-cell activation. CTLA-4 on T cells binds to B7 ligands on APCs with much higher affinity and avidity than CD28, which competitively interferes with CD28-B7 interactions, thereby preventing costimulation at the T-cell-APC interface and inhibiting activation of T cell responses [20]. Interaction of PD-1 with its ligands (PD-L1/PD-L2) can inhibit the effector stage of T-cell activation, thus suppressing the immune response [6]. The continuous interaction between inhibitory and stimulatory signals promotes adaptive responses against tumor-associated neoantigens while avoiding autoimmunity [21]. It has been confirmed that various tumors escape T-cell killing by hijacking this mechanism, and antibodies directly targeting CTLA-4, PD-1, and PD-L1 have exhibited significant anti-tumor responses. Thus far, eight ICIs inhibitors for CTLA-4 (ipilimumab), PD-1 (nivolumab, pembrolizumab, cemiplimab, and dostarlimab), and PD-L1 (durvalumab, avelumab, and atezolizumab) have been approved by the U.S. Food and Drug Administration (FDA) for the management of over 20 cancer types, the clinical indications of which are given in Table 1 [22]. Relatlimab targeting the lymphocyte-activation gene 3 (LAG-3) received its first approval by the FDA for the treatment of metastatic or unresectable melanoma in March 2022 [23]. Agents inhibiting other immune checkpoints like are still in clinical testing [24]. Abnormalities in any step of the “Cancer-Immunity Cycle” during or after treatment can result in ICI acquired resistance.

Anti-tumor immune response and ICIs. The induction of the effective anti-tumor immune response requires multiple steps: (I) The tumor-associated neoantigens are released and subsequently captured by APCs; (II) APCs present the captured antigens on MHCI molecules to T-cells, resulting in the proliferation and activation of T-cells, (III) homing and infiltration of activated T-cells. (IV) recognition of peptide MHC-I complexes and release of perforin and lytic granules to mediate tumor cell killing. Abnormalities in each of these steps during or after treatment can develop acquired resistance to ICI therapy
In contrast to primary resistance, acquired resistance is not clearly characterized across tumor types as its incidence has not been routinely reported [25]. Moreover, our understanding of acquired resistance is far from adequate and there is a desperate need to understand the mechanisms involving acquired resistance to determine the most optimal path forward for treating patients.
Potential mechanisms underlying acquired resistance to immune checkpoint inhibitors
The mechanisms of acquired resistance are indistinguishable and interdependent, and seem to overlap at least partially with the mechanisms of primary resistance. Pseudo-progression that occurs in the initial or late stage of treatment complicates the identification of resistance mechanisms. In light of Darwinian natural selection, the resistance to ICIs therapy may be pre-existing in the tumor cells before treatment, and is subsequently acquired as a result of the process of immune selection due to the genomic and epigenomic instability of tumor cells [7]. Acquired resistance may also occur at the individual tumor cell level because of the alternation in gene expression of tumor cells in response to interactions with immune cells or their products [7, 26].
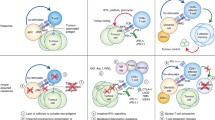
Furthermore, the mechanisms leading to resistance might vary not only by tumor types but also by patient-specific factors due to the unique genetic and clinical backgrounds of each patient. Therefore, in this section, we have shed light on the current understanding of the mechanisms concerning acquired resistance in terms of tumor cell-intrinsic and cell-extrinsic factors, based on pre-clinical animal models and clinical trials (Fig. 2).

Graphical summary that explains underlying mechanism of acquired resistance to ICIs in terms of tumor cell-intrinsic and tumor cell-extrinsic factors. Tumor intrinsic mechanisms of acquired resistance involve loss of immunogenic antigens, defects in antigen processing and presentation, and mutations of IFN‐γ/JAK signaling pathway. Tumor extrinsic mechanisms of acquired resistance mainly include activation of alternate inhibitory immune checkpoints, immunosuppressive tumor microenvironment, epigenetic modification, and dysbiosis of the gut microbiome
Tumor-intrinsic factors related to acquired resistance
Loss of immunogenic neoantigens
Neoantigens derived from somatic tumor non-synonymous mutations, which can be recognized by the immune system as “non-self”, are likely to be targets of tumor-specific T cells and elicit effective anti-tumor immune responses [21]. Tumor neoantigen burden is closely related to the immunogenicity and sensitivity to ICIs therapy [27]. Besides, tumors enriched with neoantigens have more abundant tumor-infiltrating cells (TILs) and higher levels of perforin mRNA and granzyme A, which is consistent with increased T-cell-mediated cytolytic activity [28]. Thus, mechanisms causing the loss of neoantigen expression during immunotherapy may lead to acquired resistance (Fig. 3). It has been proposed that cancer immunoediting comprises three sequential stages of elimination, equilibrium, and escape, which describe dynamic interactions between tumor cells and T cells [29]. Cancer immunoediting can lead to the partial or total elimination of neoantigens and the selection of subclones lacking neoantigen expression within the tumor, thereby conferring poor immunogenicity and acquired resistance to ICIs [29, 30]. A recent study by Anagnostou and colleagues was the first to demonstrate that acquired resistance to ICI therapy was related to the loss of mutations encoding putative mutation-associated neoantigens (MANAs) by eliminating tumor subclones or deleting chromosomal regions [31]. Specifically, whole-exome sequencing of matched resistant and pre-treatment tumors identified genomic alterations leading to the loss of 7 to 18 putative MANAs in resistant tumor clones from NSCLC patients who relapsed after an initial response to ICIs [31]. Acquired new mutations did not encode neoantigens, and the eliminated MANAs had higher predicted MHC binding affinity than either those that were gained or retained in the resistant tumors [31]. George et al. [32] identified the decreased expression of two neoantigens and loss of biallelic PTEN as the potential mechanisms of acquired resistance to ICI therapy by whole transcriptome analyses of pre-treatment and resistant tumor samples obtained from a uterine leiomyosarcoma patient. Moreover, it is arguable that the epitope of the CD19 protein sequence recognized by the chimeric antigen receptor may be selectively deleted at progression in some patients with acute lymphoblastic leukemia who demonstrated an initial response to CD19 adoptive T-cell therapy (ACT), and the pre-existing CD19 isoforms with alternative splicing may be prone to acquired resistance [33, 34].

Tumor cell-intrinsic factors contributing to acquired resistance to ICI therapy. A: Neoantigens are in partial or total eliminated conferring poor immunogenicity and acquired resistance to ICIs therapy. B: β2M mutations affect the presentation of peptide MHC-I complexes to T cells, thus leading to the tumor cells not being recognized by T cells. C: Mutations of JAK1 and JAK2 make the tumor cells insensitive to IFN-γ secreted by T-cells
The pro-inflammatory cytokines produced by TILs may induce the loss of neoantigen expression contributing to acquired resistance [35]. An ACT-treated mouse model exhibited transient tumor response following the injection of T cells targeting the melanoma differentiation antigen gp100. Subsequently, inflammatory cytokine interferon-γ (IFN-γ) secreted by TILs triggered a process of melanoma cell de-differentiation akin to epithelial-to-mesenchymal transition (EMT), leading to adaptation of melanoma cells by reducing gp100 expression and switching to a less differentiated neural crest phenotype [36]. Other inflammatory cytokines like transforming growth factor-β (TGF-β) and interleukin-6 (IL-6) have been shown to mediate the neuroendocrine differentiation of numerous types of histological cancers like lung and prostate cancer by EMT, resulting in immune escape and acquired resistance [35, 37, 38].
Defects in antigen processing and presentation
Neoantigens must be processed by APCs and presented to T cells in the form of peptide MHC-I complexes [39]. Thus, a decrease in mRNA transcription of MHC molecules, loss of genome, as well as mutations of the β-2-microglobulin (β2M) gene negatively affect antigen presentation and contribute to resistance to ICIs therapy [40] (Fig. 3). Paulson et al. [41] found a transcriptional loss of genes encoding MHC-I in two Merkel cell carcinoma patients whose tumors had relapsed after initial response to PD-1/PD-L1 inhibitors. Zaretsky and coworkers reported that acquired resistance to anti-PD-1/PD-L1 therapy was associated with a truncating mutation of the gene encoding β2M, which was related to MHC-I, and essential for its folding and transport to the surface of T cells [42, 43]. Immunohistochemical analysis of the MHC-I heavy chains in tumor samples obtained from the patient revealed that adventitial localization was lost when compared to pre-treatment tumor and adjacent stroma, despite continued production of MHC-I molecules in the recurrent tumor [43]. β2M aberrations including multiple early frameshift mutations, absence of tumor-specific β2M protein expression, and loss of heterozygosity overlapping β2M were reported to occur in about 30% of melanoma patients with progressive disease after anti-PD-1 therapy, leading to a total loss of MHC-I [44]. Knock-out of β2M in an immunocompetent lung cancer mouse model conferred resistance to PD-1 blockade in vivo, proving its role in the resistance to ICIs [40]. Tran et al. [45] described the direct loss of the chromosome 6 haplotype encoding the HLA-C*08:02 in a patient with metastatic colorectal cancer that progressed after initial response to therapy consisting of HLA-C*08:02-restricted TILs targeting mutant KRAS G12D. Other mechanisms involved in the loss of MHC-I molecules have been reported, including downregulation of the transporter associated with antigen processing 2 and low‐molecular‐weight protein 7 in MSI‐negative colorectal tumors [46]. Furthermore, the increase in PD-1+ T cell infiltration is significantly related to the increase in β2M mutations, which indicates that the resistance induced by β2M mutations is associated with PD-1+ T cell infiltration.
Mutations of IFN‐γ/JAK signaling pathway
Mutations of the janus kinases 1 and 2 (JAK1/JAK2) in the downstream signaling pathway of IFN-γ are emerging as pivotal factors in acquired resistance to ICIs therapy (Fig. 3). IFN-γ secreted by tumor-specific T cells binds to the heterodimeric IFNGR1/IFNGR2 receptor complex on tumor cells and causes the activation of JAK1 and JAK2, which in turn phosphorylate a transcription factor known as signal transducer and activator of transcription (STAT) 1. Subsequently, the translocation of phosphorylated STAT1 homodimers to the nucleus modulates the transcription of IFN-γ stimulated genes, which increases the production of chemokines, thereby attracting immune cells and directly promoting tumor cell apoptosis [47, 48]. Additionally, IFN‐γ/JAK signaling can also induce proteasome subunits and transporters associated with MHC-I as well as upregulate expression of PD-L1, which enhances antigen presentation and response to PD-1 antibodies [49, 50]. Whole-exome sequencing of tumor tissues obtained at baseline (before therapy) and after disease progression from four melanoma patients who displayed disease progression after a median 1.8 years objective response to pembrolizumab, revealed that two of the patients exhibited over 90% truncating dysfunctional mutations in JAK1 (Q503* nonsense mutation) and JAK2 (F547 splice‐site mutation), concurrent with the deletion of the wild-type allele and duplication of the mutant allele [43]. The deactivation of JAK1 and JAK2 signaling led to acquired resistance to IFN‐γ, subsequently causing damage to immune surveillance and tumor cell proliferation [43]. Several studies have shown that the loss of JAK/STAT signaling leads to resistance to CTLA-4 and PD-1 inhibitors because of the inability to upregulate PD-L1 and MHC-I expression [48, 49, 51]. Of concern, loss-of-function mutations existing at high frequency in tumors may be associated with primary resistance to ICIs therapy, with pretreatment melanoma biopsies demonstrating IFN‐γ pathway mutations in as many as 19% of the samples [48, 52]. Mutations in this pathway could additionally lead to the loss of PD-L1 expression upon IFN-γ exposure, thereby causing resistance to inhibitors of PD-1/PD-L [48]. Mutations and deletions of IFN‐γ pathway-associated proteins, like IFNGR1 and IFNGR2, as well as STAT1, may also be a reason for resistance to ICIs and need to be further explored [52].
Tumor-extrinsic factors related to resistance
Activation of alternate inhibitory immune checkpoints
Overexpression of multiple alternate immune checkpoints contributing to a severely exhausted phenotype of T-cells can exert immunosuppressive effects and lead to the failure of ICI therapy. Acquired resistance to ICIs has been described secondary to compensatory upregulation of alternative immune checkpoints like T cell immunoglobulin and mucin domain 3 (TIM-3), LAG-3, B and T lymphocyte attenuator (BTLA), V-domain Ig suppressor of T cell activation (VISTA), T cell Ig and ITIM domain (TIGIT), and so on in multiple studies (Fig. 4). A study by Thommen et al. [53] revealed that increased coexpression of CTLA-4, PD-1, TIM-3, LAG-3 as well as BTLA, was positively related to progressive T-cell exhaustion and subsequent resistance to anti-PD1 therapy in NSCLC. Their research suggested that the surface of CD8+ T cells expressing the above five receptors had serious defects causing reduced proliferation, migration, and cytokine production. Koyama et al. [54] observed that TIM-3 up-regulation in TILs of lung adenocarcinoma patients was significantly correlated with acquired resistance to anti-PD-1 antibody. Moreover, the TIM-3 expression was related to the degree of anti-PD-1 antibody binding in T cells, the positive expression of which increased with the binding degree of T cells to anti-PD-1 blockade [54]. T-cells from resistant tumors also over-expressed LAG3 and CTLA4, however, BTLA expression appeared variable. Limagne et al. [55] demonstrated that the accumulation of monocytes, bone marrow-derived suppressor cells (MDSCs), and lymphocytes expressing TIM-3 and galectin-9 was related to primary or acquired resistance following nivolumab treatment in a cohort of NSCLC patients. Compared to matched pre-treatment tumors, the frequency of VISTA expression on T cells and CD68+ macrophages were found higher in treated prostate and melanoma tumors [56]. Kakavand et al. found that twelve melanoma patients with acquired resistance to ICIs were able to obtain matched samples before and after treatment, 8 of which patients were detected with VISTA upregulation [57]. Several studies suggested that the high expression of TIGIT regulatory T cells (Tregs) positively related to increased Treg frequencies in tumors and mediate tumor resistance to ICI therapy [58, 59]. The upregulation of other immune checkpoints also potentially results in acquired resistance that needs further research.

Immunosuppressive tumor microenvironment contributing to acquired resistance to ICI therapy. Immune cells infiltrate into the TME, interacting with each other and tumor cells, harbor an immunosuppressive phenotype responsible for immune escape of tumor cells and the following acquired resistance during or after ICI treatment. These immunosuppressive cells include Tregs, TAMs, MDSCs, which express the alternate inhibitory immune checkpoints like TIM-3, LAG-3, BTLA, VISTA, and TIGIT, secrete cytokines and growth factors like IL-35, IL-10, and TGF-β, VEGF, or product IDO and adenosine, which negatively regulate anti-tumor immune immunity, remodel the extracellular matrix, and promote angiogenesis. As a result, the immunosuppressive TME promotes the tumor cell resistance to ICI therapy
Other suppressive factors in the tumor microenvironment (TME)
It is now increasingly appreciated that immunosuppressive cells including Tregs, MDSCs, tumor-associated macrophages (TAMs), and cancer-associated fibroblasts (CAFs), as well as some inhibitory cytokines, build a chronic inflammatory, pro-angiogenic, intratumoral, and immunosuppressive environment where tumor cells are liable to evade immune-mediated eradication and inhibit the therapeutic activity of ICIs [60,61,62] (Fig. 2). Tregs may be capable of suppressing the efficacy of effector T cells (TEFFs) by the secretion of some inhibitory cytokines like TGF-β, IL-10, and IL-35 [63, 64]. The response to PD-1/PD-L1 inhibitors has been shown to be correlated with an increased ratio of TEFFs to Tregs in murine models [65]. Simpson et al. [66] suggested that an increased Tregs infiltration resulted in acquired resistance to PD-1 inhibitors alone or in combination with radiotherapy. ICI treatment might cause the recruitment of Tregs to the TME and play a role in mediating resistance, as demonstrated in a murine model of claudin-low breast cancer generally known to be resistant to ICIs [67]. Timelines from clinical studies indicate that increased MDSCs in the TME promoting tumor invasion, angiogenesis, and metastasis correlate with the reduced efficacy of immunotherapies including ICIs [68, 69]. Previous clinical studies have revealed the relationship between increased infiltration of TAMs and ominous prognosis [31, 70]. TAMs can express co-inhibitory molecules, including PD-L1 and B7-H4, and release anti-inflammatory cytokines, such as IL-10 [71]. M2 macrophages were shown to directly remove anti-PD1 antibodies from the surface of PD-1+ T cells, thereby interfering with anti-PD1 therapy [70]. Thought to be one of the most abundant components in the mesenchyme of most tumors, CAFs contribute to T-cell dysfunction and immunosuppressive TME by producing several CAF-derived molecules and ligands, including IL-6, chemokine C-X-C motif ligand 12 (CXCL12), CXCL5, TGF-β, vascular endothelial growth factor (VEGF), fibronectin, and collagen, and the increased expression of immune checkpoint ligands including PD-L1, PD-L2, and FasL [72]. CAFs also promote a Dendritic cell (DC) phenotype that is unable to interact with and present antigens to CTLs [73]. Chakravarthy et al. identified a poor prognosis phenotype of CAFs driven mainly by the activation of TGF-β signaling, which was related to the PD-1 blockade resistance in bladder cancer and melanoma [74]. Increased infiltration of CAFs was related to resistance to PD-1 blockade, inhibition of which rescued the anti-tumor effects of anti-PD-1 treatment in mouse models of hepatocellular carcinoma [75]. Indole 2,3-dioxygenase (IDO), secreted by immune cells or tumor cells, can promote the secretion and activity of Tregs and MDSCs [76]. As a rate-limiting enzyme, IDO can promote the catabolism of tryptophan and generate immunosuppressive metabolites including kynurenine, thereby suppressing the function of T cells and inducing T cell apoptosis [77, 78]. Holmgaard et al. [79] reported that tumor growth was significantly delayed and OS markedly increased in IDO knockout melanoma mice treated with anti-PD1 therapy compared with that in wild-type mice. Also, the accumulation of extracellular adenosine though the HIF-1α-CD39/CD73-adenosine signaling pathway has been proven to drive an immunosuppressive TME, and promote tumor progression [80].
Influence of epigenetic modification
Epigenetic modification of tumor cells has been shown to alter the expression of immune-associated genes, thus affecting the processing and presentation of antigens, proliferation, and differentiation of T cells, T-cell exhaustion, and acquisition of phenotype of memory T cells [81] (Fig. 5). Lower levels of IFN -γ and IFN-γ-induced genes like CXCL9 contribute to decreased infiltration of CD8+ T cells and are correlated with tumor progression following anti-PD-L1 therapy [82]. A study by Peng et al. [83] revealed that cancer epigenetic silencing consisting of enhancer of zeste homolog 2 (EZH2)-mediated histone modifications and DNA methyltransferase (DNMT) -mediated DNA methylation of the gene encoding the T helper 1-type chemokines, CXCL9 and CXCL10, decreased T cells homing and infiltration, thus potentially impairing the efficacy of immunotherapy. Recent studies have suggested that the exhaustion of T cells correlated with de novo DNA methylation and may persist and be passed on to successive T cell generations [84]. Epigenetic studies have suggested that although ICIs can reinvigorate exhausted CD8+ T cells (TEX) and enhance the control of chronic infections and tumors, they have little effect on genes related to the generation of memory T-cells [85]. They found that TEX metagenes treated with PD-L1 blockade partly overlapped with TEFF, but there was little overlap with memory T cells (TMEM), indicating that acquisition of memory potential was limited after TEX reinvigoration [85]. Reinvigorated TEX failed to become TMEM after antigen clearance and they became depleted again following anti-PD-L1 treatment, even if there was neoantigens re-exposure. Although the inhibition of the PD-1 pathway led to transcriptional re-wiring and re-engagement of effector circuitry in the TEX epigenetic landscape, TEX could never acquire the phenotype of memory T cells, and this epigenetic stability of TEX might result in acquired resistance to ICIs [85]. In addition, TEX showed extensive changes in the available chromatin, which were different from those in the TEFF counterparts. Exhaustion-specific enhancers in TEX displayed different motifs at T-bet, RAR, and Sox3 marker transcription factor binding sites. The crucial differences in the profile of regulatory regions between functional CD8+ T cells and TEX were greater than those observed in gene expression, which suggests that the accessible chromatin networks related to the exhaustion state required a large rewiring [86]. A combination of ICIs and epigenetic modifiers may help to improve the durability of the response to ICIs in the future.

Epigenetic modification and acquired resistance to ICI therapy. A: Epigenetic silencing of gene of T helper 1-type chemokines CXCL9 and CXCL10 results in CD8+ T cell homing and infiltration to the TME. B: Acquisition of memory potential was limited after reinvigoration of exhausted T cells with PD-L1 blockade. Reinvigorated exhausted T cells fail to become memory T cells after antigens clearance and they will become depleted again, even if neoantigens are exposed again
Dysbiosis of gut microbiome
It is emerging that the alteration of the human gut microbiome affects tumor progression and is connected with the response to ICIs therapy [87]. Significant differences in the structure of gut microbiome were observed between patients who responded well to ICIs and those who did not [88, 89]. A landmark study conducted by Gopalakrishnan et al. demonstrated that patients who have a high diversity and abundance of gut microbiota like Faecalibacterium and Ruminococcaceae have increased antigen presentation and improved TEFF function in the periphery and TME with enhanced anti-tumor immune response, whereas patients with low diversity and high relative abundance of gut microbiota like Bacteroidales have impaired anti-tumor immune responses mediated by a decreased capacity of antigen presentation and limited intratumoral lymphoid and myeloid infiltration [90]. G Analysis of the gut microbiome composition of patients with metastatic melanoma before anti-PD-1 treatment indicated that bacterial species including Ruminococcus obeum and Roseburia intestinalis were remarkably abundant in non-responders, while Enterococcus faecium, Collinsella aerofaciens, and Bifidobacterium longum were significantly enriched in responders [91]. A prospective study that enrolled 26 metastatic melanoma patients treated with ipilimumab found that antitumor response and immune-related colitis were related to colonization by Firmicutes including butyrate-producing bacterium L2–21, Faecalibacterium prausnitzii L2–6, and Gemmiger formicilis ATCC 27749. Compared to patients with initial microbiota enriched with Bacteroidetes, patients with microbiota rich in Faecalibacterium genus and other Firmicutes had a markedly higher progression-free survival (PFS) and OS [92]. Antibiotic use and gastrointestinal immune-related adverse events (irAEs), which are common complications of ICIs therapy, disrupt the intestinal microbiome and result in acquired resistance [93]. Pre-clinical mouse models have indicated that broad-spectrum antibiotics like imipenem and colistin eliminated gut microbiota and decreased the antitumor activity of CTLA-4 antibodies, with a significant reduction in the anti-CTLA-4-mediated sensitization of tumor-infiltrating and splenic lymphocytes. Bacteroides fragilis could recover the response to CTLA-4 antibodies, while tolerogenic Bacteroides species mediated complete resistance to CTLA-4 antibodies [94]. Also, clinical data from a large cohort of patients with non-small cell lung cancer, renal cell carcinoma, or urothelial cancer suggested that patients treated with antibiotics for routine indications shortly before, during, or shortly after treatment with anti-PD1/PD-L1 mAB had significantly lower PFS and OS rates compared to patients who had not received antibiotics, suggesting that disrupted the gut microbiota via antibiotic use could potentially impair anti-tumor immune responses and resulted in acquired resistance to ICIs [89]. The impact of gut microbiome alteration further increases the complexity of responsiveness and resistance to ICIs therapy. Large prospective trials are required to define the microbial components and mechanisms underlying this effect, and to provide a basis for introducing different kinds of gut bacteria to reverse immunotherapy resistance in cancer.
Potential strategies to overcome resistance: combining immune checkpoint therapy with other therapies
The above-mentioned mechanisms of acquired resistance to ICIs do not act in isolation, and the interactions between several immunosuppressive components and the overlap between various signaling pathways together resulted in resistance. Identifying the specific resistance mechanisms in these patients is a crucial step toward effective treatment and ultimately inducing durable responses for them. It is likely that combination therapies with multiple treatment modalities have potential synergistic effects and are likely to reverse resistance. It is worth noting that efforts should be guided by an understanding of the mechanisms underpinning resistance rather than the random combination with existing drugs and therapies. In this section, we discussed potential approaches to counter the resistance and improve the efficacy of ICIs from the perspective of the resistant mechanisms mentioned above (Table 2).
Targeting neoantigen depletion
It has been demonstrated that autologous tumor vaccinations enhancing immunogenicity and stimulating adaptive immune responses with tumor-specific antigens improved sensitivity to ICI therapy [95,96,97]. NEO-PV-01, a neoantigen-based vaccine, was tested combined with nivolumab in the phase Ib clinical trial (NCT02897765) in NSCLC, melanoma, and bladder cancer demonstrated that neoantigen-specific CD4+ and CD8+ T cell responses were detected in all vaccinated patients with no serious adverse events [98]. The combined therapy of the vaccine ISA 101 and nivolumab for patients with incurable HPV-16-positive tumors achieved an overall response rate of 33% and a median OS of 17.5 months, much higher than the results obtained with nivolumab monotherapy, rendering an overall response rate of 14.3% [99]. Guo et al. reported that neoantigen-loaded monocyte-derived dendritic cell vaccines in combination with nivolumab triggered neoantigen-specific CD4+ and CD8+ T cell activation and mediated complete regression of all tumors in advanced metastatic gastric cancer [100]. The combination of oncolytic virotherapy and ICI therapy is a promising approach. Similar to self vaccination, oncolytic virustherapy could also induce tumor antigen release and provide danger signals, which consequently led to enhanced T cell priming and ameliorated resistance to anti-PD1/PDL1 therapy [101]. Talimogene laherparepvec (T-VEC) has been proven to improve the efficacy of anti-PD-1 therapy in advanced melanoma patients, with an objective response rate of 62% and a complete response rate of 33% [102]. A phase II Study (NCT01740297) suggested that the combination of T-VEC and ipilimumab shows a higher objective response rate versus ipilimumab alone without additional safety concerns in patients with advanced unresectable melanoma [103]. Consistently, a retrospective study suggested that T-VEC treatment might overcome loco-regional ICI acquired resistance in patients with stage IIIB-IV M1c melanoma [104].
Targeting defects in antigen processing and presentation
Growing evidence suggests that the correction of defects in antigen processing and presentation can greatly enhance the efficacy of ICI therapy. It was reported that the cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) antagonism controlled the growth of β2M deficient tumors, and was effective in models of either primary or acquired resistance to ICIs [105]. In a mouse model of MAPK-activated head and neck squamous cell carcinoma, trametinib, a small molecule MEK inhibitor, inhibited extracellular signal-regulated kinase phosphorylation, promoted MHC-I and PD-L1 expression, and in combination with PD-L1 blockade, overcame resistance to monotherapy, which significantly improved inhibition of tumor growth [106]. Results from a mouse model of pancreatic ductal adenocarcinoma showed that chloroquine enhanced MHC-I antigen expression, sensitizing tumors to dual immune checkpoint inhibition with anti-CTLA4 and anti-PD-1 antibodies [107]. The ASPIRE nanovaccine derived from recombinant adenovirus-infected dendritic cells, in which specific peptide-MHC-I, B7 co-stimulatory molecules, and anti-PD1 antibody were simultaneously anchored by a programmed process, could significantly enhance the delivery of antigen to lymphoid organs and induce broad-spectrum T-cell responses to eliminate established tumors [108]. Additionally, regimens targeting TLRs (e.g., TLR3, TLR9) have been shown to promote DC maturation and reasonably alleviate anti-PD1/PDL1 resistance [109].
Targeting the IFN‐γ/JAK signaling pathway
In experiments in mice, intratumoral BO-112 (a nanoplexed formulation of Poly I: C coupled to polyethylenimine) exerted anti-tumor activities in an IFN-γ and IFN-α/β dependent manner, which was observed not only in locally injected tumors but also in distant tumors. And the systemic effect was augmented by the coadministration of anti–PD-L1 antibodies [110]. A multicenter phase I clinical trial (NCT02828098) incorporating 28 patients with tumors resistance to anti–PD-1 antibodies showed the combination of BO-112 administered intratumorally with pembrolizumab or nivolumab was also well-tolerated, with 3 patients achieved partial responses, with 10 more patients having stable disease, suggesting that local BO-112 might be a strategy to revert anti-PD-1 therapy [111]. ACT was effective against JAK2 loss tumors, but not JAK1 loss tumors in an IFN signaling-deficient model of B16 murine melanoma, and overexpression of NLRC5(nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 5) restored the efficacy of ACT against B16-JAK1 loss tumors [112].
Targeting activation of alternate inhibitory immune checkpoints
The combination therapies of ICIs targeting different immune checkpoints indicate potential benefits in overcoming acquired resistance in pre-clinical studies and clinical trials. The combination of anti-CTLA-4 and anti-PD-1 treatment appears to target separate populations of T-cells and has already been approved in hepatocellular carcinoma, melanoma, renal cell carcinoma, and colorectal cancer, with a variety of trials ongoing [113, 114]. Limagne et.al suggested that TIM-3 blockade could restore in vitro the CD8 secreting property in PBMCs from lung cancer patients when used in combination with the anti-PD-1 antibody, suggesting that anti-TIM-3 reverses resistance to anti-PD-1 [55]. Phase I/II study (NCT02460224) suggested that the LAG-3 inhibitor ieramilimab plus anti-PD-1 spartalizumab were well tolerated and had anti-tumor activity, with elicited durable responses in 12 of 121 patients with solid malignancies [115]. Co-blockading PD-L1and VISTA in CT26 colon cancer and B16 melanoma mouse models showed better efficacy in inhibiting tumor growth and prolonging survival compared to targeting each molecule alone, suggesting a synergistic effect between anti-VISTA and anti-PD-L1 antibodies in inducing Teff activation [116]. Choi et al. showed that the combination of PD-1 blockade and BTLA inhibitier synergistically improved overall long-term survival compared to monotherapy, with decreased levels of Tregs and increased expression of CD4+ IFN-γ and CD8+ IFN-γ [117]. Interim data from a phase II trial (NCT03563716) demonstrated the improved efficacy of Dual inhibition of TIGIT and PD-L1 with tiragolumab and atezolizumab compared to atezolizumab alone in patients with PD-L1-positive metastatic NSCLC in terms of overall response and progression free survival without additional adverse events [118].
Targeting suppressive factors in the tumor microenvironment
It seems that immunosuppress components within the TME represent promising targets for increasing the anti-tumor efficacy of immunotherapy. A pre-clinical study showed that anti-CD25 antibody could effectively deplete tumor-infiltrating Tregs and increase ratios of effector-to-Treg, and synergized with PD-1 blockade to promote complete tumor rejection [119]. It has been suggested that CC chemokine receptor 4 (CCR4) mediated Treg recruitment into the TME, antagonism of which could suppress the frequency of CCR4 reduced Tregs and potentiated anti-tumor efficacy of ICIs [120]. A phase I study (NCT02476123) showed that combining nivolumab, with mogamulizumab (a Treg-depleting anti-CCR4 antibody) provided an acceptable safety profile and anti-tumor activity, with populations of effector Tregs reduced and CD8+ T cells in TILs increased [121]. In the tumor TME, TAMs survival depends on the CSF1/CSF1R pathway, blocking CSF1/CSF1R has been proven to significantly reduce macrophage recruitment and M2 phenotype polarization and activates CD8+ T cells, thereby sensitizing tumor to ICI and prolonged survival in pancreatic ductal adenocarcinoma and hepatocellular carcinoma [122]. Results according to mouse models of colon cancer revealed that CSF-1R kinase inhibitor PLX3397 combined with anti-PD-1 antibody and oncolytic viruses synergistically conferred significant tumor control and prolonged the survival [123]. Antagonizing CSF-1R with BLZ945 could reduce the induction of human and murine suppressive myeloid cells, when combined with PD-(L)1 blockades superiorly limited tumor progression [124]. Due to the function of modulating the differentiation of MDSCs, the combinational therapies of ATRA and immunotherapies have synergistic anti-tumor efficacy. Depleting MDSCs by all-trans-retinoic acid (ATRA) in the model of LKB1-deficient murine tumors resulted in improved anti-tumor T cell responses and increased sensitivity to PD-1 blockade [125]. Randomized phase II clinical trial (NCT02403778) reported that ATRA plus ipilimumab significantly decreased the number of MDSCs in the peripheral circulation of patients with metastatic melanoma without increased incidence of grade 3 or 4 adverse events, compared with ipilimumab monotherapy [126]. Besides, inhibition of casein kinase 2 (CK2) with BMS-211 substantially reduced the amount of polymorphonuclear MDSCs and macrophage differentiation, which was dramatically synergized with the anti-tumor efficacy of CTLA-4 blockades [127]. The phase I/II ECHO-202/KEYNOTE-037 trial (NCT02178722) revealed that epacadostat (a selective IDO1 enzyme inhibitor) plus pembrolizumab generally had a manageable safety profile and encouraging anti-tumor activity in several advanced solid tumors like melanoma, NSCLC, squamous cell carcinoma of the head and neck, renal cell carcinoma, urothelial carcinoma, and endometrial adenocarcinoma [128]. Nevertheless, epacadostat had no additional clinical benefit to pembrolizumab in the pivotal phase III study [129]. A Phase I/II trial (NCT02658890) suggested that linrodostat mesylate (an oral IDO1 inhibitor) in combination with nivolumab has demonstrated safety and preliminary evidence of clinical activity, with an ORR of 34% in the advanced bladder cancer [130]. Several preclinical studies have implicated that unleashing the immunosuppressive TME by targeting CD39/CD73 (that generate adenosine) and anti- adenosine agents (that consume adenosine) could suppress acquired resistance thus synergy ICIs efficacy [121]. Lu et.al has demonstrated that targeting CD39 on macrophages could efficiently rescue anti-PD1 resistance in hepatocellular carcinoma (HCC) [131]. The phase II trial of Oleclumab targeting CD73 in combination with Durvalumab showed prolonged PFS and increased ORR versus durvalumab alone, with manageable safety profiles [132]. Results from the phase I/II study of the A2AR antagonist NIR178 in advanced NSCLC suggested that clinical benefit was observed in immunotherapy-exposed and -naïve patients regardless of PD-L1 status, and was well tolerated [133]. A number of such combination therapies are currently ongoing in clinical trials. Moreover, vascular normalization with VEGF/VEGFR inhibitors combined with PD-(L)1 blockade has demonstrated clinical efficacy across a variety of tumor types in phase III trials and is already FDA-approved in NSCLC, renal cell carcinoma (RCC), endometrial carcinoma, and hepatocellular carcinoma. Furthermore, co-blockade of TGF-β and PD-L1 was shown to reduce TGF-β signaling in stromal cells, facilitate T-cell infiltration into the tumor centers, provoking vigorous anti-tumor immunity and tumor regression [134]. Trials evaluating the combined inhibition of TGF-β and PD-(L)1 in a variety of solid tumors are underway (NCT04390763, NCT02423343). Compared to anti-PD-L1 alone, IL-10 inhibition improved responses to anti-PD-L1 and produced more cytotoxic effector KLRG1+, IFN-γ+, and memory CD8+ T-cells, and fewer exhausted T-cells in chronic lymphocytic leukemia [135]. In contrast, pegilodecakin (pegylated IL-10) has been tested in a clinical trial (NCT02009449) and demonstrated to have a manageable toxicity profile and preliminary anti-tumor activity when in combination with pembrolizumab or nivolumab in previously treated advanced RCC [136]. Hypoxia within the TME is an intrinsic property of all solid malignant tumors, and we have detailed how to target hypoxic TME synergistically with immunotherapy in another review [80]. The induction of tertiary lymphoid structures (TLSs) that represent local and favorable sites for generating antitumor humoral and cellular immune responses has been shown to be capable of overcoming resistance to ICIs [137, 138]. A number of such combination therapies are currently ongoing in clinical trials.
Targeting epigenetic modification
Epi-drugs, defined as small-molecule inhibitors that target epigenetic regulators like DNMT, HDAC, and EZH2, have been shown to augment anti-tumor immune responses through multiple immunomodulatory activities [139, 140]. In animal models of breast, colon, and prostate cancers, inhibition of DNMT led to MHC-I upregulation, T-cell chemotaxis, and tumor infiltration of CD8+ T cells, thus enhancing the anti-tumor effects of anti-PD-1 antibodies [141]. Decitabine (a related DNMT inhibitor) stimulated the cytotoxic and infiltration responses of CD8+ T cells, which combined with anti-CTLA-4 antibody exhibited synergistic anti-tumor effects and extended survival in a syngeneic murine ovarian cancer model [142]. Panobinostat, an HDAC inhibitor, could upregulate the expression of PD-L1 and PD-L2 and improve anti-PD-1 immunotherapy in melanoma, resulting in improved inhibition of tumor growth and increased survival compared to anti-PD1 monotherapy [143]. Romidepsin was demonstrated to induce strong anti-tumor responses in a T cell-dependent manner and combinatory therapy with anti-PD-1 and induce tumor rejection in multiple lung tumor models [144]. Several preclinical models suggested that a novel orally bioavailable HDAC inhibitor HBI-8000 could augment the activity of ICIs targeting either PD-1, PD-L1, or CTLA-4, and significantly reduce tumor progression [145]. It has been shown that inhibiting EZH2 reversed acquired resistance and enhanced anti-tumor effects when in combination with anti-PD1, anti-CTLA-4, and IL-2 immunotherapy in prostate cancer and melanoma [146, 147]. Inspired by the encouraging results of preclinical studies, the combination therapy of ICIs with epigenetic drugs is being translated into clinical trials. A phase II trial (NCT02397720) revealed that the combination of azacitidine and nivolumab was safe and produced encouraging objective response rate and OS outcomes in patients with acute myeloid leukemia [148]. In phase I/Ib trial (NCT02638090), HDAC inhibitor vorinostat plus pembrolizumab was well tolerated and demonstrated preliminary anti-tumor activity, with three confirmed partial response (PR) and eleven stable disease responses among ICI-resistant metastatic NSCLC 24 patients [149]. Furthermore, preliminary results from the ENCORE-601 phase Ib/II trial revealed that entinostat, an experimental HDAC inhibitor, was well tolerated and, together with pembrolizumab, induced durable responses in melanoma patients whose disease progressed after PD-1 blockade monotherapy [150]. However, entinostat plus pembrolizumab provided a clinically meaningful benefit despite not achieving the primary response rate endpoint in the expansion cohort of ENCORE 601 that included anti-PD-(L)1-experienced patients with NSCLC [151]. Additionally, trials of EZH2 inhibitors plus pembrolizumab or ipilimumab are recruiting the patients with advanced solid tumors (NCT03854474 and NCT03525795). Together, these results provided a solid rationale for combining epigenetic agents with ICI immunotherapy.
Targeting gut microbiome dysbiosis
Manipulating the gut microbiota has been shown to enhance anti-tumor immunity and alleviate the development of acquired resistance to ICIs [94, 152]. Sivan et.al first revealed that commensal Bifidobacterium can enhance anti-tumor immunity in vivo in a non-antigen-dependent manner, which exhibited synergistic anti-tumor effectiveness with PD-L1 inhibitor [153]. It was reported that oral feeding of Bacteroides fragilis with Burkholderia cepacian or Bacteroides thetaiotaomicron enhanced the efficacy of CTLA-4 blockade via promoting DC maturation and triggering Th1 immune responses [94]. Three bacterial species (Lactobacillus johnsonii, Bifidobacterium pseudolongum, and Olsenella) were found to significantly enhanced efficacy and promote ICI therapies by raising the activation of CD4+ and CD8+ T cells [154]. A study conducted in a mouse model suggested that the presence of A. muciniphila and Akkermansia muciniphila contributed to the immunogenicity of the PD-1 blockade, and its abundance was related to clinical responses. Fecal microbiota transplantations (FMT) restored the efficacy of anti-PD-1 blockade in an IL-12-dependent manner [89]. Motivated by the encouraging results of preclinical studies, numerous clinical trials focusing on how to modulate gut microbiota composition to overcome ICI resistance are ongoing. A phase I/II clinical trial (NCT03775850) revealed that EDP1503 (an oral microbiota product) combined with pembrolizumab was well-tolerated and safe, and biomarker analysis found that EDP1503 works though upregulating the CD8+ T cells/Treg cell ratio [155]. Results of the phase I/II trial (NCT03637803) investigating the synergistic efficacy of the oral probiotic MRx0518 combined with pembrolizumab in bladder cancer, NSCLC, RCC, or melanoma have not yet been published [156]. Results from two recent clinical trials (NCT03353402 and NCT03341143) have suggested that patients with metastatic melanoma-resistant to PD-1 inhibitor after the intervention of FMT from responders had improved clinical benefits [157, 158].
Other emerging combination strategies
Besides discussed above, multiple emerging strategies combined with ICIs are underway, some of which reported early encouraging results. The stimulator of an interferon gene agonist-loaded lipid nanoparticles (STING-LNP) might represent a promising candidate for anti-PD-1-resistant tumors, which in combination with PD-1 blockade synergistically promoted anti-tumor response via the activation of NK cells to overcome anti-PD-1 resistance in a B16-F10 lung metastasis model [159]. Similarly, Li et al. reported that novel Microbubble-assisted UltraSound-guided Immunotherapy of Cancer (MUSIC) platform utilized antibody guided targeting to stimulating STING pathway in APCs, further sensitizing poorly immunogenic tumors to anti-PD-1 therapy [160]. Engineered nanoparticles carrying immunostimulatory oligonucleotides has been confirmed to synergize with anti-CTLA-4 therapy to achieve tumor suppression in animal models of melanoma, colon cancer, and breast cancer [161]. The tumor-targeted radioligand 177Lu-PSMA-617 has recently demonstrated anti-tumor activity in prostate cancer [162], and its synergy with anti–PD-1 is currently being investigated in a phase II trial (NCT03805594). In addition, the SARS-CoV-2 pandemic has enormously unlocked the potential of vaccine technology as a powerful therapeutic platform. Phase I trail (NCT02410733) suggested that BNT111 (an intravenously administered liposomal RNA vaccine), alone or in combination with PD1 blockade, mediated durable objective responses in ICI-experienced patients with unresectable melanoma [163]. Moreover, an exciting preclinical study demonstrated the vaccination of 8p4 + FK-33 nanoparticles (8FNs) incorporating different tumor antigens could inhibit tumor growth by reversing the immunosuppressive TME and potentiating anti-tumor immunity, exerting synergistic therapeutic effects with PD-1 inhibitor [164]. We hoped that the development of more emerging strategies could pave the way for overcoming resistance to ICI therapy.
Future perspectives and conclusions
Although ICI therapy has revolutionized medical oncology, the emergence of acquired resistance in a considerable proportion of patients poses a significant challenge. Currently, the true landscape of acquired resistance to ICIs remains largely uncertain, thus the uniform definition and evaluation criteria are required to be established for acquired resistance. Moreover, emerging multifaceted approaches and higher resolution investigations of interactions involving host, tumor, and/or the TME may aid in the discovery new resistant mechanisms and predict new synergistic combinations. As ongoing research into underlying biology of acquired resistance to ICI therapy, the near future will witness more and more therapeutic combinations to overcome acquired resistance, resulting in clinical benefits for more patients. However, given the heterogeneity of tumor types and the complexity of anti-tumor immune responses, a wide set of biomarkers, such as TILs status, TMB, TLS, and PD-L1 expression, is required to predict the treatment efficacy or resistance to ICIs. With the development of human bioinformatic analysis and genome sequencing, more and more patients will definitely benefit from both integrated and individualized immunotherapies.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- ICIs:
-
Immune checkpoint inhibitors
- CTLA-4:
-
Cytotoxic T lymphocyte antigen 4
- PD-1:
-
Programmed death 1
- PD-L1:
-
PD-ligand 1
- OS:
-
Overall survival
- NSCLC:
-
Non-small cell lung cancer
- SITC:
-
The Society for Immunotherapy of Cancer
- APCs:
-
Antigen-presenting cells
- MHC-I:
-
Major histocompatibility complex class I
- TCR:
-
T cell receptor
- FDA:
-
Food and Drug Administration
- LAG3:
-
Lymphocyte-activation gene 3
- TILs:
-
Tumor-infiltrating cells
- ACT:
-
Adoptive T-cell therapy
- MANAs:
-
Mutation-associated neoantigens
- IFN-γ:
-
Interferon-γ
- EMT:
-
Epithelial-to-mesenchymal transition
- TGF-β:
-
Transforming growth factor-β
- IL-6:
-
Interleukin-6
- β2M:
-
β-2-Microglobulin
- JAK1/JAK2:
-
Janus kinases 1 and 2
- STAT:
-
Signal transducer and activator of transcription
- TIM3:
-
T-cell immunoglobulin and mucin domain 3
- BTLA:
-
B and T lymphocyte attenuator
- VISTA:
-
V-domain Ig suppressor of T cell activation
- TIGIT:
-
T cell Ig and ITIM domain
- MDSCs:
-
Marrow-derived suppressor cells
- Tregs:
-
Regulatory T cells
- TAMs:
-
Tumor-associated macrophages;
- CAFs:
-
Cancer-associated fibroblasts
- BTLA:
-
B and T lymphocyte attenuator
- TEFF:
-
Effector T cells
- CXCL12:
-
Chemokine C-X-C motif ligand 12
- VEGF:
-
Vascular endothelial growth factor
- DC:
-
Dendritic cell
- IDO:
-
Indole 2,3-dioxygenase
- PFS:
-
Progression-free survival
- EZH2:
-
Zeste homolog 2
- DNMT:
-
DNA methyltransferase
- TEX:
-
Exhausted CD8+ T cells
- TMEM:
-
Memory T cells
- irAEs:
-
Immune-related adverse events
- RCC:
-
Renal cell carcinoma;
- T-VEC:
-
Talimogene laherparepvec
- cIAP1/2:
-
Cellular inhibitor of apoptosis proteins 1 and 2
- NLRC5:
-
Nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 5
- CCR4:
-
CC chemokine receptor 4
- ATRA:
-
All-trans-retinoic acid
- CK2:
-
Casein kinase 2
- HCC:
-
Hepatocellular carcinoma
- TLSs:
-
Tertiary lymphoid structures
- SCL:
-
Small cell lung cancer
- HNSCC:
-
Head and neck squamous cell carcinoma
- cHL:
-
Classical hodgkin lymphoma
- PMBCL:
-
Primary mediastinal large B cell lymphoma
- MCC:
-
Merkel cell carcinoma
- TMB:
-
Tumor mutational burden
- CSCC:
-
Cutaneous squamous cell carcinoma
- TNBC:
-
Triple-negative breast cancer
- PDAC:
-
Pancreatic ductal adenocarcinoma
- STING-LNP:
-
The stimulator of an interferon gene agonist-loaded lipid nanoparticles
- MUSIC:
-
Microbubble-assisted UltraSound-guided Immunotherapy of Cancer
- iTPNCs:
-
Immunostimulatory tandem peptide nanocomplexes
- 8FNs:
-
8P4 + FK-33 nanoparticles
References
Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223–49.
Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and Phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–94.
Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2014;372(4):320–30.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44.
Zou W, Wolchok JD, Chen L. PD-L1 (B7–H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4.
Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16(2):121–6.
Kluger HM, Tawbi HA, Ascierto ML, et al. Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J Immunother Cancer. 2020;8(1): e000398.
Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315(15):1600–9.
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535–46.
Horn L, Spigel DR, Vokes EE, et al. Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: two-year outcomes from two randomized, open-label, phase III Trials (CheckMate 017 and CheckMate 057). J Clin Oncol. 2017;35(35):3924–33.
Antonia SJ, Borghaei H, Ramalingam SS, et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol. 2019;20(10):1395–408.
Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37(4):443–55.
Morad G, Helmink BA, Sharma P, et al. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184(21):5309–37.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
Becht E, Giraldo NA, Dieu-Nosjean MC, et al. Cancer immune contexture and immunotherapy. Curr Opin Immunol. 2016;39:7–13.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
O’Donnell JS, Long GV, Scolyer RA, et al. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81.
Ribas A, Shin DS, Zaretsky J, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. 2016;4(3):194–203.
Schildberg FA, Klein SR, Freeman GJ, et al. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. 2016;44(5):955–72.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74.
Vesely MD, Zhang T, Chen L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu Rev Immunol. 2022;40:45–74.
Paik J. Nivolumab plus relatlimab: first approval. Drugs. 2022;82(8):925–31.
Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discovery. 2015;14(8):561–84.
Zhou B, Gao Y, Zhang P, et al. Acquired Resistance to Immune Checkpoint Blockades: The Underlying Mechanisms and Potential Strategies. Front Immunol. 2021;12: 693609.
Pardoll D. Cancer and the immune system: basic concepts and targets for intervention. Semin Oncol. 2015;42(4):523–38.
Kvistborg P, van Buuren MM, Schumacher TN. Human cancer regression antigens. Curr Opin Immunol. 2013;25(2):284–90.
Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–11.
O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–67.
Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–4.
Anagnostou V, Smith KN, Forde PM, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017;7(3):264–76.
George S, Miao D, Demetri GD, et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity. 2017;46(2):197–204.
Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Investig. 2016;126(10):3814–26.
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34.
Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. 2015;5(9):915–9.
Landsberg J, Kohlmeyer J, Renn M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490(7420):412–6.
Knutson KL, Lu H, Stone B, et al. Immunoediting of cancers may lead to epithelial to mesenchymal transition. J Immunol. 2006;177(3):1526–33.
Santisteban M, Reiman JM, Asiedu MK, et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Can Res. 2009;69(7):2887–95.
Ward JP, Gubin MM, Schreiber RD. The role of neoantigens in naturally occurring and therapeutically induced immune responses to cancer. Adv Immunol. 2016;130:25–74.
Gettinger S, Choi J, Hastings K, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7(12):1420–35.
Paulson KG, Voillet V, McAfee MS, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun. 2018;9(1):3868.
Bai J, Gao Z, Li X, et al. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget. 2017;8(66):110693–707.
Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29.
Sade-Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136.
Tran E, Robbins PF, Lu YC, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375(23):2255–62.
Cabrera CM, Jimenez P, Cabrera T, et al. Total loss of MHC class I in colorectal tumors can be explained by two molecular pathways: beta2-microglobulin inactivation in MSI-positive tumors and LMP7/TAP2 downregulation in MSI-negative tumors. Tissue Antigens. 2003;61(3):211–9.
Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–86.
Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201.
Fares CM, Van Allen EM, Drake CG, et al. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book. 2019;39:147–64.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Lee JH, Shklovskaya E, Lim SY, et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat Commun. 2020;11(1):1897.
Sucker A, Zhao F, Pieper N, et al. Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. 2017;8:15440.
Thommen D, Uhlenbrock F, Herzig P, et al. 66P Highly exhausted PD-1hi T cell subsets in human NSCLC are co-defined by the predominant expression of distinct inhibitory receptors and correlate with clinical outcome. J Thorac Oncol. 2016;11(4 Supplement):S83.
Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
Limagne E, Richard C, Thibaudin M, et al. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology. 2019;8(4): e1564505.
Gao J, Ward JF, Pettaway CA, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017;23(5):551–5.
Kakavand H, Jackett LA, Menzies AM, et al. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol. 2017;30(12):1666–76.
Fourcade J, Sun Z, Chauvin JM, et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight. 2018;3(14): e121157.
Chiu DK, Yuen VW, Cheu JW, et al. Hepatocellular carcinoma cells up-regulate PVRL1, stabilizing PVR and inhibiting the cytotoxic T-cell response via TIGIT to mediate tumor resistance to PD1 inhibitors in mice. Gastroenterology. 2020;159(2):609–23.
Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–8.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22.
Wang Q, Wu X. Primary and acquired resistance to PD-1/PD-L1 blockade in cancer treatment. Int Immunopharmacol. 2017;46:210–9.
Li M, Eckl J, Geiger C, et al. A novel and effective method to generate human porcine-specific regulatory T cells with high expression of IL-10, TGF-beta1 and IL-35. Sci Rep. 2017;7(1):3974.
Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64.
Viehl CT, Moore TT, Liyanage UK, et al. Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol. 2006;13(9):1252–8.
Dyck L, Wilk MM, Raverdeau M, et al. Anti-PD-1 inhibits Foxp3(+) Treg cell conversion and unleashes intratumoural effector T cells thereby enhancing the efficacy of a cancer vaccine in a mouse model. Cancer Immunol Immunother. 2016;65(12):1491–8.
Taylor NA, Vick SC, Iglesia MD, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Investig. 2017;127(9):3472–83.
Yang L, Huang J, Ren X, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23–35.
Meyer C, Cagnon L, Costa-Nunes CM, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247–57.
Arlauckas SP, Garris CS, Kohler RH, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. 2017;9(389): eaal3604.
Tie Y, Tang F, Wei Y-Q, et al. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15(1):61.
Salkeni MA, Shin JY, Gulley JL. Resistance to immunotherapy: mechanisms and means for overcoming. In: Naing A, Hajjar J, editors. Immunotherapy. Cham: Springer; 2021. p. 45–80.
Freeman P, Mielgo A. Cancer-associated fibroblast mediated inhibition of CD8+ cytotoxic T cell accumulation in tumours: mechanisms and therapeutic opportunities. Cancers. 2020;12(9):2687.
Chakravarthy A, Khan L, Bensler NP, et al. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. 2018;9(1):4692.
Yu L, Liu Q, Huo J, et al. Cancer-associated fibroblasts induce immunotherapy resistance in hepatocellular carcinoma animal model. Cell Mol Biol. 2020;66(2):36–40.
Brochez L, Chevolet I, Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur J Cancer. 2017;76:167–82.
Eleftheriadis T, Pissas G, Liakopoulos V, et al. IDO decreases glycolysis and glutaminolysis by activating GCN2K, while it increases fatty acid oxidation by activating AhR, thus preserving CD4+ Tcell survival and proliferation. Int J Mol Med. 2018;42(1):557–68.
Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin Cancer Res. 2019;25(5):1462–71.
Holmgaard RB, Zamarin D, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210(7):1389–402.
Wang B, Zhao Q, Zhang Y, et al. Targeting hypoxia in the tumor microenvironment: a potential strategy to improve cancer immunotherapy. J Exp Clin Cancer Res. 2021;40(1):24.
Gallagher SJ, Shklovskaya E, Hersey P. Epigenetic modulation in cancer immunotherapy. Curr Opin Pharmacol. 2017;35:48–56.
Herbst RS, Soria J-C, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7.
Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527(7577):249–53.
Ghoneim HE, Fan Y, Moustaki A, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170(1):142-57.e19.
Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354(6316):1160–5.
Sen DR, Kaminski J, Barnitz RA, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–9.
Saleh K, Khalife-saleh N, Kourie HR. Is gut microbiome a predictive marker to response to immune checkpoint inhibitors? Immunotherapy. 2017;9(11):865–6.
Burki TK. Gut microbiome and immunotherapy response. Lancet Oncol. 2017;18(12): e717.
Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7.
Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103.
Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–8.
Chaput N, Lepage P, Coutzac C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2019;30(12):2012.
Choi J, Lee SY. Clinical characteristics and treatment of immune-related adverse events of immune checkpoint inhibitors. Immune Netw. 2020;20(1): e9.
Vétizou M, Pitt JM, Daillère R, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–84.
Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359(6382):1355–60.
Shang S, Zhao Y, Qian K, et al. The role of neoantigens in tumor immunotherapy. Biomed Pharmacother. 2022;151: 113118.
Verma V, Shrimali RK, Ahmad S, et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol. 2019;20(9):1231–43.
Ott PA, Hu-Lieskovan S, Chmielowski B, et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell. 2020;183(2):347-62.e24.
Massarelli E, William W, Johnson F, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5(1):67–73.
Guo Z, Yuan Y, Chen C, et al. Durable complete response to neoantigen-loaded dendritic-cell vaccine following anti-PD-1 therapy in metastatic gastric cancer. NPJ Precis Oncol. 2022;6(1):34.
Ju F, Luo Y, Lin C, et al. Oncolytic virus expressing PD-1 inhibitors activates a collaborative intratumoral immune response to control tumor and synergizes with CTLA-4 or TIM-3 blockade. J Immunother Cancer. 2022;10(6): e004762.
Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170(6):1109-19.e10.
Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36(17):1658–67.
Fröhlich A, Niebel D, Fietz S, et al. Talimogene laherparepvec treatment to overcome loco-regional acquired resistance to immune checkpoint blockade in tumor stage IIIB-IV M1c melanoma patients. Cancer Immunol Immunother. 2020;69(5):759–69.
Roehle K, Qiang L, Ventre KS, et al. cIAP1/2 antagonism eliminates MHC class I-negative tumors through T cell-dependent reprogramming of mononuclear phagocytes. Sci Transl Med. 2021. https://doi.org/10.1126/scitranslmed.abf5058.
Kang SH, Keam B, Ahn YO, et al. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology. 2019;8(1): e1515057.
Yamamoto K, Venida A, Yano J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581(7806):100–5.
Liu C, Liu X, Xiang X, et al. A nanovaccine for antigen self-presentation and immunosuppression reversal as a personalized cancer immunotherapy strategy. Nat Nanotechnol. 2022;17(5):531–40.
Lei Q, Wang D, Sun K, et al. Resistance mechanisms of anti-PD1/PDL1 therapy in solid tumors. Front Cell Dev Biol. 2020;8:672.
Aznar MA, Planelles L, Perez-Olivares M, et al. Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J Immunother Cancer. 2019;7(1):116.
Márquez-Rodas I, Longo F, Rodriguez-Ruiz ME, et al. Intratumoral nanoplexed poly I: C BO-112 in combination with systemic anti-PD-1 for patients with anti-PD-1-refractory tumors. Sci Transl Med. 2020. https://doi.org/10.1126/scitranslmed.abb0391.
Kalbasi A, Tariveranmoshabad M, Hakimi K, et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci Transl Med. 2020. https://doi.org/10.1126/scitranslmed.abb0152.
Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277–90.
Sangro B, Chan SL, Meyer T, et al. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J Hepatol. 2020;72(2):320–41.
Schöffski P, Tan DSW, Martín M, et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J Immunother Cancer. 2022;10(2): e003776.
Kondo Y, Ohno T, Nishii N, et al. Differential contribution of three immune checkpoint (VISTA, CTLA-4, PD-1) pathways to antitumor responses against squamous cell carcinoma. Oral Oncol. 2016;57:54–60.
Choi J, Medikonda R, Saleh L, et al. Combination checkpoint therapy with anti-PD-1 and anti-BTLA results in a synergistic therapeutic effect against murine glioblastoma. Oncoimmunology. 2021;10(1):1956142.
Rodriguez-Abreu D, Johnson ML, Hussein MA, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J Clin Oncol. 2020;38(15_suppl):9503.
Arce Vargas F, Furness AJS, Solomon I, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. 2017;46(4):577–86.
Marshall LA, Marubayashi S, Jorapur A, et al. Tumors establish resistance to immunotherapy by regulating T(reg) recruitment via CCR4. J Immunother Cancer. 2020;8(2): e000674.
Doi T, Muro K, Ishii H, et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res. 2019;25(22):6614–22.
Zhu Y, Yang J, Xu D, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68(9):1653–66.
Shi G, Yang Q, Zhang Y, et al. Modulating the tumor microenvironment via oncolytic viruses and CSF-1R inhibition synergistically enhances anti-PD-1 immunotherapy. Mol Ther. 2019;27(1):244–60.
Mao Y, Eissler N, Blanc KL, et al. Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin Cancer Res. 2016;22(15):3849–59.
Li R, Salehi-Rad R, Crosson W, et al. Inhibition of granulocytic myeloid-derived suppressor cells overcomes resistance to immune checkpoint inhibition in LKB1-deficient non-small cell lung cancer. Can Res. 2021;81(12):3295–308.
Tobin RP, Jordan KR, Robinson WA, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int Immunopharmacol. 2018;63:282–91.
Hashimoto A, Gao C, Mastio J, et al. Inhibition of casein kinase 2 disrupts differentiation of myeloid cells in cancer and enhances the efficacy of immunotherapy in mice. Can Res. 2018;78(19):5644–55.
Mitchell TC, Hamid O, Smith DC, et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: phase i results from a multicenter, open-label phase I/II Trial (ECHO-202/KEYNOTE-037). J Clin Oncol. 2018;36(32):3223–30.
Long GV, Dummer R, Hamid O, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083–97.
Sonpavde G, Necchi A, Gupta S, et al. ENERGIZE: a Phase III study of neoadjuvant chemotherapy alone or with nivolumab with/without linrodostat mesylate for muscle-invasive bladder cancer. Future Oncol. 2020;16(2):4359–68.
Lu JC, Zhang PF, Huang XY, et al. Amplification of spatially isolated adenosine pathway by tumor-macrophage interaction induces anti-PD1 resistance in hepatocellular carcinoma. J Hematol Oncol. 2021;14(1):200.
Herbst RS, Majem M, Barlesi F, et al. COAST: An Open-Label, Phase II, Multidrug Platform Study of Durvalumab Alone or in Combination With Oleclumab or Monalizumab in Patients With Unresectable, Stage III Non-Small-Cell Lung Cancer. J Clin Oncol. 2022;40:3383.
Chiappori A, Williams CC, Creelan BC, et al. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J Clin Oncol. 2018;36(15_suppl):9089.
Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–8.
Rivas JR, Liu Y, Alhakeem SS, et al. Interleukin-10 suppression enhances T-cell antitumor immunity and responses to checkpoint blockade in chronic lymphocytic leukemia. Leukemia. 2021;35(11):3188–200.
Naing A, Wong DJ, Infante JR, et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): a multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019;20(11):1544–55.
Qi Z, Xu Z, Zhang L, et al. Overcoming resistance to immune checkpoint therapy in PTEN-null prostate cancer by intermittent anti-PI3Kα/β/δ treatment. Nat Commun. 2022;13(1):182.
Wang B, Liu J, Han Y, et al. The presence of tertiary lymphoid structures provides new insight into the clinicopathological features and prognosis of patients with breast cancer. Front Immunol. 2022;13: 868155.
Topper MJ, Vaz M, Marrone KA, et al. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. 2020;17(2):75–90.
Yi M, Zheng X, Niu M, et al. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21(1):28.
Feng S, De Carvalho DD. Clinical advances in targeting epigenetics for cancer therapy. FEBS J. 2022;289(5):1214–39.
Wang L, Amoozgar Z, Huang J, et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine ovarian cancer model. Cancer Immunol Res. 2015;3(9):1030–41.
Woods DM, Sodré AL, Villagra A, et al. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3(12):1375–85.
Zheng H, Zhao W, Yan C, et al. HDAC Inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2016;22(16):4119–32.
Bissonnette RP, Cesario RM, Goodenow B, et al. The epigenetic immunomodulator, HBI-8000, enhances the response and reverses resistance to checkpoint inhibitors. BMC Cancer. 2021;21(1):969.
Zingg D, Arenas-Ramirez N, Sahin D, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 2017;20(4):854–67.
Morel KL, Sheahan AV, Burkhart DL, et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer. 2021;2(4):444–56.
Daver N, Garcia-Manero G, Basu S, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, Phase II Study. Cancer Discov. 2019;9(3):370–83.
Gray JE, Saltos A, Tanvetyanon T, et al. Phase I/Ib study of pembrolizumab plus vorinostat in advanced/metastatic non-small cell lung cancer. Clin Cancer Res. 2019;25(22):6623–32.
Entinostat Helps Thwart Immunotherapy Resistance. Cancer discovery. 2019;9(6):685-6
Hellmann MD, Jänne PA, Opyrchal M, et al. Entinostat plus pembrolizumab in patients with metastatic NSCLC previously treated with Anti-PD-(L)1 therapy. Clin Cancer Res. 2021;27(4):1019–28.
Elkrief A, Derosa L, Zitvogel L, et al. The intimate relationship between gut microbiota and cancer immunotherapy. Gut microbes. 2019;10(3):424–8.
Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–9.
Mager LF, Burkhard R, Pett N, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 2020;369(6510):1481–9.
McHale D, Francisco-Anderson L, Sandy P, et al. P-325 Oral delivery of a single microbial strain, EDP1503, induces anti-tumor responses via gut-mediated activation of both innate and adaptive immunity. Ann Oncol. 2020;31:S195.
Pant S, Mulder I, Shah AY, et al. A phase I/II study of live biotherapeutic MRx0518 in combination with pembrolizumab in patients who have progressed on prior anti-PD-1 therapy. J Clinic Oncol. 2019;37(15):TPS2670.
Baruch EN, Youngster I, Ben-Betzalel G, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371(6529):602–9.
Davar D, Dzutsev AK, McCulloch JA, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371(6529):595–602.
Nakamura T, Sato T, Endo R, et al. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J Immunother Cancer. 2021;9(7): e002852.
Li X, Khorsandi S, Wang Y, et al. Cancer immunotherapy based on image-guided STING activation by nucleotide nanocomplex-decorated ultrasound microbubbles. Nat Nanotechnol. 2022;17(8):891–9.
Buss CG, Bhatia SN. Nanoparticle delivery of immunostimulatory oligonucleotides enhances response to checkpoint inhibitor therapeutics. Proc Natl Acad Sci U S A. 2020;117(24):13428–36.
Clinical Trials.gov. Available from: https://clinicaltrials.gov/. Accessed May 6 2023.
Sahin U, Oehm P, Derhovanessian E, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585(7823):107–12.
Xie C, You X, Zhang H, et al. A nanovaccine based on adjuvant peptide FK-13 and l-phenylalanine poly(ester amide) enhances CD8(+) t cell-mediated antitumor immunity. Adv Sci. 2023. https://doi.org/10.1002/advs.202300418.
Acknowledgements
We would like to thank Editage (www.editage.cn) for English language editing.
Funding
This research was funded in part by grants from the Health Talents Special Project of Jilin Provincial Finance Department (JLSWSRCZX2021-065), the Program of Changchun Science and Technology Bureau development Plan Project (21ZY29), the Education Department Foundation of Jilin Province (JJKH20211195KJ) and the Jilin Provincial Science and Technology Foundations (20210509003RQ and 20210402002GH).
Author information
Authors and Affiliations
Contributions
Conceptualization, BW and XJ; software, investigation, BW, QZ, and HHW; resources, YYZ and JLW; writing-original draft preparation, BW, and YH; writing—review and editing, BW, RJ, YX, and XJ; funding acquisition, XJ. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, B., Han, Y., Zhang, Y. et al. Overcoming acquired resistance to cancer immune checkpoint therapy: potential strategies based on molecular mechanisms. Cell Biosci 13, 120 (2023). https://doi.org/10.1186/s13578-023-01073-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-023-01073-9