
Abstract
Enzyme-catalyzed proximity labeling (PL) combined with mass spectrometry (MS) has emerged as a revolutionary approach to reveal the protein-protein interaction networks, dissect complex biological processes, and characterize the subcellular proteome in a more physiological setting than before. The enzymatic tags are being upgraded to improve temporal and spatial resolution and obtain faster catalytic dynamics and higher catalytic efficiency. In vivo application of PL integrated with other state of the art techniques has recently been adapted in live animals and plants, allowing questions to be addressed that were previously inaccessible. It is timely to summarize the current state of PL-dependent interactome studies and their potential applications. We will focus on in vivo uses of newer versions of PL and highlight critical considerations for successful in vivo PL experiments that will provide novel insights into the protein interactome in the context of human diseases.
Similar content being viewed by others


Background
Proteins generally form complexes, organelles or other assemblies and create interacting networks that are essential for cellular structure and functional integrity. The execution of biological function and progression of human disease are intimately tied to protein-protein interactions (PPIs), which are characterized by proximity, affinity and duration [1]. Exploration of PPIs underlying intricate cellular signaling and regulatory mechanisms has required efforts to circumvent the technical defects in many of the traditional approaches. The interactome mapping methods commonly employed are affinity purification-mass spectrometry (AP-MS) and yeast two-hybridization [2, 3]. However, these methods very often fail to reveal in vivo PPIs because they require ex vivo manipulation and subcellular fractionation for enrichment; these processes are associated with low validation rates and high false positive rates [4, 5]. Utilization of AP-based methods requires that cells first being lysed to release the bait protein for purification, which poses significant challenges to faithfully preserving its in vivo interacting status. Yeast two-hybridization assay can only suggest the potential interaction of two proteins, rather than the interaction that actually takes place in vivo [5]. Fluorescence-based techniques are most suitable for validating the candidate interacting partners of a given protein, particularly in such applications as a high-throughput drug screening platform, rather than for identifying novel partners [6, 7]. The lack of effective tools to acquire accurate information about protein distribution, protein partners, and protein complex composition remains a major challenge in these fields [8].
In the past decade, enzyme-catalyzed proximity labeling (PL) has developed as a novel alternative method to label and capture not only the proteins that interact directly with the protein of interest (POI), but also the proteins in proximity to the POI [9,10,11]. In a PL system, a promiscuous labeling enzyme is fused in frame to the POI or subcellular compartment marker proteins in living cells. Enzymatic catalyzation will covert an inert substrate into a reactive but short-lived intermediate, which will then covalently label the nearby biomolecules (proteins, RNA and DNA) in a promiscuous and proximity-dependent manner. Because in most cases the small-molecule substrate for labeling contains biotin moiety, the biotinylated proteins can be selectively enriched by affinity purification using neutravidin or streptavidin coated magnetic or agarose beads. Streptavidin shows a stronger intrinsic binding affinity toward biotin and a lower nonspecific binding than neutravidin [12, 13]. Due to higher reproducibility, purity-specificity and ease of use, magnetic beads have become the preferred support for small-scale experiments, whereas agarose beads are more economical when large amounts of purified targeting biomolecules are required. Background contamination is likely to be small because avidin-biotin interaction can withstand harsh and stringent purification conditions and the endogenous biotinylation is relatively low in mammalian cells [11]. The purified proteins are subsequently identified by high-throughput liquid chromatography coupled to mass spectrometry (LC-MS/MS). Another superior feature of PL is that it can capture transient or weak interactions that are often overlooked by conventional AP approaches. Therefore, this technique facilitates the sensitive, specific and timely detection of the interactome of a POI in vivo, which is critical for understanding its broad molecular functions quickly, simply and reliably. Accumulating publications have broadened the range of the bait proteins, from nuclear membrane proteins and transcriptional factors to ubiquitin ligases [9, 14, 15], and, recently, even to RNA-protein interactions [16,17,18,19,20].
In addition to its extensive use in cultured mammalian cells [9, 14, 21, 22], PL has been rapidly adapted for in vivo application in a wide variety of research projects and models, including yeast [23, 24], plant protoplasts [25, 26], parasites [27,28,29], mouse [30, 31], flies and worms [32]. In this review, we will focus on the evolution of powerful PL approaches and the important considerations for PL experimental design, especially the in vivo utilization of PL methods integrated with other sophisticated approaches to profile protein interactome with high confidence.
The APEX and HRP system
APEX is a monomeric 28 kDa ascorbate peroxidase that catalyzes the oxidative polymerization and local deposition of diaminobenzidine (DAB) under harsh treatment conditions; DAB can then be stained with the electron-dense OsO4 to generate strong contrast for electron microscopy (EM) imaging in mammalian organelles [33]. In living cells, exogenous biotin-phenol (BP) can be added and catalyzed by APEX in the presence of hydrogen peroxide (H2O2) to produce biotin-phenoxyl intermediate; biotin-phenoxyl can covalently react with electron-rich amino acids, such as tyrosine, in proteins in the neighborhood [34]. APEX was therefore adopted for PL (Fig. 1a, upper panel) [34,35,36]. One of the major limitations of APEX is its relatively low cellular activity and sensitivity, which may arise from its sub-optimal folding/stability, poor heme binding, or some combination of these factors [35]. Thus, in order to provide sufficient biotinylated proteins for subsequent MS identification, higher amount of total protein extracts are typically required. APEX2, which has higher catalytic activity and sensitivity, was later developed through direct evolution [35] and has been successfully used to determine interactomes in living cells [37, 38]. Because APEX2 can also directly biotinylate guanosine in RNAs, APEX-PL has been combined with RNA sequencing (APEX-seq) to determine subcellular transcriptomes [19]. Additionally, APEX2 has been tagged to human telomerase RNA to profile its interactome on a one-minute time scale [18]. Apart from the traditional APEX2 substrate biotin-phenol, a clickable substrate, alkyne-phenol (Alk-Ph), was recently shown to improve membrane permeability and enhance labeling efficiency in intact yeast cells, which enables spatially restricted proteome and transcriptome profiling in yeast [39]. These successful applications demonstrate higher spatial and temporal resolution of APEX-based PL, which is especially suitable for detection of dynamic shifts in interactomes. However, the requirement for sufficient biotoxic heme (H2O2) to confer high APEX activity limits the in vivo application of APEX-based PL in animals.

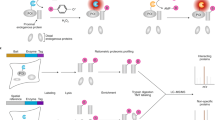
Schematic diagram of enzyme-catalytic proximity labeling approaches.
a In a standard enzyme-catalytic proximity labeling system, ascorbate peroxidase (e.g., APEX or APEX2) in the presence of H2O2 catalyzes the one-electron oxidation of biotin-phenol into a highly reactive and short-lived biotin-phenoxyl radical, which biotinylates tyrosine predominantly in nearby proteins (upper panel). In contrast biotin ligase (e.g. BioID or TurboID/miniTurbo) catalyzes the synthesis of a biotinoyl-5’-AMP intermediate from biotin and ATP and promiscuously tags lysine in nearby proteins (lower panel). These enzymes can be fused in frame to the bait protein and introduced into living cells. The biotinylation process depends on the localization of the bait protein and occurs in a proximate dependent manner. b The biotinylated proteins are first enriched and purified by streptavidin pulldown assay, then further digested into peptides and identified by quantitative LC-MS/MS. Subsequently, a specific data analysis scheme is adopted for data visualization based on the experimental purpose and design. c The chemical structure of NHS-biotin (left panel), which can biotinylate lysine in any protein without a requirement for enzymatic catalyzation (right panel)
Horseradish peroxidase (HRP) is a 44 kDa enzymatic tag with even higher activity than APEX2 that can be utilized for both EM and PL to generate a similar labeling pattern as APEX [36, 40]. The four structurally-essential disulfide bonds of HRP cannot form in a reducing environment, which precludes its applications in cytosol, nucleus and mitochondria. However, it is active in the oxidizing environment, such as secretory pathway and cell surface, and has been employed to map the proteomes of synaptic clefts in living neurons via biotinylation with biotin-xx-phenol (BxxP) and H2O2 [40]. Lately, HRP has been adapted for an antibody-guided, proximity-based labeling approach, Biotinylation by Antibody Recognition (BAR), to label proteins in primary human tissues without prior insertion of a fusion gene [41]. In a BAR system, the POI in a fixed and permeabilized tissue is initially targeted by the primary antibody, a secondary HRP-conjugated antibody is applied to generate free radicals using BP and H2O2, and the biotinylated proteins are enriched by AP and detected by MS [41]. BAR obviates the issues associated with protein fusion, but requires a highly specific monoclonal antibody that is sensitive to fixation artifacts.
The BioID and TurboID system
Initially developed and introduced in 2012, BioID (proximity-dependent biotin identification) uses a highly promiscuous mutated form of Escherichia coli (E. coli) biotin ligase, BirA*, which has become the most commonly used enzyme for PL [9, 42]. BirA* is a 35 kDa biotin protein ligase that catalyzes the synthesis of biotinoyl-5’-AMP (bioAMP) from biotin and ATP. BioAMP reacts with primary amines, predominantly provided by lysine in any protein, to achieve promiscuous protein biotinylation in a time-dependent manner (Fig. 1a, lower panel) [9]. In the cells expressing BirA* fused bait protein, the proximate proteins of the POI can be biotinylated and probed via streptavidin pulldown assay followed by mass spectrometry (Fig. 1a, b). Unlike APEX, the BioID system simply requires a supply of exogenous nontoxic biotin, which permits its in vivo application. Nevertheless, the slow kinetics of BirA*, which usually takes 18–24 h to generate sufficient biotinylated materials for proteomic analysis [9, 43, 44], precludes using it to capture an interactome snapshot within the timescale of minutes or a few hours. In addition, that BirA* has low or even undetectable catalytic activity in worms, flies, or the ER lumen of cultured mammalian cells [32] makes it problematic for in vivo application. Although newer versions of BioID, BioID2 and BASU, have been developed with improved features [17, 45,46,47], they continue to have slow catalytic kinetics that limits in vivo use. Because the stability and intracellular lifetime of reactive groups are largely associated with their working radius, it is vitally important to know the exact labeling radius of each enzyme. It was estimated that the effective biotinylation radius of BirA* is about 10–15 nm [43, 48], shorter than that of APEX, which is about 20 nm [49].
Using yeast display-based directed evolution, two engineered promiscuous mutants of BirA*, TurboID and miniTurbo, were developed. These enzymes have superb catalytic efficiency and much faster kinetics than BioID or BioID2 [32]. TurboID takes only 10 min to catalyze nearly as many biotinylated proteins as BioID/BioID2/BASU can provide in 18 hours. In addition, miniTurbo is 20% smaller than TurboID, which may minimize disturbance of fusion protein trafficking and function. It is also preferable for precise temporal control of the labeling window due to less background labeling; miniTurbo does not efficiently label proteins unless a sufficient amount of exogenous biotin is added. On the other hand, TurboID is suggested to be preferred for labeling proteins located in the mitochondrial matrix and ER lumen, and for low-abundant proteins, because it generates stronger signals for these purpose than miniTurbo or BioID2 [32].
In order to improve the spatial specificity and versatility of biotinylation in a PL system, split forms of catalytic enzymes, such as split-APEX [50], split-BioID [51, 52] and split-TurboID [53], have been created. In the split PL system, the promiscuous catalytic enzyme is split into two fragments with no activity on their own; the enzymatic activity can be reconstituted when both units are brought together in cells in a controlled way, either by a chemical molecule or through PPI. Combined with functional studies and screens, split PL is a valuable approach to map the proteomes of organelle contact sites or macromolecular complexes. Unlike split-APEX, split-TurboID does not need the addition of cofactors or co-oxidants. Demonstrated by a study targeting to probe the protein composition of endoplasmic reticulum-mitochondria contact sites, split-TurboID requires a shorter labeling time than split-BioID (4 hours versus 16 hours) and identifies a more balanced set of proteomes, while the split-BioID proteome is relatively biased toward ER membrane proteins [53]. Therefore, split-TurboID offers greater biocompatibility and less bias than split-APEX or split-BioID in mapping the composition of intracellular organelle contact sites.
In vivo application of PL
Although PL tools have been broadly used in investigations of protein interactomes in a wide range of cultured cells, it is only in recent years that this method has been applied in live animals and plants. Transgenic Drosophila or mouse lines carrying APEX or HRP-fused constructs have been reported to identify cell-type-specific proteomes [49, 54,55,56]. Nevertheless, these in vivo strategies require that living tissues must be dissected and/or perfused first, then incubated with BP or BxxP substrate and subsequently with H2O2 to activate PL biotinylation. The in vivo BioID/TurboID protocol has overcome these drawbacks. Its use in studies of the c-MYC oncoprotein provides an important example. The c-MYC plays a critical role in the initiation and progression of various types of cancer [57, 58]. For a long time, it remained a technical challenge to characterize the interaction proteins of c-MYC because it is an extremely unstable protein tightly bound to chromatin, making it very difficult to isolate MYC-containing protein complexes using traditional biochemical approaches. The BioID-based PL system yielded more than 100 high-confidence protein neighbors of c-MYC in tumor xenografts grown in mice, which has expanded the interactome of this important oncoprotein to a great extent [59]. In another instance, to capture interactomes of synaptic proteins in mouse brain, adeno-associated viral (AAV)-mediated fused synaptic proteins-BirA cortex expression and subcutaneous injection of biotin enabled the identification of a large number of proteins not previously demonstrated at the inhibitory postsynaptic density, providing a molecular prospectus for the deeper understanding of synaptic physiology [30]. A similar approach was used to identify proteome composition of nascent synapses in cortex and hippocampus of early postnatal mice [31]. The more recently developed BioID knock-in mouse model with BioID inserted at the Z-disc of titin, the giant protein determining the elasticity of myofilament, has provided new insights into sarcomere physiology [48, 60, 61]. Since the expression of titin-BioID at a physiological level leads to a significantly smaller amount of biotinylated materials for MS analysis, cryo-fractured tissue powder digested with trypsin served as the input and anti-biotin antibody facilitated retrieval of biotinylated peptides [62, 63]. Mapping of biotinylation sites to sarcomeric structures deepens our knowledge of myofilament dynamics and supports the model that myosin penetrates the Z-disc to dampen contraction. Furthermore, the proteomic investigation of heart and quadriceps muscle at ages extending from neonatal to adult has linked neonatal signaling pathways to the sarcomere [48]. Another BioID knock-in mouse line with BioID2 fused to the endogenous JPH2 coding sequence was developed to profile the cardiac dyad proteome [64]. This BioID2 knock-in strategy leads to expression levels of JPH2-BioID2 fusion protein comparable to that of the endogenous protein, but still reveals novel potential dyadic proteins that were not discovered using JPH2-HA overexpression transgenic mouse line [65]. In summary, generating biotin ligase (e.g. BioID/TurboID) fusion proteins in animal models has great potential to provide new insights into the molecular mechanisms of human disorders.
Despite attempts to establish a BioID system in Arabidopsis thaliana and TMV-infected Nicotiana benthamiana plants [66, 67], there continue to be major obstacles to the application of BioID in plants. These include their specialized cell walls and cuticle structures, low growth temperature and low endogenous production and cellular storage of biotin [68]. However, TurboID outperforms BioID and BioID2 in plant studies due to its higher catalytic activity and broader working temperature [26], even with POI of low abundance [8]. Of note, when intact plant tissue is used, it is important to include a free biotin-depletion step to reduce the amount of streptavidin beads required, whereas for mammalian cell culture washing the cells several times seems sufficient to remove excessive biotin [8]. In vivo application of TurboID has also been demonstrated in worms and flies [32], and we expect to see it soon in mouse.
Important considerations for successful in vivo PL experiments
Clearly, in vivo BioID/TurboID-based PL is an attractive tool with great potential to profile proteomes in a variety of biological situations, even for rare POIs or transient PPIs that are normally difficult to capture by standard biochemical methods. To perform successful in vivo PL investigations, certain aspects of experimental design need to be considered because they are critical to ensure reliable and meaningful results.
-
1.
Choosing the PL labeling enzyme (Table 1): As we mentioned before, APEX and HRP are not ideal for in vivo application due to the need for BP substrate and H2O2. If the goal is to capture highly dynamic processes that prioritize fine temporal control, the APEX or HRP approach can be used with dissected tissues. The latter is preferable for studies focusing on a secretory pathway or cell surface. BioID has been successfully employed in mouse with different POIs [30, 31, 48, 59]. This success boosts confidence that TurboID, which offers superior catalytic activity and much faster kinetics than BioID, will also be applicable to mouse. The weaker catalytic activity of miniTurbo may be a more advantageous choice when the tissue background has a particularly high endogenous biotin level or a restricted labeling time window is selected because of a lower utilization of endogenous biotin.
Table 1 The basic properties and features of enzymatic tags developed for enzyme-catalyzed proximity labeling approaches based on APEX and BioID -
2.
When choosing a biotin ligase, it should be considered whether the molecular size or the construction (N- or C-terminal tag) of the enzyme would interfere with the normal function of the POI or with correct targeting to the specific subcellular compartment [8, 32]. A fluorophore or antigenic tag of the fusion protein may expedite confirming its expression and subcellular localization.
-
3.
The expression level of POI-APEX or TurboID is another important factor that significantly affects the interactome analysis. Virus-mediated or transgenic overexpression of the PL enzyme-tagged POI may generate PPIs in vivo that are either not physiological or not specific, burdening the secondary validation procedure. This problem may be solved through CRISPR/Cas9-mediated knock-in to tag the PL enzyme to the endogenous POI at a physiological level [48, 64]. If the endogenous level of PL enzyme-POI yields insufficient biotinylated material for subsequent analysis, the procedure for retrieval of biotinylated peptides may require modification, such as by using cryo-fractured tissue powder digested with trypsin as an input and anti-bio tin antibody [62, 63].
-
4.
One considerable limitation of BioID/TurboID is the presence of non-specific background either due to stochastically biotinylated proteins in the same subcellular localization as the bait protein or because of proteins that nonspecifically bind to the streptavidin-coupled beads. It has been reported that a large proportion of proteins captured in the interactome of a POI are common to all samples, as is usual in affinity purification experiments [8, 31]. It is of critical to set appropriate controls to differentiate high-confidence candidates from non-specific background or contaminant proteins. A free form of biotin ligase targeted to the same subcellular compartment and expressed at the same level as the bait protein is necessary as a negative control [4].
-
5.
It is of vital importance to optimize the experimental conditions to determine the biotin concentration, the duration of biotinylation, and the amount of starting materials. Immunohistochemistry and immunoblotting are common and effective monitoring techniques: the expression patterns and subcellular localization of the biotinylated complex can be visualized by immunochemistry; and immunoblotting is able to semi-quantitatively estimate the efficiency of biotinylation and the minimum starting materials that can be detected. It is worth noting that the signal intensity of immunoblotting does not necessarily reflect the quantity of tagged protein because highly biotinylated proteins may amplify the signal by binding streptavidin at multiple binding sites [8]. Additionally, toxicity analyses in flies and worms indicate that TurboID may sequester endogenous biotin and starve cells of biotin. Therefore, exogenous biotin supplementation is not only necessary for BioID but also for the health of experimental animals. One concern about BioID is that the charge loss on primary amino acids occupied by the covalent attachment of biotin might disturb the formation of other secondary modifications, which could in turn affect the biological behavior of both the fusion protein and proximal proteins. The labeling time should be carefully optimized to determine the shortest possible period that will generate sufficient biotinylated materials but still maintain the spatial specificity of the bait protein; a longer than necessary labeling time may lead to toxicity due to the chronic biotinylation of endogenous proteins [32].
-
6.
Sometimes the strong interaction between avidin, streptavidin or neutravidin-coated beads and biotinylated proteins makes it difficult to elute the bound proteins efficiently. Instead of eluting the biotinylated proteins, digesting them on the beads directly with protease, such as trypsin or Lys-C, may increase the peptide yield [69].
-
7.
Two kinds of MS methods are normally used for final identification of target proteins, label-free and labelled quantification [49]. Introduction of stable isotope labels on the digested peptides, such as Tandam Mass Tag (TMT), or isobaric tags for relative and absolute quantification (iTRAQ), enables identical peptides from diverse samples to be distinguished within a mixture by mass spectrometry. Compared to label-free quantification, these labelled quantification techniques can significantly improve quantitative accuracy at the expense of proteome coverage [70]. When comparing markedly diverse samples, researchers must make certain to select the appropriate data normalization method [8]. Similarly, well-designed bioinformatics analysis is critical to identify high-confidence PPIs.
-
8.
Although the interactome acquired through PL is quite reliable, the ultimate proof is in vivo validation of PPI by complimentary methods together with functional readout demonstrating the interaction is significant and meaningful. Immunofluorescent-chemistry combined with super-resolution imaging can be utilized to validate co-localization of a selected protein candidate and POI; co-immunoprecipitation is an established method to validation the interaction. Development of transgenic animal models in which the expression level of interaction candidates is manipulated by CRISPR will allow in-depth morphological and functional studies [30, 31]. In combination with other state-of-art techniques, this approach will allow us to address challenging questions that are previously inaccessible.
Conclusions and future perspectives
The dynamic and transient nature of PPIs makes it challenging for conventional approaches to provide real-time in vivo information. The development and in vivo application of BioID/TurboID and its sibling PL method APEX/APEX-seq furnish a powerful toolbox for illustrating critical PPIs and protein-RNA interactions with subcellular resolution. Capitalizing on the high catalytic activity and temporal resolution, TurboID and miniTurbo are likely to be widely employed in mouse in vivo interactome studies. Split forms of the PL method will be more appropriate for studies requiring higher spatial specificity, especially for proteomic analysis of organelle contact sites or macromolecular complexes. In addition to providing interactome profiles using APEX- or BioID-PL techniques, universal protein biotinylation by N-hydroxysuccinimidobiotin (NHS-biotin) (Fig. 1c) has recently been adapted to profile the proteome of retinal ganglion cells and transportomes along the visual pathway in adult rats [71]. Another special proteome, nascent proteome, can be determined in mouse in vivo by expressing a mutant methionyl-tRNA synthetase (MetRS L274G), which allows metabolic labeling of newly-synthesized proteins with the non-canonical amino acid azidonorleucine [72]. We expect additional novel or upgraded enzyme tags to be identified in the near future. Combining these approaches will be particularly beneficial for sensitive detection of highly diverse proteomes during a defined time window in a specific cell type in vivo.
Availability of data and materials
Not applicable.
Abbreviations
- PL:
-
Proximity labeling
- PPIs:
-
Protein–protein interactions
- AP-MS:
-
Affinity purification-mass spectrometry
- BRET:
-
Bioluminescence resonance emission transfer
- POI:
-
Protein of interest
- LC–MS/MS:
-
Liquid chromatography coupled to mass spectrometry
- APEX:
-
Ascorbate peroxidase
- EM:
-
Electron microscopy
- DAB:
-
Diaminobenzidine
- BP:
-
Biotin-phenol
- H2O2 :
-
Hydrogen peroxide
- HRP:
-
Horseradish peroxidase
- BxxP:
-
Biotin-xx-phenol
- BAR:
-
Biotinylation by antibody recognition
- BioID:
-
Proximity-dependent biotin identification
- bioAMP:
-
Biotinoyl-5′-AMP
- AAV:
-
Adeno-associated viral
- TMT:
-
Tandam mass tag
- iTRAQ:
-
Isobaric tags for relative and absolute quantification
- NHS-biotin:
-
N-Hydroxysuccinimidobiotin
- MetRS L274G:
-
Mutant methionyl-tRNA synthetase
References
Rees JS, Li XW, Perrett S, Lilley KS, Jackson AP. Protein neighbors and proximity proteomics. MCP. 2015;14(11):2848–56.
Trepte P, Buntru A, Klockmeier K, Willmore L, Arumughan A, Secker C, Zenkner M, Brusendorf L, Rau K, Redel A, et al. DULIP: a dual luminescence-based co-immunoprecipitation assay for interactome mapping in mammalian cells. J Mol Biol. 2015;427(21):3375–88.
Yang F, Lei Y, Zhou M, Yao Q, Han Y, Wu X, Zhong W, Zhu C, Xu W, Tao R, et al. Development and application of a recombination-based library versus library high- throughput yeast two-hybrid (RLL-Y2H) screening system. Nucleic acids research. 2018;46(3):e17.
Gingras AC, Abe KT, Raught B. Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr Opin Chem Biol. 2019;48:44–54.
Snider J, Kotlyar M, Saraon P, Yao Z, Jurisica I, Stagljar I. Fundamentals of protein interaction network mapping. Mol Syst Biol. 2015;11(12):848.
Li P, Li J, Wang L, Di LJ. Proximity labeling of interacting proteins: application of BioID as a discovery tool. Proteomics. 2017;17:20.
Kim CK, Cho KF, Kim MW, Ting AY. Luciferase-LOV BRET enables versatile and specific transcriptional readout of cellular protein-protein interactions. eLife. 2019;8:52.
Mair A, Xu SL, Branon TC, Ting AY, Bergmann DC. Proximity labeling of protein complexes and cell-type-specific organellar proteomes in Arabidopsis enabled by TurboID. eLife. 2019;8:4.
Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012;196(6):801–10.
Kovalski JR, Bhaduri A, Zehnder AM, Neela PH, Che Y, Wozniak GG, Khavari PA. The functional proximal proteome of oncogenic RAS includes mTORC2. Mol Cell. 2019;73(4):830–44.
Trinkle-Mulcahy L. Recent advances in proximity-based labeling methods for interactome mapping. Research. 2019;8:5.
Nguyen TT, Sly KL, Conboy JC. Comparison of the energetics of avidin, streptavidin, neutrAvidin, and anti-biotin antibody binding to biotinylated lipid bilayer examined by second-harmonic generation. Anal Chem. 2012;84(1):201–8.
Jain A, Barve A, Zhao Z, Jin W, Cheng K. Comparison of avidin, neutravidin, and streptavidin as nanocarriers for efficient siRNA delivery. Mol Pharm. 2017;14(5):1517–27.
Dingar D, Tu WB, Resetca D, Lourenco C, Tamachi A, De Melo J, Houlahan KE, Kalkat M, Chan PK, Boutros PC, et al. MYC dephosphorylation by the PP1/PNUTS phosphatase complex regulates chromatin binding and protein stability. Nat Commun. 2018;9(1):3502.
Coyaud E, Mis M, Laurent EM, Dunham WH, Couzens AL, Robitaille M, Gingras AC, Angers S, Raught B. BioID-based Identification of Skp Cullin F-box (SCF)beta-TrCP1/2 E3 Ligase Substrates. MCP. 2015;14(7):1781–95.
Mukherjee J, Hermesh O, Eliscovich C, Nalpas N, Franz-Wachtel M, Maček B, Jansen RP. β-Actin mRNA interactome mapping by proximity biotinylation. Proc Natl Acad Sci USA. 2019;116(26):12863–72.
Ramanathan M, Majzoub K, Rao DS, Neela PH, Zarnegar BJ, Mondal S, Roth JG, Gai H, Kovalski JR, Siprashvili Z, et al. RNA-protein interaction detection in living cells. Nat Methods. 2018;15(3):207–12.
Han S, Zhao BS, Myers SA, Carr SA, He C, Ting AY. RNA-protein interaction mapping via MS2- or Cas13-based APEX targeting. Proc Natl Acad Sci USA. 2020;117(36):22068–79.
Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, Chang HY, Ting AY. Atlas of subcellular RNA localization revealed by APEX-SEq. Cell. 2019;178(2):473–90.
Kaewsapsak P, Shechner DM, Mallard W, Rinn JL, Ting AY. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife. 2017;6:7.
Manshouri R, Coyaud E, Kundu ST, Peng DH, Stratton SA, Alton K, Bajaj R, Fradette JJ, Minelli R, Peoples MD, et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nature communications. 2019;10(1):5125.
van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, Rocha S, Houcine A, Derua R, Verfaillie T, et al. The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with Filamin-A and F-Actin remodeling. Molecular cell. 2017;65(5):885-99.e886.
Opitz N, Schmitt K, Hofer-Pretz V, Neumann B, Krebber H, Braus GH, Valerius O. Capturing the Asc1p/receptor for activated C kinase 1 (RACK1) microenvironment at the head region of the 40S ribosome with quantitative BioID in yeast. MCP. 2017;16(12):2199–218.
Larochelle M, Bergeron D, Arcand B, Bachand F. Proximity-dependent biotinylation mediated by TurboID to identify protein-protein interaction networks in yeast. J Cell Sci. 2019;132:11.
Lin Q, Zhou Z, Luo W, Fang M, Li M, Li H. Screening of proximal and interacting proteins in rice protoplasts by proximity-dependent biotinylation. Frontiers in plant science. 2017;8:749.
Zhang Y, Song G, Lal NK, Nagalakshmi U, Li Y, Zheng W, Huang PJ, Branon TC, Ting AY, Walley JW, et al. TurboID-based proximity labeling reveals that UBR7 is a regulator of N NLR immune receptor-mediated immunity. Nat Commun. 2019;10(1):3252.
Nadipuram SM, Kim EW, Vashisht AA, Lin AH, Bell HN, Coppens I, Wohlschlegel JA, Bradley PJ. In vivo biotinylation of the toxoplasma parasitophorous vacuole reveals novel dense granule proteins important for parasite growth and pathogenesis. mBio. 2016;7:4.
Kehrer J, Frischknecht F, Mair GR. Proteomic analysis of the plasmodium berghei gametocyte egressome and vesicular bioID of osmiophilic body proteins identifies merozoite TRAP-like protein (MTRAP) as an essential factor for parasite transmission. Mol Cell Proteom. 2016;15(9):2852–62.
Dang HQ, Zhou Q, Rowlett VW, Hu H, Lee KJ, Margolin W, Li Z. Proximity interactions among basal body components in trypanosoma brucei identify novel regulators of basal body biogenesis and inheritance. Bio. 2017;8:1.
Uezu A, Kanak DJ, Bradshaw TW, Soderblom EJ, Catavero CM, Burette AC, Weinberg RJ, Soderling SH. Identification of an elaborate complex mediating postsynaptic inhibition. Science. 2016;353(6304):1123–9.
Spence EF, Dube S, Uezu A, Locke M, Soderblom EJ, Soderling SH. In vivo proximity proteomics of nascent synapses reveals a novel regulator of cytoskeleton-mediated synaptic maturation. Nat Commun. 2019;10(1):386.
Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, Ting AY. Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol. 2018;36(9):880–7.
Martell JD, Deerinck TJ, Lam SS, Ellisman MH, Ting AY. Electron microscopy using the genetically encoded APEX2 tag in cultured mammalian cells. Nat Protocols. 2017;12(9):1792–816.
Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339(6125):1328–31.
Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, Ting AY. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods. 2015;12(1):51–4.
Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol. 2012;30(11):1143–8.
Paek J, Kalocsay M, Staus DP, Wingler L, Pascolutti R, Paulo JA, Gygi SP, Kruse AC. Multidimensional tracking of GPCR signaling via peroxidase-catalyzed proximity labeling. Cell. 2017;169(2):338-49.e311.
Lobingier BT, Huttenhain R, Eichel K, Miller KB, Ting AY, von Zastrow M, Krogan NJ. An approach to spatiotemporally resolve protein interaction networks in living cells. Cell. 2017;169(2):350-60.e312.
Li Y, Tian C, Liu K, Zhou Y, Yang J, Zou P. A clickable APEX probe for proximity-dependent proteomic profiling in yeast. Cell Chem Biol. 2020;27(7):858-65 e858.
Loh KH, Stawski PS, Draycott AS, Udeshi ND, Lehrman EK, Wilton DK, Svinkina T, Deerinck TJ, Ellisman MH, Stevens B, et al. Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell. 2016;166(5):1295-307.e1221.
Bar DZ, Atkatsh K, Tavarez U, Erdos MR, Gruenbaum Y, Collins FS. Biotinylation by antibody recognition-a method for proximity labeling. Nat Methods. 2018;15(2):127–33.
Choi-Rhee E, Schulman H, Cronan JE. Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci. 2004;13(11):3043–50.
Kim DI, Birendra KC, Zhu W, Motamedchaboki K, Doye V, Roux KJ. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci USA. 2014;111(24):E2453–61.
Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21(2):228–39.
Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell. 2016;27(8):1188–96.
Birendra K, May DG, Benson BV, Kim DI, Shivega WG, Ali MH, Faustino RS, Campos AR, Roux KJ. VRK2A is an A-type lamin-dependent nuclear envelope kinase that phosphorylates BAF. Mol Biol Cell. 2017;28(17):2241–50.
Jung EM, Moffat JJ, Liu J, Dravid SM, Gurumurthy CB, Kim WY. Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat Neurosci. 2017;20(12):1694–707.
Rudolph F, Fink C, Hüttemeister J, Kirchner M, Radke MH, Lopez Carballo J, Wagner E, Kohl T, Lehnart SE, Mertins P, et al. Deconstructing sarcomeric structure-function relations in titin-BioID knock-in mice. Nat Commun. 2020;11(1):3133.
Chen CL, Hu Y, Udeshi ND, Lau TY, Wirtz-Peitz F, He L, Ting AY, Carr SA, Perrimon N. Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc Natl Acad Sci USA. 2015;112(39):12093–8.
Han Y, Branon TC, Martell JD, Boassa D, Shechner D, Ellisman MH, Ting A. Directed evolution of split APEX2 peroxidase. ACS Chem Biol. 2019;14(4):619–35.
Schopp IM, Amaya Ramirez CC, Debeljak J, Kreibich E, Skribbe M, Wild K, Béthune J. Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat Commun. 2017;8:15690.
Kwak C, Shin S, Park JS, Jung M, Nhung TTM, Kang MG, Lee C, Kwon TH, Park SK, Mun JY, et al. Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc Natl Acad Sci USA. 2020;117(22):12109–20.
Cho KF, Branon TC, Rajeev S, Svinkina T, Udeshi ND, Thoudam T, Kwak C, Rhee HW, Lee IK, Carr SA, et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc Natl Acad Sci USA. 2020;117(22):12143–54.
Mannix KM, Starble RM, Kaufman RS, Cooley L. Proximity labeling reveals novel interactomes in live Drosophila tissue. Development. 2019;146:14.
Li J, Han S, Li H, Udeshi ND, Svinkina T, Mani DR, Xu C, Guajardo R, Xie Q, Li T, et al. Cell-surface proteomic profiling in the fly brain uncovers wiring regulators. Cell. 2020;180(2):373-86.e315.
Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen BX, Kushnir A, et al. Mechanism of adrenergic Ca(V)1.2 stimulation revealed by proximity proteomics. Nature. 2020;577(7792):695–700.
Baluapuri A, Wolf E, Eilers M. Target gene-independent functions of MYC oncoproteins. Nat Rev Mol Cell Biol. 2020;21(5):255–67.
Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5(10):1024–39.
Dingar D, Kalkat M, Chan PK, Srikumar T, Bailey SD, Tu WB, Coyaud E, Ponzielli R, Kolyar M, Jurisica I, et al. BioID identifies novel c-MYC interacting partners in cultured cells and xenograft tumors. J Proteom. 2015;118:95–111.
Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res. 2004;94(4):505–13.
Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18(5):766–73.
Udeshi ND, Pedram K, Svinkina T, Fereshetian S, Myers SA, Aygun O, Krug K, Clauser K, Ryan D, Ast T, et al. Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat Methods. 2017;14(12):1167–70.
Kim DI, Cutler JA, Na CH, Reckel S, Renuse S, Madugundu AK, Tahir R, Goldschmidt HL, Reddy KL, Huganir RL, et al. BioSITe: a method for direct detection and quantitation of site-specific biotinylation. J Proteome Res. 2018;17(2):759–69.
Feng W, Liu C, Spinozzi S, Wang L, Evans SM, Chen J. Identifying the cardiac dyad proteome in vivo by a BioID2 knock-in strategy. Circulation. 2020;141(11):940–2.
Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, Beavers D, Wang G, Khalid M, Reynolds JO, Campbell HM, et al. SPEG (striated muscle preferentially expressed protein kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circulation research. 2017;120(1):110–9.
Khan M, Youn JY, Gingras AC, Subramaniam R, Desveaux D. In planta proximity dependent biotin identification (BioID). Sci Rep. 2018;8(1):9212.
Das PP, Macharia MW, Lin Q, Wong SM. In planta proximity-dependent biotin identification (BioID) identifies a TMV replication co-chaperone NbSGT1 in the vicinity of 126 kDa replicase. J Proteom. 2019;204:103402.
Bontinck M, Van Leene J, Gadeyne A, De Rybel B, Eeckhout D, Nelissen H, De Jaeger G. Recent trends in plant protein complex analysis in a developmental context. Front Plant Sci. 2018;9:640.
Tholey A, Becker A. Top-down proteomics for the analysis of proteolytic events: methods, applications and perspectives. Biochim Biophys Mol Cell Res. 2017;1864(11 Pt B):2191–9.
Megger DA, Pott LL, Ahrens M, Padden J, Bracht T, Kuhlmann K, Eisenacher M, Meyer HE, Sitek B. Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim Biophys Acta. 2014;1844(5):967–76.
Schiapparelli LM, Shah SH, Ma Y, McClatchy DB, Sharma P, Li J, Yates JR, Goldberg JL, Cline HT. The retinal ganglion cell transportome identifies proteins transported to axons and presynaptic compartments in the visual system in vivo. Cell reports. 2019;28(7):1935–47 e1935.
Alvarez-Castelao B, Schanzenbächer CT, Langer JD, Schuman EM. Cell-type-specific metabolic labeling, detection and identification of nascent proteomes in vivo. Nat Protocols. 2019;14(2):556–75.
Acknowledgements
We thank the members of the Hu lab and Dr. Alan Tessler for critical discussion and reading the manuscript.
Funding
Y.H. is supported by NIH grants EY024932, EY023295, EY028106 and EY031063 and grants from BrightFocus Foundation, Glaucoma Research Foundation, and Chan Zuckerberg Initiative. Y.X. is grateful to the China Scholarship Council for supporting her study abroad. We are grateful for an unrestricted grant from Research to Prevent Blindness and NEI P30 EY026877 to the Department of Ophthalmology.
Author information
Authors and Affiliations
Contributions
YX, XF and YH write this review together. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have consented for publication.
Competing interests
All authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, Y., Fan, X. & Hu, Y. In vivo interactome profiling by enzyme‐catalyzed proximity labeling. Cell Biosci 11, 27 (2021). https://doi.org/10.1186/s13578-021-00542-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-021-00542-3