
Abstract
Background
The mechanisms leading to retinal ganglion cell (RGC) death after optic nerve injury have not been fully elucidated. Current evidence indicates that microglial activation and M1- and M2-like dynamics may be an important factor in RGC apoptosis after optic nerve crush (ONC). Semaphorin3A (Sema3A) is a classic axonal guidance protein,which has been found to have a role in neuroinflammation processes. In this study, we investigated the contribution of microglial-derived Sema3A to progressive RGC apoptosis through regulating paradigm of M1- and M2-like microglia after ONC.
Method
A mouse ONC model and a primary microglial-RGC co-culture system were used in the present study. The expression of M1- and M2-like microglial activation markers were assessed by real-time polymerase chain reaction (RT-qPCR). Histological and Western blot (WB) analyses were used to investigate the polarization patterns of microglia transitions and the levels of Sema3A. RGC apoptosis was investigated by TUNEL staining and caspase-3 detection.
Results
Levels of Sema3A in the mouse retina increased after ONC. Treatment of mice with the stimulating factor 1 receptor antagonist PLX3397 resulted in a decrease of retinal microglia. The levels of CD16/32 (M1) were up-regulated at days 3 and 7 post-ONC. However, CD206 (M2) declined on day 7 after ONC. Exposure to anti-Sema3A antibodies (anti-Sema3A) resulted in a decrease in the number of M1-like microglia, an increase in the number of M2-like microglia, and the amelioration of RGC apoptosis.
Conclusions
An increase in microglia-derived Sema3A in the retina after ONC partially leads to a continuous increase of M1-like microglia and plays an important role in RGC apoptosis. Inhibition of Sema3A activity may be a novel approach to the prevention of RGC apoptosis after optic nerve injury.
Similar content being viewed by others


Introduction
Optic nerve injury resulting in progressive retinal ganglion cell (RGC) death is a serious and irreversible cause of blindness [1]. Retinal neuroinflammation is a leading factor limiting the recovery of RGC after primary optic nerve impairment [2, 3]. Microglia makes up a significant portion of the resident glial population in the retina and are key mediators of neuroinflammation [2, 4, 5]. In previous studies, we have investigated the essential role of microglia in triggering retinal inflammation [6, 7]. Optic nerve injury is followed by migration, activation, and proliferation of microglia [8, 9]. Activated microglia, including retinal microglia, can be divided into two major types: pro-inflammatory type M1-like microglia and anti-inflammatory type M2-like microglia [10, 11]. M1-like microglia secrete pro-inflammatory cytokines, including TNFα, IL-23, IL-1, and IL-12, which contribute to neuronal damage. M2-like microglia, activated by IL-4 or IL-13, are anti-inflammatory and promote tissue repair and wound healing [4]. Sufficient evidence has suggested that reciprocal transformation of M1-like and M2-like microglia occurs under certain conditions. This reciprocal transformation can lead to either the increase or the subsidence of neuronal inflammation [12,13,14]. However, the initiating factors governing the polarization of microglia in RGC secondary injury is still not fully elucidated.
Sema3A is involved in the negative regulation of neuronal axon and dendrite polarity [15,16,17,18,19] as well as acting as an effective regulator in some essential stages of inflammation and the immune response [16, 20,21,22]. For example, patients suffering from late-stage proliferative diabetic retinopathy have elevated levels of vitreous Sema3A, which attracts neuropilin-1(NRP-1) positive mononuclear phagocytes [23]. Sema3A also activates the transcription factor NF-κB via the TLR4 signaling pathway in macrophages to increase pro-inflammatory cytokine production and to augment inflammatory responses in a sepsis-induced cytokine storm [24]. Previous studies have confirmed that the level of Sema3A (mainly distributed in the RGC layer of the retina) increases significantly during the 3 days after optical nerve crush (ONC). Its expression can persist for 14–28 days after injury [19, 25]. Anti-Sema3A antibodies (anti-Sema3A) can rescue RGCs from apotosis that occurs after optic nerve axotomy [26]. Neuropilin 1 (Nrp1) is a receptor of Sema3A. It has been shown that levels of microglia in Nrp1-floxed mouse retina are significantly lower than in the retinas of WT mice [23]. Nrp1+ microglia are present throughout the retina during vascular development, although they are more prevalent in non-vascularized retinal tissue [27]. This suggests a potential paracrine effect of microglial Sema3A/Nrp1 signaling in retinal development and pathogenesis. However, the role of Sema3A in retinal neuroinflammation and its interaction with retinal microglia remain unclear.
In current study, we investigate the regulatory effect of Sema3A on M1- and M2-like microglia dynamics in vivo and in vitro. In a mouse ONC model, we demonstrate that a significant amount of Sema3A is secreted by retinal microglia post-ONC. The dynamic changes of M1/M2-like microglia and neuronal apoptosis were evaluated. The levels of M1/M2-like microglia and the extent of RGC apoptosis were further verified in a co-culture model of primary microglia and RGCs. We found that the increases of Sema3A expression increased the pro-inflammatory M1-like phenotype and decreased the anti-inflammatory M2-like phenotype. In consequence, as pro-inflammatory cytokine release increased, RGC underwent apoptosis. anti-Sema3A treatment in vitro and in vivo decreased the M1-like phenotype and increased the M2-like phenotype, contributed to the amelioration of RGC apoptosis.
Materials and methods
Animals and surgery
All animals were treated according to the ARVO Statement for Use of Animals in Ophthalmic and Vision Research. Experimental procedures were approved by the Institutional Animal Care and Use Committee of the Army Medical University.
C57BL/6 mice were purchased from the Army Medical University. The mice were housed at an animal care facility with a 12/12-h light/dark cycle and ad libitum access to food and water. The classic model of optic nerve crush (ONC) was performed as previously described [6, 28]. Adult C57BL/6J mice (male, aged 6–8 weeks; weight: 20–24 g) were anesthetized by intraperitoneal injection of pentobarbital sodium (30 mg/kg). The exposed optic nerve of the left eye was crushed for 10 s at a distance of 1.5 mm from the eye globe with ultrafine self-closing forceps without damage to the retinal vessels or the blood supply. The right eye was used as a sham control (Fig. 1a). The mice were killed at 3 and 7 days post-ONC. Primary microglia were cultured from newborn C57BL/6 mouse cortex for the in vitro studies.

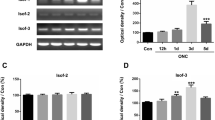
Sema3A is increased in ONC mice retina and is accompanied by microglia activation. a Schematic diagram of the ONC model demonstrating the site of ONC. b Western blot of Sema3A and Iba1 expression in retina at 3 and 7 days (3D and 7D) after ONC. β-actin was used as a loading control. c, d Quantitative western blot analysis indicates a significant up-regulation of Sema3A and Iba1 (Mean ± SEM, n = 10). e, f mRNA expression of Sema3A and its receptors in retina at 3 and 7 days post-ONC as measured by quantitative RT-qPCR. Gene expressions were normalized to β-actin. The results are representative of ten independent experiments and are shown as mean ± SEM (n = 10) (G) Representative confocal images showing immunostaining of Sema3A and Iba1 in retinal cryosections. Sema3A was mainly detected in the ganglion cell layer (GCL) and Iba1 was mainly distributed in GCL, IPL and OPL. Scale bar = 250 μm
Microglia depletion
The colony-stimulating factor 1 receptor antagonist PLX3397 was used for the pharmaceutical depletion of microglia. Male mice aged 8–10 weeks were given AIN-76 A chow containing 290 mg/kg PLX-3397 [29, 30]. Age-matched controls were given AIN-76 A chow without PLX-3397. After 3 days of diet administration, the mice underwent the ONC procedure and were sacrificed.
Intravitreous injections
Adult mice received intravitreous injections of anti-Sema3A (1 µl, neutralizing antibody) [31] in their left eyes before the ONC procedure. The right eyes were used as sham controls and received saline injections. The intravitreal injection procedure was performed as described previously [32] without elevated intraocular pressure detected in any of the eyes after surgery.
Immunostaining
Primary antibodies included anti-Sema3A (1:100, rabbit monoclonal, Abcam, ab23393), anti-CD16/32 (1:100, goat polyclonal, R&D, AF1460), anti-CD206 (1:100, mouse monoclonal, Abcam, ab8918), anti-Iba1 (1:100, rabbit monoclonal, Abcam, ab178846), and anti-P2RY12 (5 µg/ml, mouse monoclonal, Biolegend, 848001). Secondary antibodies included anti-mouse IgG, anti-goat IgG, anti-Rat IgG, and Alexa Fluor 488, 594, and 647 conjugated to anti-rabbit IgGs (1:1000, Invitrogen). Mouse retinas were fixed in 4 % paraformaldehyde and cryo-sectioned at a thickness of 10 μm. Retina cryosections and fixed primary microglia were incubated with 0.1 % Triton X-100 (Sigma-Aldrich) in PBS at room temperature for 10 min, then incubated with primary antibodies at 4 °C overnight. Secondary antibodies were applied for 1 h at room temperature. The slides were mounted after nuclei counterstaining (Hoechst 1:2000). Images were taken by an SP-8 confocal microscope (Leica, Germany). For quantification of immunofluorescence, CD16/32+, CD206+, and Iba1+ cells were counted in 3 s. (10 μm thick) of retinal tissue per mouse. The mean number of cells per field was determined. All data were expressed as mean (n = 3 mice per group) ± SEM. The sections were selected randomly and were analyzed under a Leica SP8 confocal laser scanning microscope. Quantification of cell numbers was analyzed using ImageJ software.
Whole-mounted retinal immunofluorescence
Retinas were fixed in 4% PFA for 1 h, then dissected as whole-mounts. An orientation record was maintained as previously described [28]. Briefly, the intact retinas were incubated in PBS containing 5% BSA and 3% Triton-X-100 at 4 °C overnight. Primary mouse monoclonal anti-Tuj1 antibody (Covance, Cat. MMS435P) was added (1:500) and incubated overnight at 4 °C. The retinas were incubated with secondary anti-mouse IgG antibodies conjugated to Alexa Fluor 488 overnight at 4 °C and examined by confocal microscopy with the appropriate filters (SP8, Leica, Germany).
Microglial cell culture
Mixed glial cultures were isolated from the cerebral cortices of 1-day-old C57BL/6 mice as previously described [33]. Cells were dissociated under aseptic conditions, suspended in DMEM-F12 with 10 % FBS, and seeded at a density of 62,500 cells/cm2 [34]. Cells were cultured at 37 °C and 5 % CO2 for 15 days. Mixed glial cells were then shaken at 200 rpm in a rotary incubator overnight at 37 °C to dissociate the cells. The suspended cells were collected and replanted in DMEM-F12 with 10 % FBS. The purity of the microglia was confirmed by immunostaining using anti-Iba1. Then, three independent microglial cultures were treated for 6 h with vehicle (cell culture medium) Sema3A (R&D) and 100 ng/mL of LPS (026:B6 Escherichia coli serotype, Sigma Aldrich), and primary microglia were harvested after 1 day and 3 days for further experiments.
Immunoblotting
Western blot (WB) analysis was performed on protein extracted from post-ONC mouse retinas, primary microglia, and RGCs as previously described [28]. Washed and blocked polyvinylidene fluoride membranes from WBs were incubated with primary antibodies. Primary antibodies included anti-Sema3A (1:1000, rabbit monoclonal, Abcam, ab23393), anti-CD16/32 (1:1000, goat polyclonal, R&D, AF1460), anti-CD206 (1:500, rabbit monoclonal, Abcam, ab125028), anti-Iba1 (1:1000, rabbit monoclonal, Abcam, ab178846), anti-SMI32(1:1000, rabbit monoclonal, Abcam, ab207176), and anti-Map2 (1:1000, rabbit monoclonal, CST, #4542). Membranes were then incubated with horseradish peroxidase-conjugated anti-rabbit, anti-mouse, or anti-goat IgG secondary antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, United States) in TBS-T for 1.5 h at room temperature. ECL-Plus reagent (Bioground) was applied to the membranes and chemiluminescence visualized by a Fluor-S-max imager (Bio-Rad).
RT-qPCR
Total RNA was extracted from retinas and primary microglia using the RNeasy RNA isolation kit (Qiagen). Reverse transcription reactions were performed using the PrimeScript™ RT reagent Kit (Takara) according to the company’s instructions. RT-qPCR was performed using the SYBR method (Quanta BioSciences) on a CFX96 Real-Time PCR Detection/C1000 Thermal Cycler system (Bio-Rad). The results were normalized to β-actin. Relative quantification was calculated using the 2−ΔΔCq method. The primer sequences used are listed in Table 1.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.0 software (GraphPad, La Jolla, CA, U.S.A.) and reported as mean ± S.E.M. Student’s t-test, one-way ANOVA, and least significant difference post-hoc tests were also employed. All experiments were performed independently with n > 3. P values ≤ 0.05 were considered to be statistically significant. Significant values were marked as * (P values below 0.05), ** (P values below 0.01), and *** (P values below 0.001).
Results
After optic nerve crush, Sema3A increased, accompanied by microglial activation
WB results showed that Sema3A expression in the mouse retina increased significantly and peaked at 3 days post-ONC. On day 7 post-ONC, Sema3A levels declined but remained higher than the control (Fig. 1b), consistent with previous results [19]. At 3- and 7-days post-injury, there was a concomitant increase in expression of Iba1, indicating that microglia were significantly activated (Fig. 1d). Based on the RT-qPCR analysis, there was a significant increase in mRNA expression of Sema3A and its receptor Nrp1 at 3 days and 7 days post-ONC (Fig. 1e, f). Sema3A immunofluorescence staining was primarily located in the Ganglion Cell layer (GCL) (Fig. 1g) while Iba1 was distributed throughout the GCL, inner plexiform (IPL), and outer plexiform (OPL) layers. Detailed morphological analysis at 3- and 7-days post-ONC indicated that Iba1+ cells adopted a more irregular soma shape, with increased cell body size and retracted processes.
Microglia depletion reduces Sema3A levels after ONC
Sema3A can be produced by stressed neurons, activated astrocytes, and microglia [35,36,37]. To determine the contribution of Sema3A from microglia in the mouse model, mice treated with PLX3397, which depleted microglia in retina [30, 38, 39], underwent ONC. Immunofluorescence staining revealed a significant 50 % reduction in retinal microglia after PLX3397 treatment (Fig. 2a, b). WB results showed that Sema3A and Iba1 levels in retinal microglia from PLX-3397 treated mice were both significantly lower than that in untreated mice (Fig. 2c, d), suggesting that microglia serve as one of the sources of Sema3A protein. However, the remaining microglia were activated, similar to microglia in untreated ONC mice (Fig. 2e). Immunostaining of Sema3A and Iba1 in retinal cells showed that Sema3A protein co-localized with Iba1 in microglial cells (Fig. 2f).

Sema3A is significantly reduced in microglia depleted mice retina. a Representative confocal images showing immunostaining of Iba1 (red arrows) with Hoechst nuclear staining. Scale bar = 250 μm. b Quantification of positive Iba1 in sham and microglia depletion groups (n = 4). Iba1 staining was reduced after PLX-3397 treatment. c, d Western Blot shows the expression levels of Sema3A and Iba1 in retina after microglia depletion (MD). β-actin was used as a loading control. Quantitative analysis indicates a significant down-regulation of Sema3A in MD group (Mean ± SEM, n = 5). e Representative confocal images showing immunostaining of Iba1. Scale bar = 25 μm. f Representative confocal images showing immunostaining of Sema3A (green) and Iba1(red) in retinal cryosections. Scale bar = 25 μm
M1-like microglia levels increase and M2-like microglia levels decrease in retinal tissues post-ONC, similar to cultured primary microglia treated with Sema3A
There was robust induction of the M1-like marker CD16/32 and the M2-like marker CD206 in the retina at 3 days post-ONC (Fig. 3a–c). At 7 days post-ONC, the level of CD16/32 (M1-like) was observed to be higher than that observed at 3 days post-ONC. However, the expression levels of CD206 were greatly reduced (Fig. 3a–c), which was confirmed by immunofluorescence staining (Fig. 3d). RT-qPCR revealed that cytokines secreted by M1-like microglia, including IL1-β, IL-6, iNOS, and TNF-α, were significantly up-regulated, while those of M2-like microglia, such as CD206 and Ym1, were down-regulated (Fig. 3e). Cell staining with P2ry12 co-labeled with CD16/32 and CD206 was used to distinguish resident microglia from infiltrating macrophages. The results suggested that microglia polarization after optic nerve injury mainly occurred in resident microglia (Fig. 4).

At 7 days post-ONC, activated microglia show significantly increased M1-like phenotype and decreased in M2-like phenotype. a Expression of CD16/32 and CD206 in the retina at 3 and 7 days post-ONC. b, c Quantitative analysis indicates a significant increase of CD16/32and CD206 at 3 days post-injury. Level of CD16/32 continuous to increase at 7 days post-ONC, while CD206 decreased significantly. (Mean ± SEM, n = 12). d Representative confocal images showing immunofluorescent staining of CD16/32 (red), CD206 (green) and Iba1 (white) with Hoechst (blue) nuclear staining in retina cells of sham, ONC3D, and ONC7D groups. Scale bar = 25 μm. Quantitative analysis indicates a significant increase of CD206 at 3 days post-injury, and an increase of CD32 both at 3 days and 7days post-injury. Ratio of the number of double positive CD32/Iba1 cells vs. Iba1 positive cells (bar graph). Ratio of the number of double positive CD206/Iba1 cells vs. Iba1 positive cells (bar graph). e mRNA expression of cytokines secreted by microglia in retina at 7 days post-ONC measured by RT-qPCR. Gene expressions were normalized to β-actin. The results are representative of three independent experiments and are shown as mean ± SEM (n = 3)

Microglia polarization after optic nerve injury mainly occurred in resident microglia. Representative confocal images and quantitative analysis showing immunofluorescent staining of CD206 (green), P2ry12(white) CD16/32 (red) with Hoechst (blue) nuclear staining in retina cells at 3 days and 7 days post-ONC, Scale bar = 25 μm
In vitro studies demonstrated that the treatment of primary microglial cells with Sema3A significantly increased the population of M1-like microglia (Fig. 5a). Double-immunofluorescence staining of CD16/32 and CD206 in primary microglia showed that 1 day after Sema3A and LPS treatment, CD16/32 + and CD206 + cells were both increased. However, 3 days after Sema3A and LPS treatments, an extra immunofluorescent staining in primary microglia showed increased M1-like microglia and decreased M2-like microglia, which supported the RT-qPCR results (Fig. 5b, c). These in vitro results indicate that Sema3A is involved in the pathological processes that increase the population of M1-like microglia and decrease the population of M2-like microglia after ONC.

Increased M1-like and decreased M2-like phenotypes observed in primary microglia treated with Sema3A. a mRNA expression of cytokines secreted by primary microglia treated with Sema3A. Gene expressions were normalized to β-actin. The results are representative of three independent experiments and are shown as mean ± SEM (n = 3). b, c Representative confocal images and quantitative analysis showing immunofluorescent staining of CD16/32 (red, white arrow) and CD206 (green, white arrow) with Hoechst (blue) nuclear staining in primary microglia 1 day and 3 days after Sema3A and LPS treatment. Scale bar = 100 μm
Anti-Sema3A administration significantly reduces RGCs apoptosis in vivo and in vitro
Sema3A has been shown to rescue RGCs from cell death following optic nerve axotomy [26], yet the mechanisms vary. We performed intravitreal injections of anti-Sema3A and observed that, on day 7 post-ONC, the increase in the number of M1-like polarized cells slowed but the number of M2-like cells continued to increase (Fig. 6a). In the absence of anti-Sema3A treatment, whole-mounted retinal immunofluorescence staining with anti-Tuj1 antibody indicated that the number of RGCs in the retina had decreased significantly at days 3 and 7 post-ONC. Although no significant difference in RGC numbers was detected between the ONC3D group (3-day post-ONC) and the anti-Sema3A + ONC3D group, the numbers of RGCs in the ONC7D group were significantly lower than that of the anti-Sema3A + ONC7D group (Fig. 6b). These results indicate that intravitreal treatment with anti-Sema3A reduces M1-like microglia polarization, increases M2-like level, and reduces the loss of RGCs following ONC.

Intravitreal injection of anti-Sema3A increased M2-like microglia and decreased RGCs apoptosis in the retina. a Representative confocal images and quantitative analysis showing immunofluorescent staining of CD206 (green), CD16/32 (red) and Iba1 (white) with Hoechst (blue) nuclear staining in retina from mice post-injury at 7 days with or without anti-Sema3A treatment. Scale bar = 100 μm. Quantitative analysis indicates a significant increase of CD206 with anti-Sema3A treatment and CD32 without anti-Sema3A at 7 days post-injury (Mean ± SEM, n = 5). Ratio of the number of double positive CD32/Iba1 cells vs. Iba1 positive cells (bar graph). Ratio of the number of double positive CD206/Iba1 cells vs. Iba1 positive cells (bar graph). b Representative confocal images of anti-Tuj1 and the quantitative analysis (n = 4) show reduction in RGCs (red arrows) and axons (white arrows) after ONC. anti-Sema3A alleviated RGCs apoptosis. Scale bar = 25 μm. c, d Expression of Pro-caspase3 and cleaved-caspase3 in the retina. Quantitative analysis indicates a significant increase of cleaved-caspase3 in the ONC group, which is reduced after anti-Sema3A injection (n = 6). β-actin was used as a loading control
We measured cleaved caspase-3 levels to investigate the anti-apoptotic effect of anti-Sema3A treatment on RGCs on day 7 post-ONC [28, 40]. Cleaved caspase-3 protein levels were significantly reduced in the anti-Sema3A + ONC7D group, indicating that anti-Sema3A protected RGCs from cleaved caspase-3-induced apoptosis (Fig. 6c, d). TUNEL staining and Bax/BCL-2 blotting also suggest the protection of RGCs by anti-Sema3A (Additional file 1: Fig. S1A, B).
Both Sema3A- and LPS-treated microglia induced RGCs apoptosis in an in vitro primary microglial-RGC co-culture model (Additional file 1: Fig. S1A). Anti-Sema3A + Sema3A-treated co-cultures exhibited decreased M1-like microglia levels, increased M2-like microglia levels, and decreased RGCs apoptosis (Additional file 11 Fig. S1B). In the Sema3A- and LPS-treated co-cultures, SMI32 and MAP2 proteins were significantly lower than controls. In the anti-Sema3A treated co-cultures, levels of cleaved caspase-3 were significantly reduced compared to Sema3A- and LPS-treated groups (Fig. 7d, e).

Anti-Sema3A treatment in primary microglia increased M2-like microglia and decreased RGCs apoptosis in primary microglia and RGC co-culture. a Schematic diagram of primary microglia and RGC co-culture system. b Representative confocal images showing immunofluorescent staining of CD206 (green) and CD16/32 (red) with Hoechst (blue) nuclear staining in Sema3A treated primary microglia with or without anti-Sema3A treatment. Scale bar = 100 μm. Quantitative analysis indicates a significant increase of CD206 with anti-Sema3A treatment. (Mean ± SEM, n = 6). c TUNEL staining of RGCs in control, Sema3A + anti-Sema3A, Sema3A, and LPS groups. Scale bar = 100 μm. d Quantification of TUNEL-positive cells vs. Hoechst staining (n = 4). e–h Western Blot and quantitative analysis show expression of SMI32, MAP2, pro-caspase3, and cleaved-caspase3 in RGCs co-cultured with microglia. β-actin was used as a loading control. (Mean ± SEM, n = 5)
Discussion
Visual impairment caused by optic neuropathy is a primary cause of blindness. The mechanisms of irreversible RGCs death from conditions such as optic nerve injury and glaucoma remain unclear. Previous investigations have suggested that Sema3A plays a crucial role in the development of retinal inflammation and RGCs apoptosis post-ONC [24, 26].
Sema3A is continuously expressed in the retina from development through adulthood [41, 42]. It is known that Sema3A is significantly up-regulated in the retina post-ONC [19]. Our results demonstrated that the peak expression of retinal Sema3A occurs at day 3 post-ONC and continues to be expressed for 14–28 days post-injury [43] (Fig. 1b–f). Astrocytes, endothelial cells, and activated microglia contribute to the increase of Sema3A secretion [44, 45] (Fig. 2). This is confirmed by the observation that as the number of retinal microglia declined with PLX3397 treatment, a significant decrease of Sema3A was observed in the retina.
Sema3A causes the collapse of the growth cone of regenerated axons and inhibits axon elongation by binding to the receptor complex Nrp1/PlexinA [19]. Blocking Sema3A binding to Nrp1/PlexinA effectively reduces growth cone collapse of the dorsal root ganglion after spinal cord injury and promotes nerve regeneration in a rat olfactory nerve axotomy model [46]. Emerging evidence suggests that Sema3A regulates B and T lymphocytes and contributes to the progression and development of diseases such as Systemic Lupus Erythematosus and cancer [47, 48]. Sema3A also plays a vital role in the migration and transportation of dendritic cells and the recruitment of mononuclear phagocytes [49]. Our results show that microglia are activated and the number of pro-inflammatory M1-like cells are increased with increasing retinal Sema3A. At the same time, the number of anti-inflammatory M2-like microglia is decreased, accommodating the microglial polarization M1-/M2-like dynamic response and primarily occurring in resident microglia (Figs. 3, 4 and 5). This finding extends the current understanding of the interaction between Sema3A and microglia and demonstrates the important role of Sema3A in neuroinflammation following optic nerve injury.
Anti-Sema3A inhibition of Sema3A significantly reduced the rate of M1-like cell polarization and increased the rate of M2-like microglia polarization post-ONC in the retina (Fig. 6a). Similar results were obtained in vitro using primary microglia cultured with Sema3A protein (Fig. 7). RGCs apoptosis in mouse retina post-ONC and in primary microglial-RGCs co-culture was reduced by anti-Sema3A treatment (Figs. 5, 6 and 7, Additional file 1: Fig. S1). These findings demonstrate that elevated Sema3A induces RGCs apoptosis and inhibits the regeneration of RGCs axons by regulating M1/M2-like microglia dynamics post-ONC (Figs. 1, 3, 4, 5, 6 and 7). Our results shed light on part of the RGCs apoptosis pathway induced by Sema3A post-ONC and suggest that Sema3A regulation is a possible new approach for the treatment of optic nerve injury.
Microglia can be polarized along a continuum toward an inflammatory (M1) or a non-inflammatory (M2) state and microglial reciprocal transformation may participate in the progression of neurodegenerative diseases, such as Alzheimer’s disease [14, 50]. After spinal cord injury, TNF prevents the phagocytosis-mediated conversion of M1-like to M2-like cells and mediates an increase in iron-induced changes of IL-4-polarized M2 cells to M1 cells, which is detrimental to recovery [13, 51]. Our results show that an increase in Sema3A levels post-ONC leads to a significant up-regulation of M1-like microglia and a concomitant decrease in M2-like microglia, consistent with the dynamic changes of M1/M2 phenotype after traumatic brain injury and stroke. The regulation of M1/M2-like microglia dynamics by Sema3A may play a role in M1/M2 transformation in the retina.
The signaling pathways through which Sema3A regulates M1/M2-like microglia transformation requires further study. Evidence has shown that the NF-κB signaling cascade regulates the production of pro-inflammatory mediators and contributes to the M1/M2-like microglia transition [52, 53]. Sema3A enhances LPS-induced acute kidney injury by increasing Rac1 (a key factor for activation of NF-κB) and p65 and augments LPS-induced macrophage activation and cytokine production in a plexin-A4–dependent manner [24, 54]. Previous studies confirmed that retinal TLR4 expression is increased in a mouse model of ONC [55]. TRIF knockout (KO) inactivates the NF-κB signaling pathway and reduces pro-inflammatory cytokine release by inhibiting activation of microglia in mice retina [6]. The mechanism of endogenous degeneration of RGCs remains unclear. We speculate that Sema3A/Nrp1 is involved in activating the TLR4/NF-κB signaling pathway, inducing the polarization of microglia toward the M1-like phenotype. However, the molecular pathways remain to be verified. In brief, we find that Sema3A directly affects neuron polarity and inhibits their regeneration, participates in M1/M2-like microglia dynamics regulation, and increases RGCs apoptosis. Sema3A has both direct and indirect effects on RGCs, presenting a potential therapeutic target for optic nerve injury treatment.
Conclusions
Our results provide the first evidence that retinal microglia are an important source of Sema3A protein post-ONC. Sema3A is associated with an increase in pro-inflammatory M1-like microglia, a decrease in anti-inflammatory M2-like cells, and increased RGCs apoptosis. Inhibition of Sema3A ameliorates RGCs apoptosis and promotes RGCs regeneration. Therefore, Sema3A could be a new therapeutic target for RGCs protection after optic nerve injury.
Availability of data and materials
All data in this study are included in this manuscript.
References
Maes ME, Schlamp CL, Nickells RW. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog Retin Eye Res. 2017;57:1–25.
Rathnasamy G, Foulds WS, Ling EA, Kaur C. Retinal microglia - A key player in healthy and diseased retina. Prog Neurobiol. 2019;173:18–40.
Orr MB, Gensel JC. Spinal cord injury scarring and inflammation: therapies targeting glial and inflammatory responses. Neurotherapeutics. 2018;15(3):541–53.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–68.
Silverman SM, Wong WT. Microglia in the retina: roles in development, maturity, and disease. Annu Rev Vis Sci. 2018;4:45–77.
Lin S, Liang Y, Zhang J, Bian C, Zhou H, Guo Q, et al. Microglial TIR-domain-containing adapter-inducing interferon-beta (TRIF) deficiency promotes retinal ganglion cell survival and axon regeneration via nuclear factor-kappaB. J Neuroinflammation. 2012;9:39.
Cai XF, Lin S, Geng Z, Luo LL, Liu YJ, Zhang Z, et al. Integrin CD11b deficiency aggravates retinal microglial activation and rgcs degeneration after acute optic nerve injury. Neurochem Res. 2020;45:1072–85.
Heuss ND, Pierson MJ, Roehrich H, McPherson SW, Gram AL, Li L, et al. Optic nerve as a source of activated retinal microglia post-injury. Acta Neuropathol Commun. 2018;6(1):66.
Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, et al. The role of microglia in retinal neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front Aging Neurosci. 2017;9:214.
Jin N, Gao L, Fan X, Xu H. Friend or foe? Resident microglia vs bone marrow-derived microglia and their roles in the retinal degeneration. Mol Neurobiol. 2017;54(6):4094–112.
Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53(2):1181–94.
Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/macrophage polarization dynamics following traumatic brain injury. J Neurotrauma. 2016;33(19):1732–50.
Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 2014;83(5):1098–116.
Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol Aging. 2013;34(4):1051–9.
Chan-Juan H, Sen L, Li-Qianyu A, Jian Y, Rong-Di Y. MicroRNA-30b regulates the polarity of retinal ganglion cells by inhibiting semaphorin-3A. Mol Vis. 2019;25:722–30.
Liu LN, Li XM, Ye DQ, Pan HF. Emerging role of semaphorin-3A in autoimmune diseases. Inflammopharmacology. 2018;26(3):655–65.
Roth L, Koncina E, Satkauskas S, Cremel G, Aunis D, Bagnard D. The many faces of semaphorins: from development to pathology. Cell Mol Life Sci. 2009;66(4):649–66.
Shelly M, Cancedda L, Lim BK, Popescu AT, Cheng PL, Gao H, et al. Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron. 2011;71(3):433–46.
Zhang J, Liu W, Zhang X, Lin S, Yan J, Ye J. Sema3A inhibits axonal regeneration of retinal ganglion cells via ROCK2. Brain Res. 2020;1727:146555.
Takegahara N, Takamatsu H, Toyofuku T, Tsujimura T, Okuno T, Yukawa K, et al. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat Cell Biol. 2006;8(6):615–22.
Adi SD, Eiza N, Bejar J, Shefer H, Toledano S, Kessler O, et al. Semaphorin 3A is effective in reducing both inflammation and angiogenesis in a mouse model of bronchial asthma. Front Immunol. 2019;10:550.
Podojil JR, Chiang MY, Ifergan I, Copeland R, Liu LN, Maloveste S, et al. B7-H4 modulates regulatory CD4(+) T cell induction and function via ligation of a semaphorin 3a/plexin A4/neuropilin-1 complex. J Immunol. 2018;201(3):897–907.
Dejda A, Mawambo G, Cerani A, Miloudi K, Shao Z, Daudelin JF, et al. Neuropilin-1 mediates myeloid cell chemoattraction and influences retinal neuroimmune crosstalk. J Clin Invest. 2014;124(11):4807–22.
Wen H, Lei Y, Eun SY, Ting JP. Plexin-A4-semaphorin 3A signaling is required for Toll-like receptor- and sepsis-induced cytokine storm. J Exp Med. 2010;207(13):2943–57.
Han F, Huo Y, Huang CJ, Chen CL, Ye J. MicroRNA-30b promotes axon outgrowth of retinal ganglion cells by inhibiting Semaphorin3A expression. Brain Res. 2015;1611:65–73.
Shirvan A, Kimron M, Holdengreber V, Ziv I, Ben-Shaul Y, Melamed S, et al. Anti-semaphorin 3A antibodies rescue retinal ganglion cells from cell death following optic nerve axotomy. J Biol Chem. 2002;277(51):49799–807.
Dejda A, Mawambo G, Daudelin JF, Miloudi K, Akla N, Patel C, et al. Neuropilin-1-expressing microglia are associated with nascent retinal vasculature yet dispensable for developmental angiogenesis. Invest Ophthalmol Vis Sci. 2016;57(4):1530–6.
Zhou JX, Liu YJ, Chen X, Zhang X, Xu J, Yang K, et al. Low-intensity pulsed ultrasound protects retinal ganglion cell from optic nerve injury induced apoptosis via yes associated protein. Front Cell Neurosci. 2018;12:160.
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–97.
Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG, et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci. 2018;21(3):329–40.
Yamashita N, Jitsuki-Takahashi A, Ogawara M, Ohkubo W, Araki T, Hotta C, et al. Anti-Semaphorin 3A neutralization monoclonal antibody prevents sepsis development in lipopolysaccharide-treated mice. Int Immunol. 2015;27(9):459–66.
Sidman RL, Li J, Lawrence M, Hu W, Musso GF, Giordano RJ, et al. The peptidomimetic Vasotide targets two retinal VEGF receptors and reduces pathological angiogenesis in murine and nonhuman primate models of retinal disease. Sci Transl Med. 2015;7(309):309ra165.
Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6(8):2163–78.
Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44(3):183–9.
Eixarch H, Gutierrez-Franco A, Montalban X, Espejo C. Semaphorins 3A and 7A: potential immune and neuroregenerative targets in multiple sclerosis. Trends Mol Med. 2013;19(3):157–64.
Majed HH, Chandran S, Niclou SP, Nicholas RS, Wilkins A, Wing MG, et al. A novel role for Sema3A in neuroprotection from injury mediated by activated microglia. J Neurosci. 2006;26(6):1730–8.
Costa C, Martinez-Saez E, Gutierrez-Franco A, Eixarch H, Castro Z, Ortega-Aznar A, et al. Expression of semaphorin 3A, semaphorin 7A and their receptors in multiple sclerosis lesions. Mult Scler. 2015;21(13):1632–43.
Sosna J, Philipp S, Albay R 3rd, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, et al. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener. 2018;13(1):11.
Rice RA, Spangenberg EE, Yamate-Morgan H, Lee RJ, Arora RP, Hernandez MX, et al. Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J Neurosci. 2015;35(27):9977–89.
Liu W, Li X, Chen X, Zhang J, Luo L, Hu Q, et al. JIP1 deficiency protects retinal ganglion cells from apoptosis in a rotenone-induced injury model. Front Cell Dev Biol. 2019;7:225.
Lutty GA, McLeod DS. Development of the hyaloid, choroidal and retinal vasculatures in the fetal human eye. Prog Retin Eye Res. 2018;62:58–76.
Sharma A, Pollett MA, Plant GW, Harvey AR. Changes in mRNA expression of class 3 semaphorins and their receptors in the adult rat retino-collicular system after unilateral optic nerve injury. Invest Ophthalmol Vis Sci. 2012;53(13):8367–77.
Nitzan A, Kermer P, Shirvan A, Bahr M, Barzilai A, Solomon AS. Examination of cellular and molecular events associated with optic nerve axotomy. Glia. 2006;54(6):545–56.
Molofsky AV, Kelley KW, Tsai HH, Redmond SA, Chang SM, Madireddy L, et al. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature. 2014;509(7499):189–94.
Ochsenbein AM, Karaman S, Proulx ST, Berchtold M, Jurisic G, Stoeckli ET, et al. Endothelial cell-derived semaphorin 3A inhibits filopodia formation by blood vascular tip cells. Development. 2016;143(4):589–94.
Kikuchi K, Kishino A, Konishi O, Kumagai K, Hosotani N, Saji I, et al. In vitro and in vivo characterization of a novel semaphorin 3A inhibitor, SM-216289 or xanthofulvin. J Biol Chem. 2003;278(44):42985–91.
Vadasz Z, Toubi E. Semaphorin 3A - a marker for disease activity and a potential putative disease-modifying treatment in systemic lupus erythematosus. Lupus. 2012;21(12):1266–70.
Catalano A, Caprari P, Moretti S, Faronato M, Tamagnone L, Procopio A. Semaphorin-3A is expressed by tumor cells and alters T-cell signal transduction and function. Blood. 2006;107(8):3321–9.
Takamatsu H, Takegahara N, Nakagawa Y, Tomura M, Taniguchi M, Friedel RH, et al. Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nat Immunol. 2010;11(7):594–600.
Weekman EM, Sudduth TL, Abner EL, Popa GJ, Mendenhall MD, Brothers HM, et al. Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J Neuroinflammation. 2014;11:127.
Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1beta + microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J Neurosci. 2014;34(26):8904–17.
Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158(3):995–1006.
Herwig MC, Bergstrom C, Wells JR, Holler T, Grossniklaus HE. M2/M1 ratio of tumor associated macrophages and PPAR-gamma expression in uveal melanomas with class 1 and class 2 molecular profiles. Exp Eye Res. 2013;107:52–8.
Tian X, Gan H, Zeng Y, Zhao H, Tang R, Xia Y. Inhibition of semaphorin-3a suppresses lipopolysaccharide-induced acute kidney injury. J Mol Med (Berl). 2018;96(7):713–24.
Zheng Z, Yuan R, Song M, Huo Y, Liu W, Cai X, et al. The toll-like receptor 4-mediated signaling pathway is activated following optic nerve injury in mice. Brain Res. 2012;1489:90–7.
Acknowledgements
The authors especially thank Professor Zhou Yuan-Guo for his suggestions regarding the study and the writing of the manuscript. The authors also thank Zhang Zhuo-Hang and his laboratory members at the Department of Army Occupational Disease and the Department of Ophthalmology, Daping Hospital, Army Medical University.
Funding
This work was supported by grants from the Basic Research and Scientific Frontier Foundation of Chongqing (cstc2019jcyj-msxmX0010) and the National Natural Science Foundation of China (81570840).
Author information
Authors and Affiliations
Contributions
Liu YJ carried out the experiments and drafted the manuscript. Chen X, Zhang JQ, and Zhu JY provided technical support. Lin S designed and supervised the project, revised the manuscript, and provided the funding. Ye J helped to validate the data and support the research. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experimental protocols were approved by the Animal Experimental Ethics Committee of Army Medical University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Fig. S1.
The effect of anti-Sema3A in ameliorating RGCs apoptosis. (A) Representative confocal images showing immunofluorescent staining of TUNEL (green) and Iba1 (red) with Hoechst (blue) nuclear staining in retina tissue with or without anti-Sema3A treatment at and 7 days post-injury. Scale bar = 25 μm. (B) Western Blot and quantitative analysis show expression of BCL-2, BAX expression in retina tissue at 3 days and with or without anti-Sema3A treatment at 7 days post-injury. β-actin was used as a loading control. (Mean ± SEM, n = 5)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yun-Jia, L., Xi, C., Jie-Qiong, Z. et al. Semaphorin3A increases M1-like microglia and retinal ganglion cell apoptosis after optic nerve injury. Cell Biosci 11, 97 (2021). https://doi.org/10.1186/s13578-021-00603-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-021-00603-7