
Abstract
Transcription factors directly regulate gene expression by recognizing and binding to specific DNA sequences, involving the dynamic alterations of chromatin structure and the formation of a complex with different kinds of cofactors, like DNA/histone modifying-enzymes, chromatin remodeling factors, and cell cycle factors. Despite the significance of transcription factors, it remains unclear to determine how these cofactors are regulated to cooperate with transcription factors, especially DNA/histone modifying-enzymes. It has been known that DNA/histone modifying-enzymes are regulated by post-translational modifications. And the most common and important modification is phosphorylation. Even though various DNA/histone modifying-enzymes have been classified and partly explained how phosphorylated sites of these enzymes function characteristically in recent studies. It still needs to find out the relationship between phosphorylation of these enzymes and the diseases-associated transcriptional regulation. Here this review describes how phosphorylation affects the transcription activity of these enzymes and other functions, including protein stability, subcellular localization, binding to chromatin, and interaction with other proteins.
Similar content being viewed by others
Introduction
Protein phosphorylation is crucial for various cellular processes, including cell growth, DNA damage, metabolism, inflammation. By definition, various protein kinases phosphorylate serine (Ser), threonine, and tyrosine of targeted proteins. Phosphorylation is one of the most common post-translational modifications (PTMs). It generally alters the structural conformation and interaction of proteins, by which it directly influences protein stability, cellular localization, protein/DNA binding, and enzymatic activity [1].
Approximately two-thirds of 21,000 known human proteins have been reported to be phosphorated with over 200,000 specific protein sites, and over 760,000 additional sites are predicted to be phosphorylated in several websites, including the Cell Signaling Technology PhosphoSitePlus (www.phosphosite.org) and the Kinexus PhosphoNET (www.phosphonet.ca). Among these reported proteins, there are some transcriptional factors (TFs) and the cofactors. It has been clearly defined how TFs and the cofactors organize and regulate gene transcription. In general, TFs firstly occupy DNA elements in a sequence-specific manner, and then recruit the RNA polymerases to the gene’s promoter region, which simultaneously involve chromatin remodeling complexes and histone (de)acetyltransferases for promoter accessibility [2]. It is reported that about 1,600 human proteins have been identified as transcriptional factors [3]. TFs control gene expression and chromatin status by binding specific DNA sequences, which are thus tightly controlled in normal cells [2].
TFs and the cofactors are not only regulated by upstream transcriptional activation or repression but also controlled by post-translational modifications (PTMs), such as acetylation, phosphorylation, ubiquitination, SUMOylation [4]. Phosphorylation is the most common modification of TFs and the cofactors. Phosphorylation of TFs and the cofactors positively or negatively regulates its DNA accessibility, protein stability, and protein-protein interaction to influence transcriptional activity for expression of genes. Specific phosphorylated sites of TFs and the cofactors have different influences on their functions. TFs have been cataloged based on DNA-binding specificity, which has been classified into at least nine super classes, including basic domains, zinc-coordinating DNA-binding domains, helix-turn-helix domains, alpha-Helices exposed by beta-structures, other all-alpha-helical DNA-binding domains, immunoglobulin fold, beta-Hairpin exposed by an alpha/beta-scaffold, beta-Sheet binding to DNA, beta-Barrel DNA-binding domains, and other undefined DNA-binding domains [4]. In general, once the DNA binding domain is phosphorylated, it disturbs TFs to recognize specific DNA sequences and impairs transcriptional activity, while the phosphorylation of the regulatory domain in TFs is more complicated and need more evidence to determine it. More importantly, many TFs’ cofactors generally contain a regulatory domain with enzymatic activities, such as acetylation/deacetylation, methylation/demethylation, ubiquitination, and SUMOylation [5,6,7,8]. Mostly, these kinds of TFs’ cofactors modify histones via methylation and acetylation, except for DNA methylation, as it is summarized in Fig. 1. The enzymatic activities mainly function on genomic DNA and histone proteins and the interactors. With more and more enzymatic proteins found to act as transcriptional regulators or coregulators, the regulation of PTMs in these proteins, especially these phosphorylated sites in catalytic domains, significantly determines the transcriptional activity. However, the functions of phosphorylation on these DNA/histone-modifying enzymes are more complicated. In this review, we comprehensively describe how phosphorylation affects DNA/histone-modifying enzymes with specific sites, and focus on these phosphorylated sites which are directly associated with enzymatic activation or repression.

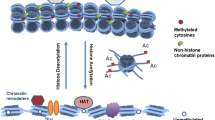
DNA/histone-modifying enzymes. DNA methylation and demethylation are dynamically regulated by DNA methyltransferases and demethylases. Histone methylation and demethylation are controlled by histone methyltransferases and demethylases. Histone acetyltransferases and deacetylases are responsible for histone acetylation and deacetylation, respectively
Regulation of DNA methyltransferases by phosphorylation
In mammals, DNA methyltransferases are responsible for DNA methylation by adding a methyl group from S-adenosyl-methionine (AdoMet) to the fifth cytosine in CpG-enriched sequences of the genome. It consists of five members: DNMT1, DNMT3a, DNMT3b, DNMT2, and DNMT3L. Among them, DNMT1 is responsible for maintaining DNA methylation by converting hemi-methylated CpG dinucleotides in daughter strands to methylated status during the DNA replication process, while DNMT3A and DNMT3B are different from DNMT1. Both of them target unmethylated DNA strands and exert de novo methylation [9]. As for another two DNA methyltransferases, DNMT2 and DNMT3L, DNMT2 has residual DNA methyltransferase activity with preference of tRNA methylation [10, 11]. It is found that DNMT2 methylates cytosine 38 in the anticodon loop of tRNA16 [12]. DNMT3L is catalytically inactive, but it interacts with DNMT3A and DNMT3B, which stimulates their methylation activity [12].
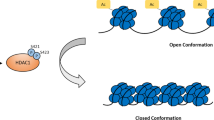
It is crucial for DNA methyltransferases to epigenetically regulate genes via DNA methylation during the development of normal and cancer cells. Therefore, post-translational modifications of DNA methyltransferases modulate protein expression of these enzymes and the level of DNA methylation. Recent studies have evidenced that phosphorylation of DNA methyltransferases at N-terminal serine/threonine residues is likely to affect their enzymatic activity. DNMT1 is a multi-domain protein with a DMAP1-binding domain, RFTS domain, a CXXC domain, two BAH domains and a C-terminal catalytic domain. The N-terminal domains of DNMT1, DMAP1-binding domain and RFTS domain, are key for its stability and subcellular localization onto DNA replication sites [13, 14]. And also, enzymatic activity of DNMT1 is also regulated by RFTS domain and CXXC domain [15, 16]. UHRF1 interacts with DNMT1 through its SRA domain, which is essential for DNA methylation maintenance [17]. It has also been reported that UHRF1 not only facilitates DNMT1 to genomic loci by recognizing H3R2 and H3K9me2/3 mark, but also stimulates DNMT1 catalytic activity via UHRF1-dependent H3 ubiquitination [18, 19]. At the same time, DNMT1 is recruited by UHRF1 and forms a macromolecular complex with other proteins during the cell cycle, including PCNA, TIP60, HDAC1, SUV39H1, HAUSP, and pRB [20]. It is noticed that PCNA partly recruits DNMT1 to the replication sites [21]. Human DNMT1 is discovered to be phosphorylated at Ser 154 by cyclin-dependent kinases (CDKs) 1, 2, and 5, which is orthologous with mouse Dnmt1 Ser152 [22]. The mutation of DNMT1 at position 154 (S154A) severely impairs its methylation activity. However, it is still unknown whether phosphorylation of Ser154 promotes the interaction of the N- and catalytic domains of DNMT1 and thus increase DNMT1 activity for DNA hypermethylation. Another site, Ser515 of human DNMT1, has been shown to be phosphorylated during the cell cycle, which enhances DNMT1 methylation activity [23]. It is suggested that phosphorylation of Ser515 in DNMT1 is helpful for an interaction between N-terminus and catalytic domains of DNMT1. Furthermore, Ser127and Ser143 of DNMT1 have also been identified to be phosphorylated by AKT and PKC [24]. AKT-mediated phosphorylation of DNMT1 at Ser143 peaks during DNA synthesis and stabilizes DNMT1 from degradation [25]. The difference of these two kinases on DNMT1 is that the phosphorylation of DNMT1 at Ser127 by PKC disturbs the DNMT1/UHRF1 interaction without affecting the DNMT1/PCNA interaction, while AKT-mediated double phosphorylation of DNMT1 at Ser127 and Ser143, repress DNMT1/PCNA and DNMT1/UHRF1 interactions. Therefore, AKT/PKC-mediated phosphorylation of DNMT1 is considered to be a hallmark determining the interaction of DNMT1 with PCNA or UHRF1. In addition, mouse Dnmt1 is also reported to be phosphorylated at Ser146 by casein kinase 1δ/ε, which disrupts the DNA-binding activity of Dnmt1 but not alter its methylation activity and interaction of Dnmt1/PCNA [26]. The phosphorylation of human DNMT1 at the N-terminal nuclear localization signal (NLS) by protein kinase B (PKB) promotes its nuclear translocation in the condition of IL6 stimulation [27]. In summary, DNMT1 phosphorylation plays a central role in its methylation activity, protein stability, and the interaction with other proteins. Aberrant phosphorylation of DNMT1 results in fibroblast activation and an increase of α-smooth muscle actin and type I collagen [28]. Previous mass spectrometry results have identified several phosphorylation sites on DNMT1. Some of the phosphorylation sites have been functionally verified, but there are still unclear for the remaining of them. It is also interesting that phosphorylation sites of DNMT1 are dependent on cell status and cell types. For example, Ser154, Ser515, and Ser714 of DNMT1 are phosphorylated in HEK293T cells, Ser127, Ser143, and Ser714 of DNMT1 are found in Jurkat cells, and Ser143 of DNMT1 is in lung cancer cells [29,30,31,32]. DNMT1 is phosphorylated at Ser154 and Ser515 in the cell cycle, which significantly affects enzymatic activity and protein stability of DNMT1 [22, 23]. Glycogen Synthase Kinase 3 (GSK3) phosphorylates DNMT1 at Ser714 to block the methylation of unmethylated DNA [33].
The de novo DNA methyltransferase DNMT3a is also reported to be phosphorylated at two key residues (Ser386 and Ser389) by casein kinase 2 (CK2), which impairs the methylation ability of DNMT3a and switches DNMT3a to localize at heterochromatin [34]. The other site, Ser255 of DNMT3a, determines its intracellular localization to regulate erythrocytic differentiation [35]. It is shown that the extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylates DNMT3a at Ser255, resulting in DNMT3a translocate into the nucleus. As for DNMT3b phosphorylation, there are still no studies on it and it needs to be further explored.
Regulation of DNA demethylases by phosphorylation
As for DNA demethylation, TET family proteins, including TET1, TET2, and TET3, can oxidize 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC), 5-carboxylcytosine (5caC), and 5-formylcytosine (5fC) during the processes of DNA replication or thymine-DNA glycosylase (TDG) and base excision repair (BER) (TDG-BER) pathway [36,37,38,39]. TET proteins-mediated DNA demethylation involves in numerous biological processes, including stem cell differentiation, metabolism, and inflammation [40,41,42]. And TET1, TET2, and TET3 display distinct expression levels in different cell types and differentiation development [43]. TET1 and TET2 are mainly expressed in mouse stem cells for active DNA demethylation on gene’ promoters and bodies, while TET3 is enriched in the oocyte and neuronal tissues [44,45,46]. The activity of TET proteins is not only directly dependent on several molecules, such as Fe(II), 2-oxoglutarate, interactors, its inhibitors, and stimulators, but also influenced by various post-translational modifications (PTMs) [36, 47]. Among these PTMs, phosphorylation of TET proteins significantly impacts their enzymatic activity, protein stability and interactions, which is reported to be inhibited by glycosyltransferase OGT-induced O-GlcNAcylation [48].
Most phosphorylated sites of TET1 are located at its N-terminus by mass spectrometry, but the functions of these sites are still unclear [48]. Until now, phosphorylation of TET2 and TET3 is the most widely studied. TET2 is considered as a tumor suppressor for its effect on the DNA 5-hydroxymethylome [49]. Phosphorylation of TET2 at Ser99 by AMP-activated kinase (AMPK) enhances its tumor suppression by stabilizing TET2. High level of glucose impairs TET2 phosphorylation in diabetic patients, which suggests an epigenetic pathway by which a hyperglycemic environment induces cancers [49]. Homologous to murine Tet2 at Ser97, phosphorylation stabilizes Tet2 and promotes its interaction with 14-3-3β [50]. Tet2 phosphorylation at Ser97 or its mimicking mutant S97E can rescue differentiation defects in C2C12 cells by upregulating expressing of Pax7. In contrast, phosphorylation of TET2 at Y1902 by fibroblast growth factor receptor 3 (FGFR3) splicing mutant FGFR3△7−9, degrades TET2 via ubiquitination and promotes hepatocellular carcinogenesis [51]. TET2 is required for lineage commitment and differentiation of stem cells. It is reported that TET2 is phosphorylated by cytokine receptor-associated JAK2 at Tyr 1939 and 1964, which facilities stem cells to differentiate to erythroid cells by interacting with the erythroid transcription factor KLF1 [52]. The dioxygenase activity of TET3 is activated by its phosphorylation at Ser1310 and Ser1379 [53]. These two sites are highly conserved within their catalytic domain and phosphorylated by cyclin-dependent kinase 5 (CDK5). Interestingly, overexpression of TET3 phosphorylation mutant (S1310A/S1379A) leads to increased expression of metabolic genes, distinct from wild-type TET3 for neuron-specific genes. Therefore, phosphorylated and unphosphorylated TET3 display different binding affinities on histones for neuronal differentiation.
The regulation of histone lysine and arginine methyltransferases by phosphorylation
Histone methylation commonly occurs by various lysine and arginine methyltransferases in a site-specific manner (Fig. 2) [54]. Lysine methyltransferases (KMTs) are responsible for mono-, di-, tri-methylated histone H3K4, H3K9, H3K27, H3K36, and H4K20, while arginine methyltransferases (PRMTs) are only for mono- or di-methylation on histone H3R2, H3R8, H4R3, and H2A [55, 56]. Specifically, several KMTs including SET1A, SET1B, ASH1, and MLL1-5, recognizes histone H3K4 for methylation; SUV39H1, SUV39H2, G9a, SETDB1, GLP, RIZ1, and CLL8 methylate histone H3K9; Histone H3K27 is the substrate of EZH2; SET2, SMYD2, and NSD1 exert their enzymatic activity on histone H3K36, and H3K36 can also be catalyzed by NSD2, SETMAR; DOT1L is for histone H3K79 methylation; SUV420H1, SUV420H2, SET7, and SET8 methylate H4K20, as shown in Table 1. The methylation activity of histone lysine and arginine methyltransferases is associated with human diseases, including prostate, breast, lung cancers and the responses to environment stress [57,58,59]. Specifically, histone methylation can regulate the tightness of the nucleosome in most case, and thus affect the access of transcription factors and RNA polymerase to their targeted genes [60]. Phosphorylation of catalytic domain of KMTs is one of the main factors to generally suppress or activate their methyltransferase activity on histones. However, only a few of KMTs have been reported to be phosphorylated (Table 1).

Different histone-modifying enzymes target different specific histone sites
KMT1A, known as SUV39H1, is phosphorylated at residues Tyr297 (mouse Tyr303 and flies Tyr308) by receptor-type tyrosine kinase ERBB4, which enhances the tri-methylation activity of histone H3K9 [61]. And the protein level of SUV39H1 peaks at the S phase and maintains from S to M phases in the cell cycle, in which SUV39H1 is phosphorylated by CDK2 at Ser391. The phosphorylation of SUV39H1 at Ser391 aims to dissociate from chromatin and prepares for histone demethylase JMJD2A occupancy [62]. KMT1C (G9a) is phosphorylated at Ser591 by ATM and colocalizes with GLP1 (also known as EHMT1) under the circumstance of DNA double-strand breaks (DSBs) [63]. The methylation activity of G9a is essential for the DNA repair pathway. It suggests that inhibition of G9a enzymatic activity by phosphorylation improves the DNA mutation rate and effects from DNA breaks. Other studies also show that G9a is phosphorylated at Ser 211 by CK2 and then directly interacts with replication protein A (RPA) on chromatins [64]. By forming the complex of RPA and G9a, it facilitates homologous recombination (HR) for DNA repair. The human mixed-lineage leukemia 1 (MLL1) is reported to be phosphorylated at Ser136 and Ser142 by CK2, which significantly enhances its interaction with a transcription coactivator LEDGF/p75 [65]. Interaction of LEDGF/p75 and MLL1 not only impacts HIV integrase for active transcription of viral genes, but also regulates acute leukemia development via MLL1 translocation fusion. It is also reported that T2724 and S2726 at taspase1-dependent cleavage domain of full MLL1 are phosphorylated by CK2, which blocks taspase1-dependent cleavage of MLL1 and results in the destabilization of MLL1 [66]. MLL1 stability is important for aggressive leukemia and phosphorylation-mediated degradation of MLL1 is likely to be a potential treatment for acute leukemia. As for Leukemogenic MLL, it takes advantage of the C-terminal SET domain to methylate histone H3 lysine 4 (H3K4), while its N-terminal domain is composed by fusing more than 60 partners and processed to generate heterodimerized MLL [67, 68]. It is verified to be phosphorylated at Ser561 by ATR in the S phase of the cell cycle and therefore promotes its dissociation from chromatin and degradation by SKP2 E3 ligase in response to genotoxic stress [69]. MLL2 (KMT2B) is phosphorylated at Thr542 by CDK2 in the late G1 phase of the cell cycle [69]. It attributes to MLL2 activation on H3K4me3 and promotes pluripotent stem cells differentiation. There are no reported phosphorylated sites of MLL3. MLL4 is reported to be phosphorylated at Ser1331 by AKT, which inhibits its methylation activity [70]. The enzymatic activity of MLL4 can be stimulated by PI3Kα inhibitors and thus it elicits a robust compensatory increase in ER-dependent transcription that limits therapeutic efficacy in ER-positive breast cancer. MLL5 is also phosphorylated and C2 targets Thr912 of MLL5 at the cell cycle [71]. It specifically occurs at the G2/M phase for entrance into mitosis through dissociation from condensed chromosomes, suggesting that subcellular localization of MLL5 is dependent on CDC2 activity. SMYD2 is reported to interact with CDK4/6 and is phosphorylated for methylation activation of H3K4 and H3K36. However, it is still not clear which sites of SMYD2 are phosphorylated. It is shown that histone dimethyltransferase WHSC1 is phosphorylated at Ser172 by activated AKT in prostate cancer, which prevents WHSC1 from degradation and subsequently activates transcription of RAC1 to drive cancer metastasis [72]. Phosphorylation of DOT1L by CDK1 at Ser1105 significantly impacts its subcellular localization and enzymatic activity In ES cells [73]. Specifically, phosphorylated DOT1L on this site cannot be present in the nucleus compared with that of wild type. And it also impairs DOT1L methylation activity so that the low methylation level of H3K79 fails to differentiate ES cells. Polycomb group proteins (PcG) PRC2-Ezh1α/β signaling pathway plays a crucial role in maintaining cell memory via H3K37me3 [74]. It is reported that Ser560, localized at the C-terminus of mouse zeste homolog 1β (Ezh1β), is phosphorylated in the cytoplasm and it promotes degradation of EZH1β by ubiquitin E3 ligase NEDD4 in the condition of oxidative stress [75]. CDK4/6-mediated phosphorylation of EZH2 at Thr345 enhances STAT3 methylation in keratinocytes, which activates STAT3 to induce the expression of a key proinflammatory transcription factor, IκBζ in psoriasis [76]. It is reported that EZH2, also called KMT6A, is phosphorylated at serine 21 by AKT, which abrogates histone H3 and reduces H3K27 trimethylation [77]. The phosphorylation of EZH2 at Ser21 functionally promotes cell growth and oncogenesis. In addition, there are still many sites identified to be phosphorylated in EZH2 and different phosphorylation sites function diversely in Table 1.
Protein arginine methyltransferases 1 (PRMTs) are a kind of enzymes, which are mainly responsible for histone arginine methylation in cells and regulate the cell cycle and cell proliferation by remodeling chromatin status. Phosphorylation of PRMTs is essential for its enzymatic activity and protein stability. It is reported that PRMT1 is phosphorylated at Ser297 in normal liver cells, while it is dephosphorylated by PP2A at this site in response to alcohol and other oxidative stresses [78]. It suggests that PRMT1 plays an important role in protecting liver patients from alcoholic disease. But Tyr291 phosphorylation inhibits PRMT1’s ability to methylate histone proteins and interacts with heterogeneous nuclear ribonucleoproteins (hnRNP A1 and hnRNP H3) in K562 cells [79]. PRMT6 is reported to be phosphorylated at Ser11 and Thr21 by CK2, which protects it from degradation and promotes RCC1 arginine methylation for tumorigenicity of glioblastoma stem cells [79].
The regulation of histone lysine and arginine demethylases by phosphorylation
Histone lysine demethylases (KDMs), in contrast to KMTs, function to remove methyl groups of methylated histone proteins. The alters on histone methylation by KDMs regulate gene expression in the processes of development and tumor growth [96]. And KDMs can be considered as tumor oncogene or tumor suppressor, which is determined by KDMs-targeted genes. KDMs are composed of eight superfamilies (KDM1-8) and targets various methylated histone proteins. KDM2A contains a JmjC domain at its N-terminus and a PHD zinc finger, a CxxC-type zinc finger at C-terminus. Thr632 at the PHD zinc finger of KDM2A is phosphorylated by ATM kinase, which impairs its chromatin-binding capacity [90]. KDM3A also undergoes phosphorylation at Ser265 by protein kinase A (PKA) and transcriptionally regulates cell-cycle genes in response to DNA damage [91]. KDM4B is phosphorylated by PKA at Ser666 and it promotes castration-resistance by weakening its binding to a splicing factor SF3B3 [92]. Protein kinase R phosphorylates KDM4C at Ser918, resulting in KDM4C ubiquitination and degradation [94]. But WNT3a can interrupt phosphorylation of KDM4C via GSK3-dependent protein kinase R inactivation. Phosphorylation of KDM5B at Ser1456 by cyclin-dependent kinase 1 (CDK1) abolishes its binding to the promoters of several pluripotency genes [95]. It is reported that the phosphorylation of KDM8 is decreased in KDM4C knockdown cells, but the specific phosphorylated sites are unknown [97]. Until now, it remains controversial about the existence of arginine demethylases [98]. Recently, several lysine demethylases, including KDM3A, KDM4E, KDM5C, have been reported to demethylate methylarginine in vitro [99]. However, there are no phosphorylated sites reported on these three enzymes. In summary, the effects of phosphorylation on these demethylases are not enough. Further investigation is necessary to explore the function of phosphorylation on these lysine/arginine demethylases.
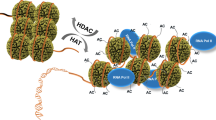
The regulation of histone acetyltransferases by phosphorylation
Histone acetyltransferases (HATs) are major players in epigenetically modulating gene transcription via acetylation on histones (Fig. 2). They recognize and transfer an acetyl group from acetyl CoA to acetylate lysine residues of histones, which generally occurs at gene promoters. And substrates of HATs are not only histones, but also other non-histone proteins, such as p53, GATA1, and erythroid Kruppel-like factor (EKLF) [100,101,102]. Abnormal HATs expression directly or indirectly leads to many human diseases, such as cancers and neurodegenerative disease. Phosphorylation of HATs plays a pivotal role in its enzymatic activity and protein stability (Table 2). HAT1, also called KAT1, is reported to be phosphorylated by active adenosine monophosphate (AMP)-activated protein kinase (AMPK) at Ser190, which enhances its acetylation activity on histone H4 and inhibits DNMT1 binding to relaxed chromatin [103]. GCN5 (KAT2A) plays a dual role in the deeding-to fasting transition and is phosphorylated by PKA at Ser275 [104]. In the fasted condition, GCN5 induces gluconeogenesis and acetylates PGC1α to repress the activity of PGC1α. In order to switch to the fed state, GCN5 is phosphorylated by PKA in the complex of GCN5-CITED2-PKA and then turn to acetylate histone H3.
Until now, P300 is the most studied protein about its functions influenced by phosphorylation. Among these phosphorylated sites we have summarized, phosphorylation of Ser1834 by AKT, Ser2271, Ser2279, Ser2291, and Ser2315 by mTOR, and Ser2279, Ser2315, and Ser2366 by extracellular signal-regulated kinase 2 (ERK2), significantly enhances P300 acetylation activity [105,106,107]. P300 phosphorylation by AKT at Ser1834 enhances its acetylation activity on its adaptor factor, alteration/deficiency in activation 3 (ADA3), and it involves in growth factor-associated cell cycle by epidermal growth factor receptor (EGFR) activation [105]. MTOR-mediated phosphorylation of P300 at its C-terminal domain prevents the catalytic domain from binding to the RING domain, which reduces starvation-induced cell autophagy and lipogenesis [108]. P300 phosphorylation by ERK2 at the C terminus is essential for its recruitment to the promoter region of keratin 16 and cooperatively interacts with SP1 and c-Jun for upregulation of keratin 16 [109]. And also, ERK2-induced P300 phosphorylation stimulates acetylation of the nuclear factor of activated T-cells c1 (NFATc1) and activation of the myosin heavy chain 1 (MYHC1) expression during the transformation of skeletal muscle fiber type [110]. Other phosphorylated sites, such as Ser106 by ataxia-telangiectasia mutate (ATM) involved in DNA damage, Ser1038/2039 by CDK1 stabilizing P300, and Ser89 by CK2 or SIK2 influencing downstream gene transcription [111,112,113]. In addition, the HAT activity of P300 is also dynamic in the differentiation of mouse F9 embryonal carcinoma cells. It is reported that the HAT activity of P300 is only dependent on P300 phosphorylation in differentiated F9 cells, although P300 is also strongly phosphorylated in undifferentiated F9 cells [114].
AKT also phosphorylates the acetyltransferase p300/CBP-associated factor (PCAF) and increases its acetylation on high-mobility group AT-hook 2 (HMGA2) at lysine 26 (K26) for esophageal squamous cell carcinoma growth [106]. The phosphorylation of CBP at Ser436 by a typical protein kinase C (aPKC), homologous to P300 phosphorylation G422S functions to increase new neurons’ survival and thus promotes hippocampal neurogenesis [107]. It is explained that maybe p300 phosphorylation weakens its interaction with CREB. Insulin also suppresses the formation of the complex of CREB-CBP by phosphorylating CBP, resulting in aberrant hepatic glucose production (HGP) [115]. And it is confirmed again that p300 prefers binding to unphosphorylated CBP. As for TIP60 phosphorylation, only two sites, Ser86 and Ser90, have been reported. Phosphorylation of TIP60 at Ser86 by GSK3 promotes its enzymatic activity on histone H4 and P53 acetylation, resulting in apoptosis [116]. While phosphorylation at Ser90 of TIP60 by CDK9 enhances its binding to chromatin [117]. It is also found that TIP60 phosphorylation at Ser90 by cyclin B/CDC2 can arrest cells at the G2/M phase of the cell cycle [118]. Other KATs, including KAT7 and KAT13A, are also reported to be phosphorylated. Specifically, KAT7 (HBO1) is phosphorylated at Ser57 by polo-like kinase 1 (PIK1) to drive the transition of the cell cycle from G1 to S phase, and then HBO1 is phosphorylated by CDK1 at Thr85 and Thr88 during mitosis [116]. Significantly, it is evidenced that HBO1 phosphorylation by CDK1 provides a docking site for PIK1 binding. The protein stability of KAT7 is also regulated by phosphorylation. Protein kinase D1 (PDK1) can directly phosphorylate KAT7 at Thr97 and prevent KAT7 from ubiquitination-mediated degradation [119]. The predicted phosphorylation sites of KAT13A are numerous, but we have only summarized experimentally-verified sites in Table 2, as well as other KATs proteins.
The regulation of histone deacetylases by phosphorylation
Histone acetylation and deacetylation are dynamic processes dependent on cell states. Histone acetylation and deacetylation are performed by KAT family proteins and histone deacetylases (HDACs). There are four classes of HDACs based on their homologous protein sequences: the class I RPD3-like proteins including HDAC1, HDAC2, HDAC3, and HDAC8; class II HDA1-like proteins are HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10; class III sirt2-like proteins containing SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7; class IV protein is HDAC11. Many HDACs, which belong to class I, II, IV, require a zinc ion to deacetylate acetylated lysine, but for the class III HDACs, the proteins require NAD+ as a cofactor for the enzyme activity. In addition, other transcription factors, such as TCF1 and LEF1, is discovered recently to own the ability of deacetylating H3K9ac and H3K27ac [128]. But here, we do not discuss these two proteins without known phosphorylation sites.
In class I HDACs, HDAC1 is phosphorylated by various protein kinases and the specific sites directly influence HDAC1 subcellular localization, enzymatic activity and protein stability (Table 3). One of the most studied sites is Ser421 of HDAC1. It is reported to be targeted by different protein kinases. When it is phosphorylated by CK2, along with phosphorylation of Ser423, enhances its deacetylation activity on histones and also leads to nuclear accumulation of HDAC1 in response to neurotoxic [129, 130]. Other studies have found that Ser421 of HDAC1 can be phosphorylated by nemo-like kinase (NLK), which negatively regulates the WNT signaling pathway by inactivation of β-Catenin/LEF1 transcription [131, 132]. In the zebrafish central nervous system, HDAC1 is phosphorylated at Ser406 by Aurora A/B kinase, which affects its enzymatic activity and occurs during mitosis [133]. Among other sites, only Tyr72 is discovered to be phosphorylated by EGFR, and its phosphorylation maintains HDAC1 stability and its anti-apoptosis in tumors [134]. Different from HDAC1 phosphorylation at Ser421 on its enzymatic activity, Ser394, Ser422, and Ser424 of HDAC2 can be phosphorylated by CK2 and inhibits its deacetylation activity [135,136,137]. Furthermore, P21 is a key factor for vascular remodeling and its expression is regulated by a complex of HDAC2, retinoic acid receptor (RAR), and kruppel-like factor 5 (KIF5). Phosphorylation of HDAC2 dissociates it from RAR and deacetylates KIF5, resulting in transcriptional activation of P21 [137]. Single phosphorylation of HDAC2 at Ser394 enhances its interaction with HSP70 and fails to dephosphorylate HDAC2 by weakening the binding of PP2CA [138]. For HDAC3 phosphorylation, it is interesting that phosphorylated Ser424 increases HDAC3 activity, but several protein kinases are reported to be involved in, including CK2, TANK-binding kinase (TBK1), leucine-rich repeat kinase 2 (LRRK2), c-Jun N-terminal kinase (JNK), and PTEN-induced putative kinase 1 (PINK1) [139,140,141,142,143]. The other site, Ser374, is phosphorylated by homeodomain-interacting protein kinase 2 (HIPK2), which inhibits the enzymatic activity of HDAC3 [144]. Similarly, the enzymatic activity of HDAC8 is repressed by PKA-mediated phosphorylation at Ser39 [145, 146]. In addition, Ser39, Ser43, and Ser63 of HDAC8 are shown to be phosphorylated by activated AMPK under the condition of glucose deprivation in cancer cells, connecting cancer survival with glycogen pathway via overexpression of phosphoglucomutase 1 (PGM1) [147].
Phosphorylation by various serine/threonine kinases alters the subcellular localization of HDACs. HDAC4, as one of class II HDACs, is commonly verified to be phosphorylated at Ser246, Ser467, and Ser632 by calmodulin kinase II (CaMKII), which lead to nuclear export of HADC4 to the cytoplasm [148,149,150]. Another site, Ser740 of HDAC4, can be phosphorylated to prevent HDAC4 from degradation and it is phosphorylated by PKA [151]. It has been known that 14-3-3 binding sites of HDACs is crucial for nuclear-cytoplasmic shutting. Phosphorylation of HDAC5 at Ser259 and Ser498 by protein kinase D (PKD), as well as Ser218 and Ser448 of HDAC9, contributes to the formation of 14-3-3 binding sites. But it is only verified at cardiomyocytes and myocyte-like cells [152, 153]. The two sites of HDAC5, Ser259 and Ser498, are also reported by other kinases, including AMPK, AKT, CaMKII in cells with different types [154,155,156,157]. Although kinases-phosphorylated HDACs at specific sites partly promote the binding of 14-3-3 to HDACs and stabilize HDACs, the subcellular localization of HDACs is not strictly controlled by phosphorylation. Different from other HDACs, Ser178-phosphorylated HDAC7 exists in both the nucleus and the cytoplasm, while Ser344 and Ser479-phosphorylated HDAC7 is only localized in the nucleus [157]. The enzymatic activity of HDAC6 is reported to be significantly affected by phosphorylation in previous studies. Different kinases-mediated phosphorylation of HDAC6 at Ser21, Ser22, Ser458, Ser1035 activates its ability to deacetylate histones [158,159,160,161]. Only Tyr570 of HDAC6 is phosphorylated to inhibit its deacetylation activity [162].
SIRT (1–7) belongs to the class III HDACs with nicotine adenine dinucleotide (NAD+) as a cofactor. It is originally discovered in yeast and characterized to deacetylate histones for transcription repression [183]. Structurally, all SIRT proteins share a highly conserved catalytic domain which forms a clef for the substrates and nicotinamide. Due to that SIRT3, SIRT4 and SIRT5 localize in mitochondria, the roles of these phosphorylated proteins on histone are not clarified. Therefore, only SIRT1, SIRT2, SIRT6, and SIRT7 are discussed (Table 4). Phosphorylated SIRT1 is involved in multiple cellular processes, including metabolism, DNA repair, apoptosis, inflammation, and aging. It is reported that mammalian sterile 20-like kinase 1 (MST1) overexpression leads to DNA damage-induced apoptosis by upregulating P53 acetylation [184]. P53 acetylation accounts for inhibiting the deacetylation of SIRT1 by MST1-mediated phosphorylation in the C-terminus of SIRT1. But SIRT1 phosphorylation at Ser47 by mTOR can rescue cells from DNA damage-induced senescent by elevating p65/RelA NF-κB acetylation [185]. The phosphorylation of two sites of SIRT1, Ser47and Thr522, have been reported to promote SIRT1 deacetylation activity, while only phosphorylated Ser47 of SIRT1 induces ubiquitination of SIRT1 for degradation. For SIRT2, its phosphorylation at Ser327, Ser331, Ser335, Ser368, Ser372, and Ser473, notably represses its catalytic activity on histones with different kinases [186,187,188,189]. SIRT6 phosphorylation only affects its protein stability on Ser338, chromatin binding on Thr294 and mono-ADP ribosylation activity on Ser10 [190,191,192]. And until now, there is no evidence for SIRT7 phosphorylation.
It is worth noting that several enzymes, including EZH2, P300, KAT7, KAT13A, and almost of HDACs, own a relatively large number of functional phosphorylation sites in the summarized tables. EZH2 is comprised of SANT domain, CXC domain, and catalytic SET domain. The phosphorylated sites in the N-terminal domain of EZH2 by various protein kinases not only inhibits its methylation activity, but also promotes its ubiquitination for degradation. Only one site, Thr350, can be phosphorylated and activates enzymatic activity of EZH2. And phosphorylation of Ser646 in the region of SET domain of EZH2 promotes its degradation in malignant lymphoma [89]. Even it is reported that CXC domain of EZH2 is conformational flexible and structurally block SET domain for substrate binding, there is still unknown whether phosphorylation of these sites is dependent on the dynamic conformational plasticity to regulate EZH2 enzymatic activity or not [193]. Most phosphorylated sites in C-terminal of P300, ranging from 1830 to 2370 amino acids of P300 protein sequence, have been evidenced to enhance its acetylation activity, while there is complex for sites in the N-terminus of P300. Phosphorylation of Ser12 by MAPK, activates P300 catalytic activity in cancers, and phosphorylated Ser106 can stabilize P300 in response to DNA damage. It is well studied that Ser89 of P300 is phosphorylated by different kinases with diverse biological functions. AMPK can phosphorylate Ser89 of P300 to repress its acetylation activity, and the interactions between P300 and chromatin/proteins is determined by PKC/SIK2-mediated phosphorylation of P300 on Ser89. However, it is still confusing that how different kinases phosphorylate the same site of P300 and what kinds of changes on P300 itself lead to obvious distinctions of these functions. Therefore, the interaction network on these questions should be further studied. Similar with P300, HDACs phosphorylation has also been questioned. It has been found that Ser 424 of HDAC3 can be phosphorylated by five kinases, including CK2, TBK1, LRRK2, C-JUN, AND PINK1. All of these kinases enhance the deacetylation activity of HDAC3 by phosphorylating Ser424, while it is verified with experiments in distinct disease models, such as triple-negative breast cancer, Parkinson’s disease, and innate antiviral immunity. Combined with these single studies, it is complicated to some extent and there are no explanations on their internal relations.
Conclusions and perspectives
In this review, we specifically discuss how phosphorylation regulates two kinds of transcription cofactors, including DNA methylation/demethylation-related enzymes and histone-modifying enzymes (the model in Fig. 3). Many transcription factors occur transcriptional activation or repression via phosphorylation-enhanced enzymatic activity or phosphorylation-inhibited enzymatic activity. And also, phosphorylation controls the protein stability of TFs, nuclear localization, and the interaction with chromatin or other proteins. Precise mechanisms by which phosphorylation enhances or represses the transcriptional activity of these two kinds of enzymes are still poorly understood. Maybe it occurs either: (i) by a converted structural conformation that covers or uncovers a docking site for histones, (ii) by the removal of an inhibitory protein or molecule resulted from phosphorylation, (iii) by directly blocking the histone binding sites in the catalytic domain of TFs, (iv) by recruiting other proteins to tighten or relax chromatin.

The model of transcriptional regulation of DNA/histone-modifying enzymes by phosphorylation
Since the discovery of histone PTMs, it has been evidenced to be dynamic and reversible [213]. DNA and histone modifications occur through “writer” enzymes, including DNMTs, KMTs, and KATs. The opposite enzymes are known as “erasers” for eliminating histone modifications, which contain TETs, KDMs, HDACs. Other enzymes, some of which are called “readers”, can recognize and bind specifically modified histones and possibly interact with “writers” or “erasers”. DNA and histone modifications significantly affect chromatin structure and transcription regulation of genes. The phosphorylation of histone-modifying enzymes has opened up a new perspective for molecular mechanisms of transcription regulation. In general, phosphorylation-dependent enzymatic activation or inactivation of TFs, and even phosphorylation-mediated protein degradation or dissociation from chromatin, usually lead to aberrant gene expression and this effect is likely to be more severe in diseases, including cancers and mental disorders. In our review, we summarized all studied phosphorylated sites of DNA/histone-modifying enzymes described in previous researches. Interestingly, one serine residue (or threonine and tyrosine residue), which plays a crucial role in TFs functions, is reported to be phosphorylated by distinct kinases and regulates different genes’ expression during multiple subcellular processes, including cell differentiation, DNA damage and repair, inflammation, and metabolism. Substrate specificity is characterized by a protein kinase because the phosphorylation of a protein is determined by a targeted phospho-acceptor site in a consensus motif of a substrate [214]. However, one or two sites or motif cannot completely determine substrate specificity. Indeed, the interaction between TFs and protein kinases is also important for efficient substrate phosphorylation. Moreover, a specific protein kinase-dependent phosphorylation of TFs is also verified in one type of cell line. It is not clear whether different kinases compete for binding to one TFs and how to phosphorylate TFs at one site in an orderly fashion. And how different cell types influence TFs phosphorylation. Maybe phosphorylation involves TFs in the different signaling pathways which corporately regulate their cellular functions. These questions are still unresolved, so the further detailed study is needed to uncover the network of the known molecular mechanisms. Therefore, a comprehensive and systematic study on TFs phosphorylation will possibly expand our understanding of the transcriptional regulation of these DNA/histone-modifying enzymes via new advanced technologies. In addition, deletion of specific kinases that target one transcription factor, or insertion of mutations of phosphorylated sites on transcription factors in a mouse model, will enhance our comprehension and genetically prove the importance of phosphorylation in regulating themselves and overall functions of TFs.
Availability of data and materials
Not applicable.
Abbreviations
- 5caC:
-
5-Carboxylcytosine
- 5fC:
-
5-Formylcytosine
- 5hmC:
-
5-Hydroxymethylcytosine
- 5mC:
-
5-Methylcytosine
- ADA3:
-
Alteration/Deficiency in Activation 3
- AdoMet:
-
S-adenosyl-methionine
- AMPK:
-
MP-activated kinase
- AMPK:
-
(AMP)-activated protein kinase
- ATM:
-
Ataxia-telangiectasia mutate
- BER:
-
Base excision repair
- CaMKII:
-
Calmodulin kinase II
- CDK1:
-
Cyclin-dependent kinase 1
- CDK5:
-
Cyclin-dependent kinase 5
- CDKs:
-
Cyclin-dependent kinases
- CK2:
-
Casein kinase 2
- DSBs:
-
Double-strand breaks
- EGFR:
-
Epidermal Growth Factor Receptor
- EKLF:
-
Kruppel-like factor
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- ERK2:
-
Extracellular signal-regulated kinase 2
- ETS:
-
E-twenty-six
- EZH2:
-
Zeste homolog 2
- FGFR3:
-
Fibroblast growth factor receptor 3
- H3K4:
-
Histone H3 lysine 4
- HATs:
-
Histone acetyltransferases
- HDACs:
-
Histone deacetylases
- HGP:
-
Hepatic glucose production
- HIPK2:
-
Homeodomain-interacting protein kinase 2
- HMGA2:
-
High-mobility group AT-hook 2
- HR:
-
Homologous recombination
- HSC:
-
Hematopoietic stem cell
- JNK:
-
C-Jun N-terminal kinase
- KDMs:
-
Histone lysine demethylases
- KIF5:
-
Kruppel-like factor 5
- KLF4:
-
Kruppel like factor 4
- KMTs:
-
Lysine methyltransferases
- LRRK2:
-
Leucine-rich repeat kinase 2
- MK:
-
Megakaryocyte
- MLL1:
-
Mixed lineage leukemia 1
- MST1:
-
Mammalian sterile 20-like kinase 1
- MYHC1:
-
Myosin heavy chain 1
- NFATc1:
-
Nuclear factor of activated T-cells c1
- NLK:
-
Nemo-like kinase
- NLS:
-
Nuclear localization signal
- PcG:
-
Polycomb group proteins
- PDK1:
-
Protein kinase D1
- PGM1:
-
Phosphoglucomutase 1
- PIK1:
-
Polo-like kinase 1
- PINK1:
-
PTEN-induced putative kinase 1
- PKA:
-
Protein kinase A
- PKB:
-
Protein kinase B
- PKC:
-
Protein kinase C
- PKD:
-
Protein kinase D
- PRMTs:
-
Protein arginine methyl transferases 1
- PTMs:
-
Post-translational modifications
- RAR:
-
Retinoic acid receptor
- RPA:
-
Replication protein A
- Ser:
-
Serine
- TBK1:
-
TANK-binding kinase
- TDG:
-
Thymine-DNA glycosylase
- TFs:
-
Transcriptional factors
- Thr:
-
Threonine
- Tyr:
-
Tyrosine
References
Xun L, Matthias W, Janet T, Maja K. Elucidating human phosphatase-substrate networks. Sci Signaling. 2013;6:10. https://doi.org/10.1126/scisignal.2003203.
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. 2018;175:598–9. https://doi.org/10.1016/j.cell.2018.09.045.
Cramer P. Organization and regulation of gene transcription. Nature. 2019;573:45–54. https://doi.org/10.1038/s41586-019-1517-4.
Lam Dai Vu, Gevaert K, De Smet I. Protein language: post-translational modifications talking to each other. Trends Plant Sci. 2018;23:1068–80. https://doi.org/10.1016/j.tplants.2018.09.004.
Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–59. https://doi.org/10.1128/MMBR.64.2.435-459.2000.
Zhang Yi, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–60. https://doi.org/10.1101/gad.927301.
Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69. https://doi.org/10.1146/annurev.biochem.75.103004.142422.
Nathan D, Ingvarsdottir K, Sterner DE, Bylebyl GR, Dokmanovic M, Dorsey JA, et al. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev. 2006;20:966–76. https://doi.org/10.1101/gad.1404206.
Liang G, Chan MF, Tomigahara Y, Tsai YC, Gonzales FA, Li En, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22:480–91. https://doi.org/10.1128/MCB.22.2.480-491.2002.
Hermann A, Schmitt S, Jeltsch A. The human Dnmt2 has residual DNA-(cytosine-C5) methyltransferase activity*. J Biol Chem. 2003;278:31717–21. https://doi.org/10.1074/jbc.M305448200.
Mary GG, Finn Kirpekar A, Maggert Keith A, Jeffrey Y, Hsieh C-L, Zhang X, et al. Methylation of tRNAAsp by the DNA Methyltransferase Homolog Dnmt2. Science. 2006;311:395–8. https://doi.org/10.1126/science.1120976.
Shanmugam R, Fierer J, Kaiser S, Helm M, Jurkowski TP, Jeltsch A. Cytosine methylation of tRNA-Asp by DNMT2 has a role in translation of proteins containing poly-Asp sequences. Cell Discov. 2015;1:15010–15010. https://doi.org/10.1038/celldisc.2015.10.
Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–77. https://doi.org/10.1038/77023.
Ding F, Richard Chaillet J. In vivo stabilization of the Dnmt1 (cytosine-5)- methyltransferase protein. Proc Natl Acad Sci U S A. 2002;99:14861–6. https://doi.org/10.1073/pnas.232565599.
Pradhan M, Estève P-O, Chin HG, Samaranayke M, Kim G-D, Pradhan S. CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry. 2008;47:10000–9. https://doi.org/10.1021/bi8011725.
Song J, Rechkoblit O, Bestor TH, Patel DJ. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011;331:1036–40. https://doi.org/10.1126/science.1195380.
Sharif J, Muto M, Takebayashi S-I, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–12. https://doi.org/10.1038/nature06397.
Arita K, Isogai S, Oda T, Unoki M, Sugita K, Sekiyama N, et al. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc Natl Acad Sci U S A. 2012;109:12950–5. https://doi.org/10.1073/pnas.1203701109.
Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K, et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013;502:249–53. https://doi.org/10.1038/nature12488.
Bronner C, Thierry Chataigneau B, Schini-Kerth V, Landry Y. The “Epigenetic Code Replication Machinery”, ECREM: a promising drugable target of the epigenetic cell memory. Curr Med Chem. 2007;14:2629–41. https://doi.org/10.2174/092986707782023244.
Chuang Linda SH, Ian H-I, Koh T-W, Ng H-H, Guoliang Xu, Li Benjamin FL. Human DNA-(Cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277:1996–2000. https://doi.org/10.1126/science.277.5334.1996.
Lavoie G, St-Pierre Y. Phosphorylation of human DNMT1: implication of cyclin-dependent kinases. Biochem Biophys Res Commun. 2011;409:187–92. https://doi.org/10.1016/j.bbrc.2011.04.115.
Goyal R, Rathert P, Laser H, Gowher H, Jeltsch A. Phosphorylation of serine-515 activates the Mammalian maintenance methyltransferase Dnmt1. Epigenetics. 2007;2:155–60. https://doi.org/10.4161/epi.2.3.4768.
Hervouet E, Lalier L, Debien E, Cheray M, Geairon A, Rogniaux H, et al. Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis from human and mice glial cells. PLoS ONE. 2010;5:e11333–e11333. https://doi.org/10.1371/journal.pone.0011333.
Estève P-O, Chang Y, Samaranayake M, Upadhyay AK, Horton JR, Feehery GR, et al. A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat Struct Mol Biol. 2011;18:42–8. https://doi.org/10.1038/nsmb.1939.
Sugiyama Y, Hatano N, Sueyoshi N, Suetake I, Tajima S, Kinoshita E, et al. The DNA-binding activity of mouse DNA methyltransferase 1 is regulated by phosphorylation with casein kinase 1δ/ε. Biochem J. 2010;427:489–97. https://doi.org/10.1042/BJ20091856.
Hodge DR, Cho E, Copeland TD, Guszczynski TAD, Yang E, Seth AK, et al. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics. 2007;4:387–98.
Tan X, Xingbo Xu, Zeisberg EM, Zeisberg M. High inorganic phosphate causes DNMT1 phosphorylation and subsequent fibrotic fibroblast activation. Biochem Biophys Res Commun. 2016;472:459–64. https://doi.org/10.1016/j.bbrc.2016.01.077.
Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2199–204. https://doi.org/10.1073/pnas.0611217104.
Gauci S, Helbig AO, Slijper M, Krijgsveld J, Heck AJR, Mohammed S. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal Chem. 2009;81:4493–501. https://doi.org/10.1021/ac9004309.
Viveka Mayya H, Deborah L, Hwang S-I, Rezaul K, Linfeng W, Eng Jimmy K, et al. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signaling. 2009;2:46. https://doi.org/10.1126/scisignal.2000007.
Tsai C-F, Wang Y-T, Chen Y-R, Lai C-Y, Lin P-Y, Pan K-T, et al. Immobilized metal affinity chromatography revisited: pH/acid control toward high selectivity in phosphoproteomics. J Proteome Res. 2008;7:4058–69. https://doi.org/10.1021/pr800364d.
Sharma V, Jayadev Joshi IJu, Yeh YD, Blankenberg D, Wald D, et al. Re-expression of ERα and AR in receptor negative endocrine cancers via GSK3 inhibition. Front Oncol. 2022;12:824594–824594. https://doi.org/10.3389/fonc.2022.824594.
Deplus R, Blanchon L, Rajavelu A, Boukaba A, Defrance M, Luciani J, et al. Regulation of DNA methylation patterns by CK2-mediated phosphorylation of Dnmt3a. Cell Rep. 2014;8:743–53. https://doi.org/10.1016/j.celrep.2014.06.048.
Po-Shu Tu, Lin E-Y, Chen H-W, Chen S-W, Lin T-A, Gau J-P, et al. The extracellular signal-regulated kinase 1/2 modulates the intracellular localization of DNA methyltransferase 3A to regulate erythrocytic differentiation. Am J Transl Res. 2020;12:1016–30.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. https://doi.org/10.1126/science.1170116.
He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. https://doi.org/10.1126/science.1210944.
Ito S, Shen Li, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. https://doi.org/10.1126/science.1210597.
Xiaoji Wu, Zhang Yi. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18:517–34. https://doi.org/10.1038/nrg.2017.33.
Lee HJ, Hore TA, Reik W. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell. 2014;14:710–9. https://doi.org/10.1016/j.stem.2014.05.008.
Zhang P, Tianjiao Chu N, Dedousis BS, Mantell IS, Li L, et al. DNA methylation alters transcriptional rates of differentially expressed genes and contributes to pathophysiology in mice fed a high fat diet. Mol Metab. 2017;6:327–39. https://doi.org/10.1016/j.molmet.2017.02.001.
Cong B, Zhang Q, Cao X. The function and regulation of TET2 in innate immunity and inflammation. Protein Cell. 2021;12:165–73. https://doi.org/10.1007/s13238-020-00796-6.
Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38:e181–e181. https://doi.org/10.1093/nar/gkq684.
Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2014;111:1361–6. https://doi.org/10.1073/pnas.1322921111.
Tian-Peng Gu, Guo F, Yang H, Hai-Ping Wu, Gui-Fang Xu, Liu W, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–10. https://doi.org/10.1038/nature10443.
Yufei Xu, Chao Xu, Kato A, Tempel W, Abreu JG, Bian C, et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell. 2012;151:1200–13. https://doi.org/10.1016/j.cell.2012.11.014.
Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–6. https://doi.org/10.1038/nature12362.
Bauer C, Göbel K, Nagaraj N, Colantuoni C, Wang M, Müller U, et al. Phosphorylation of TET proteins is regulated via O-GlcNAcylation by the O-linked N-acetylglucosamine transferase (OGT). J Biol Chem. 2015;290:4801–12. https://doi.org/10.1074/jbc.M114.605881.
Di Wu, Di Hu, Chen H, Shi G, Fetahu IS, Feizhen Wu, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature. 2018;559:637–41. https://doi.org/10.1038/s41586-018-0350-5.
Zhang T, Guan X, Choi UL, Dong Q, Lam MMT, Zeng J, et al. Phosphorylation of TET2 by AMPK is indispensable in myogenic differentiation. Epigenetics Chromatin. 2019;12:32–32. https://doi.org/10.1186/s13072-019-0281-x.
Jin Z, Feng H, Liang J, Jing X, Zhao Q, Zhan L, et al. FGFR3(△7-9) promotes tumor progression via the phosphorylation and destabilization of ten-eleven translocation-2 in human hepatocellular carcinoma. Cell Death Dis. 2020;11:903–903. https://doi.org/10.1038/s41419-020-03089-2.
Jeong JJ, Xiaorong Gu, Nie Ji, Sundaravel S, Liu H, Kuo W-L, et al. Cytokine-regulated phosphorylation and activation of TET2 by JAK2 in hematopoiesis. Cancer Discov. 2019;9:778–95. https://doi.org/10.1158/2159-8290.CD-18-1138.
Rao VK, Swarnaseetha A, Tham G-H, Lin W-Q, Han B-B, Benoukraf T, et al. Phosphorylation of Tet3 by cdk5 is critical for robust activation of BRN2 during neuronal differentiation. Nucleic Acids Res. 2020;48:1225–38. https://doi.org/10.1093/nar/gkz1144.
Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell. 2002;109:801–6. https://doi.org/10.1016/s0092-8674(02)00798-5.
Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. https://doi.org/10.1146/annurev-biochem-051710-134100.
Bedford MT, Richard S. Arginine methylation: an emerging regulatorof protein function. Mol Cell. 2005;18:263–72. https://doi.org/10.1016/j.molcel.2005.04.003.
Chiang K, Zielinska AE, Shaaban AM, Sanchez-Bailon MP, Jarrold J, Clarke TL, et al. PRMT5 is a critical regulator of breast cancer stem cell function via histone methylation and FOXP1 expression. Cell Rep. 2017;21:3498–513. https://doi.org/10.1016/j.celrep.2017.11.096.
Kexin Xu. In: Kaneda A, Tsukada Y-I, editors. DNA and Histone Methylation as Cancer Targets. Cham: Springer International Publishing; 2017. p. 489–529.
Chen Y, Liu X, Li Y, Quan C, Zheng L, Huang K. Lung cancer therapy targeting histone methylation: opportunities and challenges. Comput Struct Biotechnol J. 2018;16:211–23. https://doi.org/10.1016/j.csbj.2018.06.001.
Cheng X. Structural and functional coordination of DNA and histone methylation. Cold Spring Harb Perspect Biol. 2014;6: a018747. https://doi.org/10.1101/cshperspect.a018747.
Nakajo H, Ishibashi K, Aoyama K, Kubota S, Hasegawa H, Yamaguchi N, et al. Role for tyrosine phosphorylation of SUV39H1 histone methyltransferase in enhanced trimethylation of histone H3K9 via neuregulin-1/ErbB4 nuclear signaling. Biochem Biophys Res Commun. 2019;511:765–71. https://doi.org/10.1016/j.bbrc.2019.02.130.
Park SH, Seung Eun Yu, Chai YG, Jang YK. CDK2-dependent phosphorylation of Suv39H1 is involved in control of heterochromatin replication during cell cycle progression. Nucleic Acids Res. 2014;42:6196–207. https://doi.org/10.1093/nar/gku263.
Yang Q, Zhu Q, Xiaopeng Lu, Yipeng Du, Cao L, Shen C, et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc Natl Acad Sci U S A. 2017;114:E6054–63. https://doi.org/10.1073/pnas.1700694114.
Sharma S, Čermáková K, De Rijck J, Demeulemeester J, Fábry M, El Ashkar S, et al. Affinity switching of the LEDGF/p75 IBD interactome is governed by kinase-dependent phosphorylation. Proc Natl Acad Sci U S A. 2018;115:E7053–62. https://doi.org/10.1073/pnas.1803909115.
Zibo Zhao Lu, Wang AG, Volk NW, Birch KL, Stoltz ET, et al. Regulation of MLL/COMPASS stability through its proteolytic cleavage by taspase1 as a possible approach for clinical therapy of leukemia. Genes Dev. 2019;33:61–74. https://doi.org/10.1101/gad.319830.118.
Liu H, Cheng EHY, Hsieh JJD. MLL fusions: pathways to leukemia. Cancer Biol Ther. 2009;8:1204–11. https://doi.org/10.4161/cbt.8.13.8924.
Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23:1490–9. https://doi.org/10.1038/leu.2009.33.
Liu H, Takeda S, Kumar R, Westergard TD, Brown EJ, Pandita TK, et al. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature. 2010;467:343–6. https://doi.org/10.1038/nature09350.
Amar MS, Dustin WP, Valeriya VAS, Kenneth M, Daniel WA. In: Turksen K, editor. Stem Cell Heterogeneity: Methods and Protocols. New York: Springer; 2016. p. 153–69.
Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017;355:1324–30. https://doi.org/10.1126/science.aah6893.
Li LX, Zhou JX, Wang X, Zhang H, Harris PC, Calvet JP, et al. Cross-talk between CDK4/6 and SMYD2 regulates gene transcription, tubulin methylation, and ciliogenesis. Sci Adv. 2020;6:e3154. https://doi.org/10.1126/sciadv.abb3154.
Li Ni, Xue W, Yuan H, Dong B, Ding Y, Liu Y, et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J Clin Invest. 2017;127:1284–302. https://doi.org/10.1172/JCI91144.
Michowski W, Chick JM, Chu C, Kolodziejczyk A, Wang Y, Suski JM, et al. Cdk1 controls global epigenetic landscape in embryonic stem cells. Mol Cell. 2020;78:459-476.e413. https://doi.org/10.1016/j.molcel.2020.03.010.
Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013;20:1147–55. https://doi.org/10.1038/nsmb.2669.
Liu P, Shuaib M, Zhang H, Nadeef S, Orlando V. Ubiquitin ligases HUWE1 and NEDD4 cooperatively control signal-dependent PRC2-Ezh1α/β-mediated adaptive stress response pathway in skeletal muscle cells. Epigenetics Chromatin. 2019;12:78–78. https://doi.org/10.1186/s13072-019-0322-5.
Müller A, Dickmanns A, Resch C, Schäkel K, Hailfinger S, Dobbelstein M, et al. The CDK4/6-EZH2 pathway is a potential therapeutic target for psoriasis. J Clin Invest. 2020;130:5765–81. https://doi.org/10.1172/JCI134217.
Tai-Lung Cha P, Binhua Z, Xia W, Yadi Wu, Yang C-C, Chen C-T, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–10. https://doi.org/10.1126/science.1118947.
Schonfeld M, Villar MT, Artigues A, Weinman SA, Tikhanovich I. Arginine methylation of hepatic hnRNPH suppresses complement activation and systemic inflammation in alcohol-fed mice. Hepatol Commun. 2021;5:812–29. https://doi.org/10.1002/hep4.1674.
Huang T, Yang Y, Song X, Wan X, Bingli Wu, Sastry N, et al. PRMT6 methylation of RCC1 regulates mitosis, tumorigenicity, and radiation response of glioblastoma stem cells. Mol Cell. 2021;81:1276-1291.e1279. https://doi.org/10.1016/j.molcel.2021.01.015.
Ginjala V, Rodriguez-Colon L, Ganguly B, Gangidi P, Gallina P, Al-Hraishawi H, et al. Protein-lysine methyltransferases G9a and GLP1 promote responses to DNA damage. Sci Rep. 2017;7:16613–16613. https://doi.org/10.1038/s41598-017-16480-5.
Liu J, Wang XN, Cheng F, Liou Y-C, Deng L-W. Phosphorylation of mixed lineage leukemia 5 by CDC2 affects its cellular distribution and is required for mitotic entry. J Biol Chem. 2010;285:20904–14. https://doi.org/10.1074/jbc.M109.098558.
Li B, Yan J, Phyu T, Fan S, Chung T-H, Mustafa N, et al. MELK mediates the stability of EZH2 through site-specific phosphorylation in extranodal natural killer/T-cell lymphoma. Blood. 2019;134:2046–58. https://doi.org/10.1182/blood.2019000381.
Yan J, Li B, Lin B, Lee PT, Chung T-H, Tan J, et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood. 2016;128:948–58. https://doi.org/10.1182/blood-2016-01-690701.
Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, et al. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J Biol Chem. 2017;292:6269–80. https://doi.org/10.1074/jbc.M116.764407.
Wan L, Kexin Xu, Wei Y, Zhang J, Han T, Fry C, et al. Phosphorylation of EZH2 by AMPK suppresses PRC2 methyltransferase activity and oncogenic function. Mol Cell. 2018;69:279-291.e275. https://doi.org/10.1016/j.molcel.2017.12.024.
Ko H-W, Lee H-H, Huo L, Xia W, Yang C-C, Hsu JL, et al. GSK3β inactivation promotes the oncogenic functions of EZH2 and enhances methylation of H3K27 in human breast cancers. Oncotarget. 2016;7:57131–44. https://doi.org/10.18632/oncotarget.11008.
Nie L, Wei Y, Zhang F, Hsu Y-H, Chan L-C, Xia W, et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat Commun. 2019;10:5114–5114. https://doi.org/10.1038/s41467-019-13105-5.
Wan J, Zhan J, Li S, Ma Ji, Weizhi Xu, Liu C, et al. PCAF-primed EZH2 acetylation regulates its stability and promotes lung adenocarcinoma progression. Nucleic Acids Res. 2015;43:3591–604. https://doi.org/10.1093/nar/gkv238.
Sahasrabuddhe AA, Chen X, Chung F, Velusamy T, Lim MS, Elenitoba-Johnson KSJ. Oncogenic Y641 mutations in EZH2 prevent Jak2/β-TrCP-mediated degradation. Oncogene. 2015;34:445–54. https://doi.org/10.1038/onc.2013.571.
Cao LL, Wei F, Du Y, Song B, Wang D, Shen C, et al. ATM-mediated KDM2A phosphorylation is required for the DNA damage repair. Oncogene. 2016;35:402–402. https://doi.org/10.1038/onc.2015.311.
Baker M, Petasny M, Taqatqa N, Bentata M, Kay G, Engal E, et al. KDM3A regulates alternative splicing of cell-cycle genes following DNA damage. RNA. 2021;27:1353–62. https://doi.org/10.1261/rna.078796.121.
Duan L, Chen Z, Jun Lu, Liang Y, Wang M, Roggero CM, et al. Histone lysine demethylase KDM4B regulates the alternative splicing of the androgen receptor in response to androgen deprivation. Nucleic Acids Res. 2019;47:11623–36. https://doi.org/10.1093/nar/gkz1004.
Fu LN, Wang YQ, Tan J, Xu J, Gao QY, Chen YX, et al. Role of JMJD2B in colon cancer cell survival under glucose-deprived conditions and the underlying mechanisms. Oncogene. 2018;37:389–402. https://doi.org/10.1038/onc.2017.345.
Chen Y, Fang R, Yue C, Chang G, Li P, Guo Q, et al. Wnt-induced stabilization of KDM4C is required for Wnt/β-catenin target gene expression and glioblastoma tumorigenesis. Cancer Res. 2020;80:1049–63. https://doi.org/10.1158/0008-5472.CAN-19-1229.
Ju Yeh I, Esakov E, Lathia JD, Miyagi M, Reizes O, Montano MM. Phosphorylation of the histone demethylase KDM5B and regulation of the phenotype of triple negative breast cancer. Sci Rep. 2019;9:17663–17663. https://doi.org/10.1038/s41598-019-54184-0.
Sterling J, Menezes SV, Abbassi RH, Munoz L. Histone lysine demethylases and their functions in cancer. Int J Cancer. 2021;148:2375–88. https://doi.org/10.1002/ijc.33375.
Lin C-Y, Wang B-J, Chen B-C, Tseng J-C, Jiang SS, Tsai KK, et al. Histone demethylase KDM4C stimulates the proliferation of prostate cancer cells via activation of AKT and c-Myc. Cancers (Basel). 2019;11:1785. https://doi.org/10.3390/cancers11111785.
Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. https://doi.org/10.1038/nrc3409.
Walport LJ, Hopkinson RJ, Chowdhury R, Schiller R, Ge W, Kawamura A, et al. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat Commun. 2016;7:11974–11974. https://doi.org/10.1038/ncomms11974.
Tang Yi, Zhao W, Chen Y, Zhao Y, Wei Gu. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–26. https://doi.org/10.1016/j.cell.2008.03.025.
Zheng W-W, Dong X-M, Yin R-H, Fei-Fei Xu, Ning H-M, Zhang M-J, et al. EDAG positively regulates erythroid differentiation and modifies GATA1 acetylation through recruiting p300. Stem Cells. 2014;32:2278–89. https://doi.org/10.1002/stem.1723.
Sengupta T, Chen K, Milot E, Bieker JJ. Acetylation of EKLF is essential for epigenetic modification and transcriptional activation of the beta-globin locus. Mol Cell Biol. 2008;28:6160–70. https://doi.org/10.1128/MCB.00919-08.
Marin TL, Gongol B, Zhang F, Martin M, Johnson DA, Xiao H, et al. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Science Signaling. 2017;10:e7478. https://doi.org/10.1126/scisignal.aaf7478.
Sakai M, Tujimura-Hayakawa T, Yagi T, Yano H, Mitsushima M, Unoki-Kubota H, et al. The GCN5-CITED2-PKA signalling module controls hepatic glucose metabolism through a cAMP-induced substrate switch. Nat Commun. 2016;7:13147–13147. https://doi.org/10.1038/ncomms13147.
Srivastava S, Mohibi S, Mirza S, Band H, Band V. Epidermal growth factor receptor activation promotes ADA3 acetylation through the AKT-p300 pathway. Cell Cycle. 2017;16:1515–25. https://doi.org/10.1080/15384101.2017.1339846.
Yue Wu, Wang X, Feifei Xu, Zhang Lu, Wang T, Xueli Fu, et al. The regulation of acetylation and stability of HMGA2 via the HBXIP-activated Akt-PCAF pathway in promotion of esophageal squamous cell carcinoma growth. Nucleic Acids Res. 2020;48:4858–76. https://doi.org/10.1093/nar/gkaa232.
Syal C, Seegobin M, Sarma SN, Gouveia A, Hsu K, Niibori Y, et al. Ectopic expression of aPKC-mediated phosphorylation in p300 modulates hippocampal neurogenesis, CREB binding and fear memory differently with age. Sci Rep. 2018;8:13489–13489. https://doi.org/10.1038/s41598-018-31657-2.
Wan W, You Z, Yinfeng Xu, Zhou Li, Guan Z, Peng C, et al. mTORC1 phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis. Mol Cell. 2017;68:323-335.e326. https://doi.org/10.1016/j.molcel.2017.09.020.
Chen Y-J, Wang Y-N, Chang W-C. ERK2-mediated C-terminal serine phosphorylation of p300 is vital to the regulation of epidermal growth factor-induced keratin 16 gene expression*. J Biol Chem. 2007;282:27215–28. https://doi.org/10.1074/jbc.M700264200.
Jang ER, Choi JD, Jeong G, Lee J-S. Phosphorylation of p300 by ATM controls the stability of NBS1. Biochem Biophys Res Commun. 2010;397:637–43. https://doi.org/10.1016/j.bbrc.2010.05.060.
Wuchao Yuan L, Gambee JE. Phosphorylation of p300 at Serine 89 by Protein Kinase C. J Biol Chem. 2000;275:40946–51. https://doi.org/10.1074/jbc.M007832200.
Zhang Z-N, Gong L, Lv S, Li J, Tai X, Cao W, et al. SIK2 regulates fasting-induced PPARα activity and ketogenesis through p300. Sci Rep. 2016;6:23317–23317. https://doi.org/10.1038/srep23317.
Wang S-A, Hung C-Y, Chuang J-Y, Chang W-C, Hsu T-I, Hung J-J. Phosphorylation of p300 increases its protein degradation to enhance the lung cancer progression. Biochimica et Biophysica Acta Mol Cell Res. 2014;1843:1135–49. https://doi.org/10.1016/j.bbamcr.2014.02.001.
Brouillard F, Cremisi CE. Concomitant increase of histone acetyltransferase activity and degradation of p300 during retinoic acid-induced differentiation of F9 cells. J Biol Chem. 2003;278:39509–16. https://doi.org/10.1074/jbc.M307123200.
He L, Naik K, Meng S, Cao J, Sidhaye AR, Ma A, et al. Transcriptional co-activator p300 maintains basal hepatic gluconeogenesis. J Biol Chem. 2012;287:32069–77. https://doi.org/10.1074/jbc.M112.385864.
Zhao-Qiu Wu, Liu X. Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proc Natl Acad Sci U S A. 2008;105:1919–24. https://doi.org/10.1073/pnas.0712063105.
Brauns-Schubert P, Schubert F, Wissler M, Weiss M, Schlicher L, Bessler S, et al. CDK9-mediated phosphorylation controls the interaction of TIP60 with the transcriptional machinery. EMBO Rep. 2018;19:244–56. https://doi.org/10.15252/embr.201744311.
Sin TK, Zhang G, Zhang Z, Zhu JZ, Zuo Y, Frost JA, et al. Cancer-induced muscle wasting requires p38β MAPK activation of p300. Cancer Res. 2021;81:885–97. https://doi.org/10.1158/0008-5472.CAN-19-3219.
Liang Y, Yuanyuan Su, Chenzhong Xu, Zhang Na, Liu D, Li G, et al. Protein kinase D1 phosphorylation of KAT7 enhances its protein stability and promotes replication licensing and cell proliferation. Cell Death Discovery. 2020;6:89. https://doi.org/10.1038/s41420-020-00323-w.
Zhang Y, Qiu J, Wang X, Zhang Y, Xia M. AMP-activated protein kinase suppresses endothelial cell inflammation through phosphorylation of transcriptional coactivator p300. Arterioscler Thromb Vasc Biol. 2011;31:2897–908. https://doi.org/10.1161/ATVBAHA.111.237453.
Schwartz C, Beck K, Mink S, Schmolke M, Budde B, Wenning D, et al. Recruitment of p300 by C/EBPbeta triggers phosphorylation of p300 and modulates coactivator activity. EMBO J. 2003;22:882–92. https://doi.org/10.1093/emboj/cdg076.
Charvet C, Wissler M, Brauns-Schubert P, Wang S-J, Tang Yi, Sigloch FC, et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol Cell. 2011;42:584–96. https://doi.org/10.1016/j.molcel.2011.03.033.
Lemercier C, Legube G, Caron C, Louwagie M, Garin J, Trouche D, et al. Tip60 acetyltransferase activity is controlled by phosphorylation. J Biol Chem. 2003;278:4713–8. https://doi.org/10.1074/jbc.M211811200.
Niida H, Matsunuma R, Horiguchi R, Uchida C, Nakazawa Y, Motegi A, et al. Phosphorylated HBO1 at UV irradiated sites is essential for nucleotide excision repair. Nat Commun. 2017;8:16102–16102. https://doi.org/10.1038/ncomms16102.
Duong MT, Akli S, Macalou S, Biernacka A, Debeb BG, Yi M, et al. Hbo1 is a cyclin E/CDK2 substrate that enriches breast cancer stem-like cells. Cancer Res. 2013;73:5556–68. https://doi.org/10.1158/0008-5472.CAN-13-0013.
Gupta A, Hunt CR, Hegde ML, Chakraborty S, Chakraborty S, Udayakumar D, et al. MOF phosphorylation by ATM regulates 53BP1-mediated double-strand break repair pathway choice. Cell Rep. 2014;8:177–89. https://doi.org/10.1016/j.celrep.2014.05.044.
Moore NL, Weigel NL. Regulation of progesterone receptor activity by cyclin dependent kinases 1 and 2 occurs in part by phosphorylation of the SRC-1 carboxyl-terminus. Int J Biochem Cell Biol. 2011;43:1157–67. https://doi.org/10.1016/j.biocel.2011.04.009.
Xing S, Li F, Zeng Z, Zhao Y, Shuyang Yu, Shan Q, et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol. 2016;17:695–703. https://doi.org/10.1038/ni.3456.
Zhu Y, Vidaurre OG, Adula KP, Kezunovic N, Wentling M, Huntley GW, et al. Subcellular distribution of HDAC1 in neurotoxic conditions is dependent on serine phosphorylation. J Neurosci. 2017;37:7547–59. https://doi.org/10.1523/JNEUROSCI.3000-16.2017.
Mary KH, Pflum JK, Tong WS. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem. 2001;276:47733–41. https://doi.org/10.1074/jbc.M105590200.
Masoumi KC, Daams R, Sime W, Siino V, Ke H, Levander F, et al. NLK-mediated phosphorylation of HDAC1 negatively regulates Wnt signaling. Mol Biol Cell. 2017;28:346–55. https://doi.org/10.1091/mbc.E16-07-0547.
Daams R, Sime W, Leandersson K, Sitnicka E, Massoumi R. Deletion of Nemo-like Kinase in T Cells Reduces Single-Positive CD8 + Thymocyte Population. J Immunol. 2020;205:1830. https://doi.org/10.4049/jimmunol.2000109.
Loponte S, Segré CV, Senese S, Miccolo C, Santaguida S, Deflorian G, et al. Dynamic phosphorylation of Histone Deacetylase 1 by Aurora kinases during mitosis regulates zebrafish embryos development. Sci Rep. 2016;6:30213–30213. https://doi.org/10.1038/srep30213.
Bahl S, Ling H, Acharige NPN, Santos-Barriopedro I, Mary KH, Pflum and Edward Seto. EGFR phosphorylates HDAC1 to regulate its expression and anti-apoptotic function. Cell Death Dis. 2021;12:469–469. https://doi.org/10.1038/s41419-021-03697-6.
Khan DH, He S, Jenny Yu, Winter S, Cao W, Seiser C, et al. Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. J Biol Chem. 2013;288:16518–28. https://doi.org/10.1074/jbc.M112.440446.
Adenuga D, Rahman I. Protein kinase CK2-mediated phosphorylation of HDAC2 regulates co-repressor formation, deacetylase activity and acetylation of HDAC2 by cigarette smoke and aldehydes. Arch Biochem Biophys. 2010;498:62–73. https://doi.org/10.1016/j.abb.2010.04.002.
Zheng B, Han M, Shu Y-N, Li Y-J, Miao S-B, Zhang X-H, et al. HDAC2 phosphorylation-dependent Klf5 deacetylation and RARα acetylation induced by RAR agonist switch the transcription regulatory programs of p21 in VSMCs. Cell Res. 2011;21:1487–508. https://doi.org/10.1038/cr.2011.34.
Yoon S, Kim M, Min H-K, Lee Y-U, Kwon D-H, Lee M, et al. Inhibition of heat shock protein 70 blocks the development of cardiac hypertrophy by modulating the phosphorylation of histone deacetylase 2. Cardiovasc Res. 2019;115:1850–60. https://doi.org/10.1093/cvr/cvy317.
Zhang Y, Zheng X, Tan H, Yilu Lu, Tao D, Liu Y, et al. PIWIL2 suppresses Siah2-mediated degradation of HDAC3 and facilitates CK2α-mediated HDAC3 phosphorylation. Cell Death Dis. 2018;9:423–423. https://doi.org/10.1038/s41419-018-0462-8.
Tang J-L, Yang Qi, Chong-Hui Xu, Zhao He, Liu Y-L, Liu C-Y, et al. Histone deacetylase 3 promotes innate antiviral immunity through deacetylation of TBK1. Protein Cell. 2021;12:261–78. https://doi.org/10.1007/s13238-020-00751-5.
Han KA, Shin WH, Jung S, Seol W, Seo H, Ko C, et al. Leucine-rich repeat kinase 2 exacerbates neuronal cytotoxicity through phosphorylation of histone deacetylase 3 and histone deacetylation. Hum Mol Genet. 2017;26:1–18. https://doi.org/10.1093/hmg/ddw363.
Hanigan TW, Aboukhatwa SM, Taha TY, Frasor J, Petukhov PA. Divergent JNK phosphorylation of HDAC3 in triple-negative breast cancer cells determines HDAC inhibitor binding and selectivity. Cell Chem Biol. 2017;24:1356-1367.e1358. https://doi.org/10.1016/j.chembiol.2017.08.015.
Choi H-K, Choi Y, Kang H, Lim E-J, Park S-Y, Lee H-S, et al. PINK1 positively regulates HDAC3 to suppress dopaminergic neuronal cell death. Hum Mol Genet. 2015;24:1127–41. https://doi.org/10.1093/hmg/ddu526.
Zhang F, Qi L, Feng Q, Zhang B, Li X, Liu C, et al. HIPK2 phosphorylates HDAC3 for NF-κB acetylation to ameliorate colitis-associated colorectal carcinoma and sepsis. Proc Natl Acad Sci. 2021;118: e2021798118. https://doi.org/10.1073/pnas.2021798118.
Nakayama T, Akagawa K. Transcription regulation mechanism of the syntaxin 1A gene via protein kinase A. Biochemical Journal. 2017;474:2465–73. https://doi.org/10.1042/BCJ20170249.
Lee H, Sengupta N, Villagra A, Rezai-Zadeh N, Seto E. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol Cell Biol. 2006;26:5259–69. https://doi.org/10.1128/MCB.01971-05.
Li Y, Liang R, Sun M, Li Z, Sheng H, Wang J, et al. AMPK-dependent phosphorylation of HDAC8 triggers PGM1 expression to promote lung cancer cell survival under glucose starvation. Cancer Lett. 2020;478:82–92. https://doi.org/10.1016/j.canlet.2020.03.007.
Ohnuki Y, Umeki D, Mototani Y, Jin H, Cai W, Shiozawa K, et al. Role of cyclic AMP sensor Epac1 in masseter muscle hypertrophy and myosin heavy chain transition induced by β2-adrenoceptor stimulation. J Physiol. 2014;592:5461–75. https://doi.org/10.1113/jphysiol.2014.282996.
Cohen TJ, Choi M-C, Kapur M, Lira VA, Yan Z, Yao T-P. HDAC4 regulates muscle fiber type-specific gene expression programs. Mol Cells. 2015;38:343–8. https://doi.org/10.14348/molcells.2015.2278.
Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–64. https://doi.org/10.1172/JCI27438.
Shimizu E, Nakatani T, He Z, Partridge NC. Parathyroid hormone regulates histone deacetylase (HDAC) 4 through protein kinase A-mediated phosphorylation and dephosphorylation in osteoblastic cells. J Biol Chem. 2014;289:21340–50. https://doi.org/10.1074/jbc.M114.550699.
He T, Huang J, Chen L, Han G, Stanmore D, Krebs-Haupenthal J, et al. Cyclic AMP represses pathological MEF2 activation by myocyte-specific hypo-phosphorylation of HDAC5. J Mol Cell Cardiol. 2020;145:88–98. https://doi.org/10.1016/j.yjmcc.2020.05.018.
Zhang H, Shao Z, Alibin CP, Acosta C, Anderson HD. Liganded peroxisome proliferator-activated receptors (PPARs) preserve nuclear histone deacetylase 5 levels in endothelin-treated Sprague-Dawley rat cardiac myocytes. PLoS ONE. 2014;9:e115258–e115258. https://doi.org/10.1371/journal.pone.0115258.
Zhao J-X, Yue W-F, Zhu M-J, Min Du. AMP-activated protein kinase regulates beta-catenin transcription via histone deacetylase 5. J Biol Chem. 2011;286:16426–34. https://doi.org/10.1074/jbc.M110.199372.
Pietruczuk P, Jain A, Simo-Cheyou ER, Anand-Srivastava MB, Srivastava AK. Protein kinase B/AKT mediates insulin-like growth factor 1-induced phosphorylation and nuclear export of histone deacetylase 5 via NADPH oxidase 4 activation in vascular smooth muscle cells. J Cell Physiol. 2019;234:17337–50. https://doi.org/10.1002/jcp.28353.
Meng Z-X, Gong J, Chen Z, Sun J, Xiao Y, Wang L, et al. Glucose sensing by skeletal myocytes couples nutrient signaling to systemic homeostasis. Mol Cell. 2017;66:332-344.e334. https://doi.org/10.1016/j.molcel.2017.04.007.
Gao C, Li X, Minh Lam Yu, Liu SC, Kao H-Y. CRM1 mediates nuclear export of HDAC7 independently of HDAC7 phosphorylation and association with 14–3-3s. FEBS Lett. 2006;580:5096–104. https://doi.org/10.1016/j.febslet.2006.08.038.
Lagman J, Sayegh P, Lee CS, Sulon SM, Jacinto AZ, Sok V, et al. G protein-coupled receptor kinase 5 modifies cancer cell resistance to paclitaxel. Mol Cell Biochem. 2019;461:103–18. https://doi.org/10.1007/s11010-019-03594-9.
Mazzetti S, De Leonardis M, Gagliardi G, Calogero AM, Basellini MJ, Madaschi L, et al. Phospho-HDAC6 gathers into protein aggregates in Parkinson’s disease and atypical Parkinsonisms. Front Neurosci. 2020;14:624–624. https://doi.org/10.3389/fnins.2020.00624.
Watabe M, Nakaki T. CK2 as anti-stress factor. Commun Integr Biol. 2012;5:278–80. https://doi.org/10.4161/cib.19473.
Williams KA, Zhang Mu, Xiang S, Chen Hu, Jheng-Yu Wu, Zhang S, et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J Biol Chem. 2013;288:33156–70. https://doi.org/10.1074/jbc.M113.472506.
Deribe YL, Wild P, Chandrashaker A, Curak J, Mirko S, Kalaidzidis Y, et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signaling. 2009;2:84. https://doi.org/10.1126/scisignal.2000576.
Zhang M, Yang X, Zimmerman RJ, Wang Q, Ross MA, Granger JM, et al. CaMKII exacerbates heart failure progression by activating class I HDACs. J Mol Cell Cardiol. 2020;149:73–81. https://doi.org/10.1016/j.yjmcc.2020.09.007.
Karwowska-Desaulniers P, Ketko A, Kamath N, Mary Pflum KH. Histone deacetylase 1 phosphorylation at S421 and S423 is constitutive in vivo, but dispensable in vitro. Biochem Biophys Res Commun. 2007;361:349–55. https://doi.org/10.1016/j.bbrc.2007.06.167.
Yang Qi, Tang J, Pei R, Gao X, Guo J, Chonghui Xu, et al. Host HDAC4 regulates the antiviral response by inhibiting the phosphorylation of IRF3. J Mol Cell Biol. 2019;11:158–69. https://doi.org/10.1093/jmcb/mjy035.
Wang AH, Kruhlak MJ, Wu J, Bertos NR, Vezmar M, Posner BI, et al. Regulation of histone deacetylase 4 by binding of 14–3-3 proteins. Mol Cell Biol. 2000;20:6904–12. https://doi.org/10.1128/MCB.20.18.6904-6912.2000.
Sinnett-Smith J, Ni Y, Wang J, Ming M, Young SH, Rozengurt E. Protein kinase D1 mediates class IIa histone deacetylase phosphorylation and nuclear extrusion in intestinal epithelial cells: role in mitogenic signaling. Am J Physiol Cell Physiol. 2014;306:C961–71. https://doi.org/10.1152/ajpcell.00048.2014.
Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, et al. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat Med. 2012;18:783–90. https://doi.org/10.1038/nm.2709.
Popov S, Kyriaki Venetsanou P, Chedrese J, Pinto V, Takemori H, Franco-Cereceda A, et al. Increases in intracellular sodium activate transcription and gene expression via the salt-inducible kinase 1 network in an atrial myocyte cell line. Am J Physiol-Heart Circul Physiol. 2012;303:H57–65. https://doi.org/10.1152/ajpheart.00512.2011.
Khai Huynh Q. Evidence for the phosphorylation of serine259 of histone deacetylase 5 by protein kinase Cδ. Arch Biochem Biophys. 2011;506:173–80. https://doi.org/10.1016/j.abb.2010.12.005.
Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, et al. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2010;107:15467–72. https://doi.org/10.1073/pnas.1000462107.
Harrison BC, Huynh K, Lundgaard GL, Helmke SM, Benjamin Perryman M, Mckinsey TA. Protein kinase C-related kinase targets nuclear localization signals in a subset of class IIa histone deacetylases. FEBS Lett. 2010;584:1103–10. https://doi.org/10.1016/j.febslet.2010.02.057.
Park CH, Lee JH, Lee MY, Lee JH, Lee BH, Kwang-Seok Oh. A novel role of G protein-coupled receptor kinase 5 in urotensin II-stimulated cellular hypertrophy in H9c2UT cells. Mol Cell Biochem. 2016;422:151–60. https://doi.org/10.1007/s11010-016-2814-y.
Dodge-Kafka KL, Gildart M, Li J, Thakur H, Kapiloff MS. Bidirectional regulation of HDAC5 by mAKAPβ signalosomes in cardiac myocytes. J Mol Cell Cardiol. 2018;118:13–25. https://doi.org/10.1016/j.yjmcc.2018.03.001.
Sato T, Verma S, Castro CD, Andrade MO, Campbell N, Wang JS, et al. A FAK/HDAC5 signaling axis controls osteocyte mechanotransduction. Nat Commun. 2020;11:3282–3282. https://doi.org/10.1038/s41467-020-17099-3.
Ran J, Liu M, Feng J, Li H, Ma H, Song T, et al. ASK1-mediated phosphorylation blocks HDAC6 ubiquitination and degradation to drive the disassembly of photoreceptor connecting cilia. Dev Cell. 2020;53:287-299.e285. https://doi.org/10.1016/j.devcel.2020.03.010.
Wang S, Li X, Parra M, Verdin E, Bassel-Duby R, Olson EN. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc Natl Acad Sci U S A. 2008;105:7738–43. https://doi.org/10.1073/pnas.0802857105.
Ha CH, Jhun BS, Kao H-Y, Jin Z-G. VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler Thromb Vasc Biol. 2008;28:1782–8. https://doi.org/10.1161/ATVBAHA.108.172528.
Chengzhuo Gao Yu, Liu ML, Kao H-Y. Histone deacetylase 7 (HDAC7) regulates myocyte migration and differentiation. Biochim Biophys Acta. 2010;1803:1186–97. https://doi.org/10.1016/j.bbamcr.2010.06.008.
Parra M, Mahmoudi T, Verdin E. Myosin phosphatase dephosphorylates HDAC7, controls its nucleocytoplasmic shuttling, and inhibits apoptosis in thymocytes. Genes Dev. 2007;21:638–43. https://doi.org/10.1101/gad.1513107.
Jensen ED, Gopalakrishnan R, Westendorf JJ. Bone morphogenic protein 2 activates protein kinase D to regulate histone deacetylase 7 localization and repression of Runx2. J Biol Chem. 2009;284:2225–34. https://doi.org/10.1074/jbc.M800586200.
Welker KR, Leng CA, Castañeda CD, Islam B, Haider SM, Christianson DW, et al. Phosphorylation of histone deacetylase 8: structural and mechanistic analysis of the phosphomimetic S39E mutant. Biochemistry. 2019;58:4480–93. https://doi.org/10.1021/acs.biochem.9b00653.
Imai S-I, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. https://doi.org/10.1038/35001622.
Yuan F, Xie Qi, Junbing Wu, Bai Y, Mao B, Dong Y, et al. MST1 promotes apoptosis through regulating Sirt1-dependent p53 deacetylation. J Biol Chem. 2011;286:6940–5. https://doi.org/10.1074/jbc.M110.182543.
Back JH, Rezvani HR, Zhu Y, Guyonnet-Duperat V, Athar M, Ratner D, et al. Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent Inhibition of sirtuin 1. J Biol Chem. 2011;286:19100–8. https://doi.org/10.1074/jbc.M111.240598.
Liu S, Zhou Z, Zhang L, Meng S, Li S, Wang X. Inhibition of SIRT2 by targeting GSK3β-mediated phosphorylation alleviates SIRT2 toxicity in SH-SY5Y cells. Front Cell Neurosci. 2019;13:148–148. https://doi.org/10.3389/fncel.2019.00148.
Naini SM, Sheridan AM, Force T, Shah JV, Bonventre JV. Group IVA cytosolic phospholipase A2 regulates the G2-to-M transition by modulating the activity of tumor suppressor SIRT2. Mol Cell Biol. 2015;35:3768–84. https://doi.org/10.1128/MCB.00184-15.
Nahhas F, Dryden SC, Abrams J, Tainsky MA. Mutations in SIRT2 deacetylase which regulate enzymatic activity but not its interaction with HDAC6 and tubulin. Mol Cell Biochem. 2007;303:221–30. https://doi.org/10.1007/s11010-007-9478-6.
Kang WK, Kim YH, Kang HA, Kwon K-S, Kim J-Y. Sir2 phosphorylation through cAMP-PKA and CK2 signaling inhibits the lifespan extension activity of Sir2 in yeast. Elife. 2015;4: e09709. https://doi.org/10.7554/eLife.09709.
Thirumurthi U, Shen J, Xia W, Labaff AM, Wei Y, Li C-W, et al. MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci Signaling. 2014;7:71. https://doi.org/10.1126/scisignal.2005076.
Gao T, Li M, Guanqun Mu, Hou T, Zhu W-G, Yang Y. PKCζ phosphorylates SIRT6 to mediate fatty acid β-oxidation in colon cancer cells. Neoplasia. 2019;21:61–73. https://doi.org/10.1016/j.neo.2018.11.008.
Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, et al. JNK phosphorylates SIRT6 to stimulate DNA double-strand break repair in response to oxidative stress by recruiting PARP1 to DNA breaks. Cell Rep. 2016;16:2641–50. https://doi.org/10.1016/j.celrep.2016.08.006.
Hong Wu, Zeng H, Dong A, Li F, He H, Senisterra G, et al. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS ONE. 2013;8:e83737–e83737. https://doi.org/10.1371/journal.pone.0083737.
Wen L, Chen Z, Zhang F, Cui X, Sun W, Geary GG, et al. Ca2+/calmodulin-dependent protein kinase kinase β phosphorylation of Sirtuin 1 in endothelium is atheroprotective. Proc Natl Acad Sci U S A. 2013;110:E2420–7. https://doi.org/10.1073/pnas.1309354110.
Tatomir A, Rao G, Boodhoo D, Vlaicu SI, Beltrand A, Anselmo F, et al. Histone deacetylase SIRT1 mediates C5b–9-induced cell cycle in oligodendrocytes. Front Immunol. 2020;11:619–619. https://doi.org/10.3389/fimmu.2020.00619.
Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, De Cabo R, et al. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS ONE. 2009;4:e8414–e8414. https://doi.org/10.1371/journal.pone.0008414.
Wang S, Yang Y, He X, Yang L, Wang J, Xia S, et al. Cdk5-mediated phosphorylation of Sirt1 contributes to podocyte mitochondrial dysfunction in diabetic nephropathy. Antioxid Redox Signal. 2020;34:171–90. https://doi.org/10.1089/ars.2020.8038.
Senthil KKJ, Gokila VM, Mau J-L, Lin C-C, Chu F-H, Wei C-C, et al. A steroid like phytochemical Antcin M is an anti-aging reagent that eliminates hyperglycemia-accelerated premature senescence in dermal fibroblasts by direct activation of Nrf2 and SIRT-1. Oncotarget. 2016;7:62836–61. https://doi.org/10.18632/oncotarget.11229.
Gao Z, Zhang J, Kheterpal I, Kennedy N, Davis RJ, Ye J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J Biol Chem. 2011;286:22227–34. https://doi.org/10.1074/jbc.M111.228874.
Choi SE, Kwon S, Seok S, Xiao Z, Lee K-W, Kang Y, et al. Obesity-linked phosphorylation of SIRT1 by casein kinase 2 inhibits its nuclear localization and promotes fatty liver. Mol Cell Biol. 2017;37:e00006-00017. https://doi.org/10.1128/MCB.00006-17.
Wang W, Li F, Yuanming Xu, Wei J, Zhang Y, Yang H, et al. JAK1-mediated Sirt1 phosphorylation functions as a negative feedback of the JAK1-STAT3 pathway. J Biol Chem. 2018;293:11067–75. https://doi.org/10.1074/jbc.RA117.001387.
Lee C-W, Wong L-Y, Tse E-T, Liu H-F, Leong V-L, Lee J-F, et al. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012;72:4394–404. https://doi.org/10.1158/0008-5472.CAN-12-0429.
Guo X, Kesimer M, Tolun G, Zheng X, Qing Xu, Jing Lu, et al. The NAD(+)-dependent protein deacetylase activity of SIRT1 is regulated by its oligomeric status. Sci Rep. 2012;2:640–640. https://doi.org/10.1038/srep00640.
Utani K, Haiqing Fu, Jang S-M, Marks AB, Smith OK, Zhang Ya, et al. Phosphorylated SIRT1 associates with replication origins to prevent excess replication initiation and preserve genomic stability. Nucleic Acids Res. 2017;45:7807–24. https://doi.org/10.1093/nar/gkx468.
Shan P, Fan G, Sun L, Liu J, Wang W, Chen Hu, et al. SIRT1 functions as a negative regulator of eukaryotic poly(A)RNA transport. Curr Biol. 2017;27:2271-2284.e2275. https://doi.org/10.1016/j.cub.2017.06.040.
Huang Y, Jianlin Lu, Zhan Li, Wang M, Shi R, Yuan X, et al. Resveratrol-induced Sirt1 phosphorylation by LKB1 mediates mitochondrial metabolism. J Biol Chem. 2021;297:100929–100929. https://doi.org/10.1016/j.jbc.2021.100929.
Lee HR, Shin HK, Park SY, Kim HY, Lee WS, Rhim BY, et al. Attenuation of β-amyloid-induced tauopathy via activation of CK2α/SIRT1: Targeting for cilostazol. J Neurosci Res. 2014;92:206–17. https://doi.org/10.1002/jnr.23310.
Dehennaut V, Loison I, Pinte S, Leprince D. Molecular dissection of the interaction between HIC1 and SIRT1. Biochem Biophys Res Commun. 2012;421:384–8. https://doi.org/10.1016/j.bbrc.2012.04.026.
Conrad E, Polonio-Vallon T, Meister M, Matt S, Bitomsky N, Herbel C, et al. HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 2016;23:110–22. https://doi.org/10.1038/cdd.2015.75.
Choi YH, Kim H, Lee SH, Jin Y-H, Lee KY. Src regulates the activity of SIRT2. Biochem Biophys Res Commun. 2014;450:1120–5. https://doi.org/10.1016/j.bbrc.2014.06.117.
North BJ, Verdin E. Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J Biol Chem. 2007;282:19546–55. https://doi.org/10.1074/jbc.M702990200.
Hussein UK, Ahmed AG, Song Y, Kim KM, Moon YJ, Ahn A-R, et al. CK2α/CSNK2A1 induces resistance to doxorubicin through SIRT6-mediated activation of the DNA damage repair pathway. Cells. 2021;10:1770. https://doi.org/10.3390/cells10071770.
Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A. 1964;51:786–94. https://doi.org/10.1073/pnas.51.5.786.
Zhang H, Zha X, Tan Yi, Hornbeck PV, Mastrangelo AJ, Alessi DR, et al. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J Biol Chem. 2002;277:39379–87. https://doi.org/10.1074/jbc.M206399200.
Acknowledgements
We thank Yanfa Sun for revision and suggestions for preparing this manuscript. We also thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This research has been supported by School of Biomedical Engineering, Shenzhen University Health Science Center with Initial Scientific Research Fund.
Author information
Authors and Affiliations
Contributions
PZ prepared the original draft. SM discussed and revised the manuscript. PZ supervised the project and contributed to the final version of the manuscript. All authors have made a direct and intellectual contribution to the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors without a commercial or financial relationship, declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, P., Malik, S. The phosphorylation to acetylation/methylation cascade in transcriptional regulation: how kinases regulate transcriptional activities of DNA/histone-modifying enzymes. Cell Biosci 12, 83 (2022). https://doi.org/10.1186/s13578-022-00821-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-022-00821-7