
Abstract
Determination of the viability, ratio of dead and live cell populations, of Sulfolobus acidocaldarius is still being done by tedious and material-intensive plating assays that can only provide time-lagged results. Although S. acidocaldarius, an extremophilic Archaeon thriving at 75 °C and pH 3.0, and related species harbor great potential for the exploitation as production hosts and biocatalysts in biotechnological applications, no industrial processes have been established yet. One hindrance is that during development and scaling of industrial bioprocesses timely monitoring of the impact of process parameters on the cultivated organism is crucial—a task that cannot be fulfilled by traditional plating assays. As alternative, flow cytometry (FCM) promises a fast and reliable method for viability assessment via the use of fluorescent dyes. In this study, commercially available fluorescent dyes applicable in S. acidocaldarius were identified. The dyes, fluorescein diacetate and concanavalin A conjugated with rhodamine, were discovered to be suitable for viability determination via FCM. For showing the applicability of the developed at-line tool for bioprocess monitoring, a chemostat cultivation on a defined growth medium at 75 °C, pH 3.0 was conducted. Over the timeframe of 800 h, this developed FCM method was compared to the plating assay by monitoring the change in viability upon controlled pH shifts. Both methods detected an impact on the viability at pH values of 2.0 and 1.5 when compared to pH 3.0. A logarithmic relationship between the viability observed via plating assay and via FCM was observed.
Key points
-
Development of a flow cytometry (FCM) method for viability determination of S. acidocaldarius using the fluorescent dyes fluorescein diacetate and concanavalin A conjugated with rhodamine.
-
Applicability of the developed method was shown via viability monitoring during a continuous cultivation with triggered pH shifts.
-
A logarithmic trend was observed between the developed FCM method and the state-of-the-art method, plating assay.
Similar content being viewed by others

Introduction
Flow cytometry (FCM) has shown to be a powerful technique for analyzing a broad spectrum of cell parameters on a single cell level (Díaz et al. 2010; Adan et al. 2017; Maciorowski et al. 2017; Robinson 2018). It has been used for decades for the characterization of organisms from all domains of life (Scheper et al. 1987; Forment et al. 2012; Vees et al. 2020) and most applications of FCM are based upon fluorescence labelling. An important application of FCM in biotechnology is the determination of live and dead cells in a cultivation which is a key parameter for bioprocess development and control (Rieseberg et al. 2001). Successful applications of FCM have been published for the well-known mesophilic industrial production host Escherichia coli (Wurm et al. 2017; Kopp et al. 2020) and for Pichia pastoris (Hohenblum et al. 2003), highlighting how bioprocess development can benefit from FCM-based viability monitoring. These studies showed that measuring the ratio of dead cells to the total cell count can be used for monitoring the effect of process parameters (Wurm et al. 2017) and on the other hand the effect of different cultivation strategies (Hohenblum et al. 2003; Kopp et al. 2020) on process performance.
In recent years, growing interest in more exotic biotechnological production hosts can be observed. Members of the order Sulfolobales have been proposed as potential key organisms for future bioprocesses, harboring unique metabolic pathways and many valuable products such as extremozymes, highly stable lipids in their cell membrane as well as antibiotic complexes (Quehenberger et al. 2017; Rastädter et al. 2020). Sulfolobus acidocaldarius has emerged as an important model organism for the phylum Crenarchaeota due to its stable and fully sequenced genome (Chen et al. 2005) and its well-developed toolbox for genetic engineering (Wagner et al. 2012; Peng et al. 2017). It was only recently considered as an organism of high potential in biotechnological applications (Zeldes et al. 2015; Quehenberger et al. 2017; Schocke et al. 2019). Hence, physiological strain characterization is only in its beginnings (Rastädter et al. 2021) and no at-line methods for viability determination for fast identification of critical process parameters exist for S. acidocaldarius. Although, in the literature a number of fluorescence based, culture-independent detection methods for Sulfolobales species can be found (Brock et al. 1972; Bernander and Poplawski 1997; Hjort and Bernander 1999; Bernander 2007; Han et al. 2017), these methods do not assess the cell viability. Therefore, the current state-of-the-art method for determining the viability of a population is still via plating assay (Lindström and Sehlin 1989; Han et al. 2017). The outcome is time-delayed and depends on various factors for accurate results, such as correct dilution of cell suspension prior to plating, correct counting as well as subjective definition of a colony.
Generally, a cell is determined viable if it is capable of reproduction. While this property is directly assessed by plating assays that yield colony forming units (CFUs) as a response, for fast at-line estimation of viability it is necessary to find proxies that can be determined independently of time-consuming incubation periods. Since metabolic activity is required for cell division, measuring the activity of representative housekeeping enzymes, like esterases, can be used as an indirect, though at-line way for determining the viability (Nyström 2001; Cangelosi and Meschke 2014). Following this principle, FCM analysis combined with fluorescent dyes, such as fluorescein diacetate (FDA) (Xiao et al. 2011), can evaluate the cell’s metabolic activity, thereby allowing the estimation of the viability of a microbial population on a cellular level.
This study aims to develop an at-line method for bioprocess monitoring of the viability of S. acidocaldarius by using FCM in combination with suitable fluorescent dyes as a tool for efficient bioprocess monitoring. This method is then compared to the state-of-the-art method, plating assay, during a continuous bioreactor cultivation with triggered changes in viability via shifts in the pH value.
Material and methods
Fluorescence microscopy
A Leica DMI 8 fluorescence microscope (Leica Microsystems, Germany) equipped with a mercury light source was used for screening of fluorescence dyes. Three filter configurations were available: Filter 1, excitation (ex.) 532–558 nm/emission (em.) 570–640 nm); Filter 2, ex. 450–490 nm / em. 500–550 nm; Filter 3, ex. 600–660 nm / em. 662–738 nm. Undiluted cell suspensions were applied on glass slides and after addition of fluorescent dyes samples were incubated in the dark over a range of 5–50 min before investigation with the microscope.
Screening of fluorescent dyes
Eight different fluorescent dyes were investigated regarding their suitability for cell staining of S. acidocaldarius (Table 1).
Strain and bioreactor setup
Sulfolobus acidocaldarius DSM 639, obtained at German Collection of Microorganisms and Cell Cultures (DSMZ, Germany), was grown continuously in a 2 L Biostat A-plus bioreactor (Sartorius, Germany). The culture was stirred at 350 rpm, supplied with 0.23 vvm (0.45 L/h) pressurized air and kept at a constant temperature of 75 °C. The pH was measured with an Easyferm Plus Electrode (Hamilton, USA) and the dissolved oxygen (dO2) was monitored by a VisiFerm DO225 probe (Hamilton, USA). pH was adjusted by automatic addition of 4.8% (v/v) H2SO4. CO2 and O2 concentrations in the exhaust gas were measured using a gas analyzing unit (Müller Systems AG, Switzerland). The cultivation was monitored and controlled using the Lucullus process control system (SecureCell, Switzerland). The batch phase was started with an initial OD600 of 0.275 in 1.5 L of Vienna Defined (VD) Medium (Quehenberger et al. 2019) with modified concentrations of carbon sources (2 g/L monosodium glutamate (MSG), 1 g/L d-glucose). During the fed-batch phase an exponential feed was applied, starting with 14.8 g/h and a growth rate of 0.035 h−1. After reaching 2 L working volume a dilution rate of 0.03 h−1 was established and the chemostat phase was started. Feed used during fed-batch and chemostat phases contained a 5-times concentrated VD Medium with modified carbon source concentrations (9.5 g/L MSG, 4.5 g/L d-glucose and 0.5 g/L NZ-amine (Sigma, USA, a protein hydrolysate containing all 20 amino acids). For maintaining a constant volume during chemostat cultivation, cell broth was pumped out of the reactor via a bleed tube at a fixed height.
Biomass for the following viability experiments was harvested during the chemostat phase. To trigger changes in viability, the pH was changed from the standard value of 3.0 to 2.0 and 1.5, respectively.
Biomass determination
Optical density (OD600) was determined via a spectrophotometer (ONDA V-10 PLUS, XS instruments, Italy) at 600 nm against a blank of deionized water.
Plating assay: viability determination
Since plating is the state-of-the-art method for viability determination with S. acidocaldarius (Lindström and Sehlin 1989; Han et al. 2017), this method was used as benchmark in the present study. The generated response of the plating method are CFUs, consequently this method assesses the capability of replication. 400 mL 1.2% gelrite and 400 mL 2× concentrated brock medium (Brock et al. 1972; Quehenberger et al. 2019) supplemented with 0.4 g CaCl2 and 0.76 g MgCl2 were prepared aseptically. Glucose with a final concentration of 2 g/L and 1 g/L g NZ-Amine served as the carbon sources. After their preparation the two solutions were mixed and the pH was set to 3.0 with 4.8% H2SO4 prior to pouring á 20 mL into 94 × 16 mm petri dishes (Greiner Bio-One, Austria). Depending on the OD600 and on the expected CFUs the cell broth after sampling was diluted 10–4 to 5·10–6 with VD Medium without carbon sources. 50 µL of this suspension was pipetted onto the plate and dispersed. After 6 days at 75 °C incubation, the CFUs were determined. Each sampling point was plated in four replicates. The specific viability, Vplating [CFU/ml/OD600], was calculated as follows: \({V}_{plating}= \frac{CFU*dilution factor}{O{D}_{600}*{Volume}_{plated}}\).
Flow cytometry: viability determination
At each sampling point, 300 µL of cell broth were centrifuged in a 2 mL Eppendorf tube at 10,000×g, 4 °C and for 10 min. The cell pellet was washed twice and resuspended with 0.2 µm-filtered 10 mM phosphate buffered saline (PBS), pH 5.5. In between the washing steps, a five-minute centrifugation step at 20,000×g and 4 °C was carried out. Then, 600 µL of PBS buffer were added to the 300 µL washed cell suspension to yield a 1:3 dilution. 4.5 µL fluorescein diacetate (FDA, 5 g/L in acetone, Sigma- Aldrich, USA) were added to the obtained 900 µL. The sample was then incubated for 10 min at 37 °C in a thermoblock. After incubation, another centrifugation step at 20,000×g, 4 °C and for 5 min occurred to reduce background fluorescence caused by released fluorescein in the supernatant. The cell pellet was resuspended in 900 µL PBS buffer.
After FDA staining, the sample was diluted 1:3000 with PBS buffer to a final OD of 0.002 to 0.003. 50 µL concanavalin A (ConA)-rhodamine (5 g/L, Vector Laboratories, USA) were centrifuged prior to use to remove any possible protein aggregates which may form during storage. Shortly before measuring, 1 µL of ConA-rhodamine was added to 1 mL diluted FDA-stained cell suspension. The measurement was performed using a Cyflow Cube 8 flow cytometer (Sysmex, Germany), equipped with a 488 nm blue and 532 nm green laser. Emission spectra were obtained with fluorescence channels FL1, 536/40 nm bandpass, and FL4, 610/30 nm bandpass filter. Additionally, a forward scatter (FSC) and side scatter (SSC) detection was available. As all cells are stained by ConA-rhodamine, the emission spectra were acquired by using the FL4 as a trigger parameter. Opensource software FCSalyzer (Mostböck) was used for data visualization. The viability identified via the FCM was termed VFCM and was determined as follows: \({V}_{FCM}=\frac{viable cells}{all cells}\).
To obtain non-viable cells, the cells were killed via suffocation at high temperature. This was done by pouring 1 mL of cell suspension in a 2 mL Eppendorf tube which was then sealed with parafilm. The tube was put in a 100 mL Erlenmeyer flask filled with water and closed off with aluminum foil. The flask was transferred to a 75 °C-oil bath and was incubated over night while shaking. By doing this procedure, the remaining oxygen in the cell suspension is consumed resulting in depletion of ATP. Eventually, this leads to acidification of the cytosolic pH, which can no longer be maintained at the physiological value of 6.5 due to inactivity of proton pumps and simultaneous influx of H+ ions form the surrounding medium. The respective non-viable cell suspension sample was then treated like mentioned above. For calibration purposes and to determine the sensitivity of the method, the non-viable and viable samples after FDA staining were mixed in the respective ratios.
Results
Method development
Screening of fluorescence dyes
Compared to mammalian cells and Bacteria, for Archaea only a limited number of fluorescent dyes are commercially available (Johnson and Spence 2010). Nevertheless, SYTO 9 and acridine orange (AO) have already been tested for the use in Archaea and have been reported to successfully stain Sulfolobus cells (Brock et al. 1972; Leuko et al. 2004) and detect dead/all cells in haloarchaea (Leuko et al. 2004). In this study, 8 fluorescent dyes, listed in Table 1, were tested regarding their applicability for viability determination of S. acidocaldarius. Only 5 of the 8 investigated dyes led to fluorescent cells under the fluorescence microscope: AO, FDA, SYTO 9, ConA-rhodamine and propidium iodide. Since the viability determination of a population via the fluorescence microscope is time consuming and highly operator dependent, and hence would defeat the purpose of finding an at-line monitoring tool, a fluorescence microscope-based viability assay was not pursued within this study. Instead, an FCM-based approach was chosen as this method generates results within minutes after the sample is prepared.
Cell staining with FCM
FCM is described as a reproducible method for acquiring optical and fluorescence information of cultures on a single cell level (Rieseberg et al. 2001; Díaz et al. 2010). Generally, to determine viability of a cell culture via FCM, at least one fluorescent stain that highlights a specific characteristic such as viable or non-viable is needed. Cells can either be distinguished from the background noise due to size in the forward scatter or by using an additional fluorescent dye that specifically stains all cells. By combining these characteristics, two populations can be identified. Out of the five dyes that stained cells in the fluorescence microscope, only AO, FDA and ConA-rhodamine also showed a fluorescent signal when investigated with the flow cytometer. AO permeates cells regardless of their membrane integrity and subsequently leads to fluorescence emission when bound to intracellular nucleic acid (Martens-Habbena and Sass 2006). FDA staining is based on development of fluorescence upon cleavage of the FDA molecule by cellular esterases. The produced fluorescein accumulates inside the cell, resulting in green fluorescence when excited. Consequently, compromised cells with a reduced amount of esterase activity exhibit a reduced fluorescence signal compared to healthy cells (Jones and Senft 1985). Although, complementary staining properties are given in case of AO (stains all cells) and FDA (only living cells) it is not feasible to use the two dyes simultaneously in a single assay, since both dyes have similar emissions maxima (526 vs. 520 nm) and consequently, due to spectral overlap their signals cannot be discriminated. ConA-rhodamine was used instead, which binds to α-linked mannose present in the core oligosaccharides in the membrane glycoproteins (Fontaniella et al. 2004). In Sulfolobales mannose is present in the glycoslylated S-layer protein SlaA and flagellin FlaB (Elferink et al. 2001; Peyfoon et al. 2010; Zolghadr et al. 2010; Meyer et al. 2011). ConA conjugated with rhodamine (stains all cells), which has an emissions maximum of 575 nm, can be used in combination with FDA. Hence, the rhodamine emission in FL4 (610/30 nm bandpass) determined all cells, while the fluorescence signal originating from FDA hydrolysis in FL1 (236/40 nm bandpass) identified the viable cells. Fluorescence microscope images, showing cells stained with FDA and ConA-rhodamine and an overlay of both figures can be seen in the Additional file 1. Thereby it is shown that all cells exhibit red fluorescence resulting from ConA-rhodamine (Additional file 1: Figure S1B). However, viable cells additionally exhibit green fluorescent from FDA staining (Additional file 1: Figure S1A, C). In this paper, for better readability metabolic inactive/active cells according to the FCM are described as non-viable/viable.
Determination of viability via FCM (VFCM)
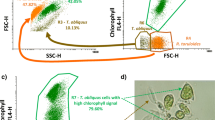
By setting the trigger parameter to FL4, only particles that are ConA-rhondamine stained were recorded, thereby reducing particle events. While PBS buffer spiked with ConA-rhodamine still generated background noise, caused by unspecific fluorescence and aggregates (Fig. 1A), the cell population (gated in Fig. 1B) could be clearly distinguished from this background via the FSC and SSC plot due to the cell size and form. The cell gate was then used for determining the viability in FL1 (showing FDA stained cells) versus FL4 plot (Fig. 1C). On the left side with low FL1 intensity the non-viable cells can be seen and on the right side the viable cells.

Gate definition for viability evaluation of Sulfolobus acidocaldarius. A density plot of side scatter versus forward scatter for ConA-rhodamine in PBS buffer, showing the background, color code: red-high to purple-low; B density plot of side scatter versus forward scatter for cells stained with FDA and ConA-rhodamine, showing the cell gate; C density plot of FL1 (536/40 nm bandpass) versus FL4 (610/30 nm bandpass) of cells gated in B; D statistics of FL1 vs. FL4 shown in C
Sensitivity analysis and comparison with state-of-the-art
Cells were killed by O2 depletion and then mixed with viable cells in certain ratios. Since the sample of viable cells already contained around 7% non-viable cells, the amount was subtracted from the calculated percentage of live cells based on the mixed ratios. The trend between measured viable cells and calculated viable cells (according to the mixed ratios) in the measured populations shows a linear correlation between the determined viability (VFCM) and the percentage of calculated viable cells in a sample over the range of 2.5–92.7% (Fig. 2A). For comparison with the state-of-the-art method plating (VFCM vs. Vplating), the same mixtures of viable and non-viable cells were also cultivated on gelrite plates and the CFUs were determined 6 days later. The relationship between the CFUs of the plated viable/non-viable mixtures and the VFCM shows a logarithmic trend (Fig. 2B).

A Sensitivity analysis of viability according to mixed ratios [%] vs. VFCM [%]. Viability according to mixed ratios [%] were obtained by mixing different ratios of non-viable and viable cell populations. VFCM cells [%] were measured by the flow cytometer and evaluated according to Fig. 1. B Comparison of state-of-the-art method log[Vplating (CFU/mL/OD600)] versus VFCM [%]
Applicability test of FCM method
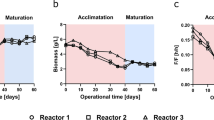
For showing the applicability of the developed FCM method, continuously cultivated cells were subjected to stress induced by pH changes. The viability was monitored by FCM (VFCM) and plating (Vplating) throughout these pH changes. VFCM and Vplating decreased in response to the shift towards more acidic conditions (Fig. 3). This effect was less prominent after the pH value was changed from the pH optimum of 3.0 (Rastädter et al. 2021) to 2.0 and more severe when a pH value of 1.5 was applied. After shifting back to the pH optimum of 3.0, the cells recovered in both cases and VFCM and Vplating resumed the same values as before the pH shift. The relation between VFCM and VCFU is shown in Fig. 4. Similar to the relationship in the sensitivity analysis where cells were killed via oxygen depletion (Fig. 2B) also here a logarithmic relationship was observed as the pH shifts apparently impacted Vplating more severely than VFCM.

Comparison of viability measurements of Sulfolobus acidocaldarius in response to the shift in pH value, observed over time [h]. The viability was determined by flow cytometry (VFCM [%]) and by plating assay (Vplating [%]). Both pH shifts from 3.0 to 2.0 and to 1.5, respectively caused a drop in both viability-determining methods as well as in OD600. In this case, Vplating measured at the beginning of the experiment was set to 100%, and for determining Vplating [%], the CFUs of each sample were then divided by this initial value

Logarithmic trend of VFCM [%] versus log (Vplating) with a correlation factor of R2 = 0.90
Discussion
FCM methods applied to other organism have shown to be a robust tool for getting an insight into the viability of a cell population (Xiao et al. 2006; Wurm et al. 2017; Vees et al. 2020). Additionally, it could be shown that the impact of process parameters such as feeding strategy (Xiao et al. 2006; Vees et al. 2020) and cultivation temperature (Wurm et al. 2017) can be monitored.
Despite significant background signals for all cells during ConA-rhodamine staining (Fig. 1A), a clear differentiation between the background and cells can be made when gating based on forward/side scatter, thereby adding information on characteristic shape/size of the cells (Fig. 1B). The resulting trend in the sensitivity analysis (Fig. 2A) showed a linear range between 2.45 and 92.72% of VFCM. A logarithmic correlation between state-of-the-art method plating assay and the developed FCM method was observed (Fig. 2B). A possible explanation for the logarithmic nature of the correlation could be that these methods investigate viability differently. Plating on gelrite plates [CFU/mL/OD600] and FDA/ConA-rhodamine staining combined with flow cytometry are based on different mechanisms to determine cell viability. With one method, the number of proliferable cells upon transfer to a different growth environment (gelrite plates) is obtained, while the other method examines the metabolic activity of each cell as an at-line measurement (Cangelosi and Meschke 2014). Apparently, cells harvested during the shift to pH 2.0 were already significantly impaired in their ability to form colonies in the gelrite plate assay. This circumstance was neither mirrored in the limited drop of OD600 nor in the moderate reduction of VFCM during this shift to pH 2.0, while all three parameters (Vplating, VFCM, OD600) dropped significantly when the pH was reduced even further to a value of 1.5.
Although it is the current standard method for viability determination (Lindström and Sehlin 1989; Han et al. 2017), the plating assay applied in this paper harbors limitations that can explain possible divergence in the viability when compared to VFCM. First, cells could retain metabolic activity while being unable to divide (Rollins and Colwell 1986; Nyström 2001; Cangelosi and Meschke 2014). The inability of proliferation despite metabolic activity could explain the drastic changes in Vplating upon pH value changes while the VFCM of the culture only moderately decreased (Fig. 3). This is also mirrored in the logarithmic relationship between VFCM and Vplating (Figs. 2B and 4). Suffocation (Fig. 2B) as well as a pH drop (Fig. 4) lead to a decrease of cytosolic pH as a result of too low proton pump activity (Anemüller et al. 1985; Gleissner et al. 1997; Baker-Austin and Dopson 2007). Due to the cytosolic pH decrease, adaption to this new condition is necessary and apparently at the beginning of this process the cells are metabolically active, but reproduction is stalled (Fig. 3). Secondly, in the plating assay the standard medium pH value is 3.0 (Wagner et al. 2012; Bischof et al. 2019). However, the pH value of the preceding cultivation conditions might differ from this pH optimum of 3.0. Hence, the cells have to overcome another pH adaption in order to grow, which could cause the lower viability in comparison to VFCM.
The time-delay for obtaining the results after sampling is still the biggest limitation, making plating assay-based bioprocess monitoring unfeasible. Additionally, wrong dilutions of the cell suspension prior to plating could lead either to too many colonies or too little to count, which both causes inaccurate results—a fatal error that cannot be corrected, since the actual sampling time already lies at least 6 days in the past. Incorrect dilutions during a FCM measurement can easily be detected and a re-run of the sample is possible immediately. In the evaluation process of plating, the timing for counting the colonies on the plate is of essence. If it is done too late two colonies can merge and appear as one, on the other hand small colonies can be overlooked in an early evaluation.
The collective disadvantages of plating show the need for a robust and at-line FCM method with high statistical significance due to high sample size and high event numbers to determine the viability in a cell population. The developed FCM method harbors many applications in the rising field of S. acidocaldarius bioprocessing. It can be used to determine which process parameters such as stirrer speed or aeration rate are critical and that within less than 1 h after sampling, compared to 6 days that are necessary to yield a result in form of visible colonies when applying a plating assay. Further, identifying stress parameters in the upscaling to an industrial process is facilitated. Thereby, this FCM method for bioprocess monitoring harbors the potential to pave the way for the first industrial process with S. acidocaldarius or related Crenarchaeota.
Availability of data and materials
Raw data that support the findings of this study are available from the corresponding author upon request.
Abbreviations
- AO:
-
Acridine orange
- CFUs:
-
Colony forming units
- ConA:
-
Concanavalin A
- em.:
-
Emission
- ex.:
-
Excitation
- FCM:
-
Flow cytometry
- FDA:
-
Fluorescein diacetate
- FSC:
-
Forward scatter
- OD600 :
-
Optical density
- PBS:
-
Phosphate buffered saline
- SSC:
-
Side scatter
- VFCM :
-
Specific viability determined via flow cytometry
- Vplating :
-
Specific viability determined via plating assay
References
Adan A, Alizada G, Kiraz Y, Baran Y, Nalbant A (2017) Flow cytometry: basic principles and applications. Crit Rev Biotechnol 37:163–176. https://doi.org/10.3109/07388551.2015.1128876
Anemüller S, Lübben M, Schäfer G (1985) The respiratory system of Sulfolobus acidocaldarius, a thermoacidophilic archaebacterium. FEBS Lett 193:83–87. https://doi.org/10.1016/0014-5793(85)80084-3
Baker-Austin C, Dopson M (2007) Life in acid: pH homeostasis in acidophiles. Trends Microbiol 15:165–171. https://doi.org/10.1016/j.tim.2007.02.005
Bernander R (2007) The cell cycle of Sulfolobus. Mol Microbiol 66:557–562. https://doi.org/10.1111/j.1365-2958.2007.05917.x
Bernander R, Poplawski A (1997) Cell cycle characteristics of thermophilic archaea. J Bacteriol 179:4963–4969. https://doi.org/10.1128/jb.179.16.4963-4969.1997
Bischof LF, Haurat MF, Hoffmann L, Albersmeier A, Wolf J, Neu A, Pham TK, Albaum SA, Jakobi T, Schouten S, Neumann-Schaal M, Wright PC, Kalinowski J, Siebers B, Albers S-V (2019) Early Response of Sulfolobus acidocaldarius to Nutrient Limitation. Front Microbiol. https://doi.org/10.3389/fmicb.2018.03201
Brock TD, Brock KM, Belly RT, Weiss RL (1972) Sulfolobus: a new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Archiv Mikrobiol 84:54–68. https://doi.org/10.1007/BF00408082
Cangelosi GA, Meschke JS (2014) Dead or alive: molecular assessment of microbial viability. Appl Environ Microbiol 80:5884–5891. https://doi.org/10.1128/AEM.01763-14
Chen L, Brügger K, Skovgaard M, Redder P, She Q, Torarinsson E, Greve B, Awayez M, Zibat A, Klenk H-P, Garrett RA (2005) The genome of Sulfolobus acidocaldarius, a Model organism of the Crenarchaeota. J Bacteriol 187:4992–4999. https://doi.org/10.1128/JB.187.14.4992-4999.2005
Díaz M, Herrero M, García LA, Quirós C (2010) Application of flow cytometry to industrial microbial bioprocesses. Biochem Eng J 48:385–407. https://doi.org/10.1016/j.bej.2009.07.013
Elferink MGL, Albers S-V, Konings WN, Driessen AJM (2001) Sugar transport in Sulfolobus solfataricus is mediated by two families of binding protein-dependent ABC transporters. Mol Microbiol 39:1494–1503. https://doi.org/10.1046/j.1365-2958.2001.02336.x
Fontaniella B, Millanes A-M, Vicente C, Legaz M-E (2004) Concanavalin A binds to a mannose-containing ligand in the cell wall of some lichen phycobionts. Plant Physiol Biochem 42:773–779. https://doi.org/10.1016/j.plaphy.2004.09.003
Forment JV, Walker RV, Jackson SP (2012) A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry A 81A:922–928. https://doi.org/10.1002/cyto.a.22155
Gleissner M, Kaiser U, Antonopoulos E, Schäfer G (1997) The archaeal SoxABCD complex is a proton pump in Sulfolobus acidocaldarius. J Biol Chem 272:8417–8426. https://doi.org/10.1074/jbc.272.13.8417
Han W, Xu Y, Feng X, Liang YX, Huang L, Shen Y, She Q (2017) NQO-induced DNA-less cell formation is associated with chromatin protein degradation and dependent on A0A1-ATPase in Sulfolobus. Front Microbiol 8:1480. https://doi.org/10.3389/fmicb.2017.01480
Hjort K, Bernander R (1999) Changes in cell size and DNA content in Sulfolobus cultures during dilution and temperature shift experiments. J Bacteriol 181:5669–5675
Hohenblum H, Borth N, Mattanovich D (2003) Assessing viability and cell-associated product of recombinant protein producing Pichia pastoris with flow cytometry. J Biotechnol 102:281–290. https://doi.org/10.1016/S0168-1656(03)00049-X
Johnson I, Spence MTZ (2010) The molecular probes handbook. Life Technologies Corporation, Carlsbad
Jones KH, Senft JA (1985) An improved method to determine cell viability by simultaneous staining with fluorescein diacetate-propidium iodide. J Histochem Cytochem 33:77–79. https://doi.org/10.1177/33.1.2578146
Kopp J, Kittler S, Slouka C, Herwig C, Spadiut O, Wurm DJ (2020) Repetitive fed-batch a promising process mode for biomanufacturing with E. Coli. Front Bioeng Biotechnol. 8:573607. https://doi.org/10.3389/fbioe.2020.573607
Leuko S, Legat A, Fendrihan S, Stan-Lotter H (2004) Evaluation of the LIVE/DEAD BacLight Kit for detection of extremophilic archaea and visualization of microorganisms in environmental hypersaline samples. Appl Environ Microbiol 70:6884–6886. https://doi.org/10.1128/AEM.70.11.6884-6886.2004
Lindström EB, Sehlin HM (1989) High efficiency of plating of the thermophilic sulfur-dependent Archaebacterium Sulfolobus acidocaldarius. Appl Environ Microbiol 55:3020–3021
Maciorowski Z, Chattopadhyay PK, Jain P (2017) Basic multicolor flow cytometry. Curr Protoc Immunol. https://doi.org/10.1002/cpim.26
Martens-Habbena W, Sass H (2006) Sensitive determination of microbial growth by nucleic acid staining in aqueous suspension. Appl Environ Microbiol 72:87–95. https://doi.org/10.1128/AEM.72.1.87-95.2006
Meyer BH, Zolghadr B, Peyfoon E, Pabst M, Panico M, Morris HR, Haslam SM, Messner P, Schäffer C, Dell A, Albers S-V (2011) Sulfoquinovose synthase – an important enzyme in the N-glycosylation pathway of Sulfolobus acidocaldarius. Mol Microbiol 82:1150–1163. https://doi.org/10.1111/j.1365-2958.2011.07875.x
Mostböck S FCSalyzer. In: SourceForge. https://sourceforge.net/projects/fcsalyzer/. Accessed 1 Sep 2021
Nyström T (2001) Not quite dead enough: on bacterial life, culturability, senescence, and death. Arch Microbiol 176:159–164. https://doi.org/10.1007/s002030100314
Peng N, Han W, Li Y, Liang Y, She Q (2017) Genetic technologies for extremely thermophilic microorganisms of Sulfolobus, the only genetically tractable genus of crenarchaea. Sci China Life Sci 60:370–385. https://doi.org/10.1007/s11427-016-0355-8
Peyfoon E, Meyer B, Hitchen PG, Panico M, Morris HR, Haslam SM, Albers S-V, Dell A (2010) The S-layer glycoprotein of the crenarchaeote Sulfolobus acidocaldarius is glycosylated at multiple sites with chitobiose-linked N-glycans. Archaea. https://doi.org/10.1155/2010/754101
Quehenberger J, Shen L, Albers S-V, Siebers B, Spadiut O (2017) Sulfolobus—a potential key organism in future biotechnology. Front Microbiol 8:2474. https://doi.org/10.3389/fmicb.2017.02474
Quehenberger J, Albersmeier A, Glatzel H, Hackl M, Kalinowski J, Spadiut O (2019) A defined cultivation medium for Sulfolobus acidocaldarius. J Biotechnol 301:56–67. https://doi.org/10.1016/j.jbiotec.2019.04.028
Rastädter K, Wurm DJ, Spadiut O, Quehenberger J (2020) The cell membrane of Sulfolobus spp.—Homeoviscous adaption and biotechnological applications. Int J Mol Sci 21:3935. https://doi.org/10.3390/ijms21113935
Rastädter K, Wurm DJ, Spadiut O, Quehenberger J (2021) Physiological characterization of Sulfolobus acidocaldarius in a controlled bioreactor environment. Int J Environ Res Public Health 18:5532. https://doi.org/10.3390/ijerph18115532
Rieseberg M, Kasper C, Reardon KF, Scheper T (2001) Flow cytometry in biotechnology. Appl Microbiol Biotechnol 56:350–360. https://doi.org/10.1007/s002530100673
Robinson JP (2018) Overview of flow cytometry and microbiology. Curr Protoc Cytom 84:e37. https://doi.org/10.1002/cpcy.37
Rollins DM, Colwell RR (1986) Viable but nonculturable stage of Campylobacter jejuni and its role in survival in the natural aquatic environment. Appl Environ Microbiol 52:531–538
Scheper T, Hitzmann B, Rinas U, Schügerl K (1987) Flow cytometry of Escherichia coli for process monitoring. J Biotechnol 5:139–148. https://doi.org/10.1016/0168-1656(87)90010-1
Schocke L, Bräsen C, Siebers B (2019) Thermoacidophilic Sulfolobus species as source for extremozymes and as novel archaeal platform organisms. Curr Opin Biotechnol 59:71–77. https://doi.org/10.1016/j.copbio.2019.02.012
Vees CA, Veiter L, Sax F, Herwig C, Pflügl S (2020) A robust flow cytometry-based biomass monitoring tool enables rapid at-line characterization of S. cerevisiae physiology during continuous bioprocessing of spent sulfite liquor. Anal Bioanal Chem 412:2137–2149. https://doi.org/10.1007/s00216-020-02423-z
Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J, Albers S-V (2012) Versatile genetic tool box for the crenarchaeote Sulfolobus acidocaldarius. Front Microbiol. https://doi.org/10.3389/fmicb.2012.00214
Wurm DJ, Marschall L, Sagmeister P, Herwig C, Spadiut O (2017) Simple monitoring of cell leakiness and viability in Escherichia coli bioprocesses—a case study. Eng Life Sci 17:598–604. https://doi.org/10.1002/elsc.201600204
Xiao X, Han Z, Chen Y, Liang X, Li H, Qian Y (2011) Optimization of FDA–PI method using flow cytometry to measure metabolic activity of the cyanobacteria, Microcystis aeruginosa. Phys Chem Earth 36:424–429. https://doi.org/10.1016/j.pce.2010.03.028
Xiao A, Zhou X, Zhou L, Zhang Y (2006) Improvement of cell viability and hirudin production by ascorbic acid in Pichia pastoris fermentation. Appl Microbiol Biotechnol 72:837–844. https://doi.org/10.1007/s00253-006-0338-1
Zeldes BM, Keller MW, Loder AJ, Straub CT, Adams MWW, Kelly RM (2015) Extremely thermophilic microorganisms as metabolic engineering platforms for production of fuels and industrial chemicals. Front Microbiol. https://doi.org/10.3389/fmicb.2015.01209
Zolghadr B, Klingl A, Koerdt A, Driessen AJM, Rachel R, Albers S-V (2010) Appendage-mediated surface adherence of Sulfolobus solfataricus. J Bacteriol 192:104–110. https://doi.org/10.1128/JB.01061-09
Acknowledgements
Not applicable.
Funding
J.Q. acknowledges funding from the Austrian Science Fund (FWF) via the project “CO2 fixation in extreme conditions” (Project Nr.: I 4508-B). K.R. acknowledges funding from L'ORÉAL Austria and the Austrian Academy of Sciences (ÖAW) via the L’ORÉAL Austria Fellowship Programme 2021. The authors acknowledge the TU Wien Bibliothek for financial support through its Open Access Funding Program.
Author information
Authors and Affiliations
Contributions
Conceptualization, KR, DJW, OS and JQ; investigation, KR, AT and JQ; writing—original draft preparation, KR; writing—review and editing, AT, JQ, DJW and OS; visualization, KR; supervision, JQ; funding acquisition, OS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no conflict of interest. D.J.W. and J.Q. disclose their employment at the company NovoArc GmbH which is engaged in lipid research and commercialization of archaeal lipids.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig. S1. Fluorescence microscope. Mixture of dead and alive cells of Sulfolobus acidocaldarius stained with fluorescein diacetate (FDA) and concanvalin A conjugated with rhodamine and investigated with a Leica DMI 8 fluorescence microscope (Leica Microsystems, Germany). A: Image acquired via the equipped filter 2 (excitation (ex.) 450-490 nm / emission (em.) 500-550 nm). B: Image acquired via the equipped filter 1 (ex. 532-558 nm /em. 570-640 nm). C: overlay of figures A and B showing metabolically active cells as yellow dots (overlay of red and green)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rastädter, K., Tramontano, A., Wurm, D.J. et al. Flow cytometry-based viability staining: an at-line tool for bioprocess monitoring of Sulfolobus acidocaldarius. AMB Expr 12, 107 (2022). https://doi.org/10.1186/s13568-022-01447-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-022-01447-1