
Abstract
Mycoplasma bovis is responsible for various inflammatory diseases in cattle. The prevention and control of M. bovis are complicated by the absence of effective vaccines and the emergence of multidrug-resistant strains, resulting in substantial economic losses worldwide in the cattle industry. Lipoproteins, vital components of the Mycoplasmas cell membrane, are deemed potent antigens for eliciting immune responses in the host upon infection. However, the functions of lipoproteins in M. bovis remain underexplored due to their low sequence similarity with those of other bacteria and the scarcity of genetic manipulation tools for M. bovis. In this study, the lipoprotein LppA was identified in all examined M. bovis strains. Utilizing immunoelectron microscopy and Western blotting, it was observed that LppA localizes to the surface membrane. Recombinant LppA demonstrated dose-dependent adherence to the membrane of embryonic bovine lung (EBL) cells, and this adhesion was inhibited by anti-LppA serum. In vitro binding assays confirmed LppA’s ability to associate with fibronectin, collagen IV, laminin, vitronectin, plasminogen, and tPA, thereby facilitating the conversion of plasminogen to plasmin. Moreover, LppA was found to bind and enhance the accumulation of Annexin A2 (ANXA2) on the cell membrane. Disrupting LppA in M. bovis significantly diminished the bacterium’s capacity to adhere to EBL cells, underscoring LppA’s function as a bacterial adhesin. In conclusion, LppA emerges as a novel adhesion protein that interacts with multiple host extracellular matrix proteins and ANXA2, playing a crucial role in M. bovis’s adherence to host cells and dissemination. These insights substantially deepen our comprehension of the molecular pathogenesis of M. bovis.
Similar content being viewed by others

Introduction
Mycoplasma bovis, a significant pathogen linked with bovine respiratory disease syndrome, instigates intense inflammatory responses, manifesting as pneumonia, mastitis, arthritis, otitis, and keratoconjunctivitis. Globally widespread, M. bovis inflicts considerable economic losses on the cattle industry [1]. M. bovis infection is implicated in various chronic diseases in feedlots, representing a formidable obstacle in the health management protocols of farms [2].
The adhesion to and invasion of host cells are crucial phases in the M. bovis infection process. M. bovis adheres to bovine tracheobronchial epithelial cells, which aids colonization of the bacteria within the host’s lungs [2]. Moreover, this pathogen can adhere to and penetrate diverse bovine cell types, potentially leading to an array of clinical diseases in cattle [3,4,5]. M. bovis engages with the extracellular matrix (ECM) and plasminogen, enhancing its adhesion, invasion, and spread within the host [6]. Nonetheless, the exact M. bovis factors integral to these interactions are yet to be distinctly understood. Variable surface protein (Vsp) has been recognized as a crucial adhesion-associated gene in M. bovis, known for provoking a robust immune reaction in the host [7, 8]. However, ongoing alterations in the expression, structure, and antigenicity of the Vsp protein result in an ineffective immune response, allowing M. bovis to consistently dodge the host’s antibody defenses [9, 10]. Although numerous M. bovis adhesion-related proteins, including NOX, MbfN, FBA, TrmFO, α-Enolase, MilA, P27, VpmaX, P26, and Mbov-0503, have been uncovered in recent times [11,12,13,14,15,16,17,18,19,20,21], devising an efficacious M. bovis vaccine remains problematic. Consequently, an immediate demand exists to pinpoint and investigate other potential candidate targets for vaccine development.
Membrane-associated proteins, especially lipoproteins, play a pivotal role in host–bacteria interactions [16, 22]. Presumed to be exposed to the extracellular environment, these proteins potentially facilitate not only the adhesion and invasion of pathogens into host cells but also the elicitation of immune responses [23]. Lipoproteins serve as key functional proteins for Mycoplasma, integral to various life processes of the microorganism, and stand as promising candidates for vaccine development [16]. Studies have identified lipoprotein LppA as a principal T-cell antigen in Mycoplasma mycoides subsp. mycoides [24]. An adenoviral vector expressing lipoprotein A (Ad5-LppA) elicited robust specific humoral and cell-mediated immune responses in mice, suggesting its potential as a vaccine against the severe respiratory disease contagious bovine pleuropneumonia [25]. However, the role of LppA in M. bovis adhesion and invasion remains unclarified. Most pathogenic microorganisms target ECM components from the host for adhesion and colonization [16, 26]. Mycoplasma can bind to ECM components such as collagen, laminin, fibronectin, plasminogen, and the glycosaminoglycan heparin [17, 27,28,29]. This interaction facilitates the adherence of pathogenic bacteria to host cells and fosters their subsequent invasion. Mycoplasma spp. is one of the smallest self-replicating organisms with reduced genomes. The lack of efficient genetic manipulation tools for these distinct organisms has impeded the characterization of virulence-related genes and the understanding of pathogenic mechanisms in these bacteria.
The present study investigated the role of LppA in M. bovis infection, thereby augmenting our comprehension of the pathogenic mechanisms at play in M. bovis. The study findings unveiled that LppA engages with host ECM components, enhances plasminogen activation through tissue plasminogen activator (tPA), and prompts the enrichment of ANXA2 protein on the cell membrane. These collective actions regulate the adhesion of M. bovis to host cells.
Materials and methods
Cells and strains
Embryonic bovine lung (EBL) cells and the HEK-293T cell line (commonly referred to as 293T) were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco, USA), enriched with 10% fetal bovine serum (Gibco), and maintained at 37 °C in a 5% CO2 atmosphere. EBL cells, derived from the lung tissue of fetal cattle at approximately seven-months of gestation, serve as a significant cellular model for investigating viral and bacterial diseases in cattle [30]. The 293T cell line originates from HEK 293 cells and stably expresses the SV40 large T antigen [31]; it is frequently utilized for protein expression and the examination of protein–protein interactions in vitro. M. bovis was propagated in a pleuropneumonia-like organism (PPLO) medium supplemented with 20% horse serum (Gibco), following established protocols [32]. Cultivation of mutant strains required an additional 100 μg/mL of kanamycin, while the complemented strain, produced by transforming the ΔLppA mutant with the pIRR45-LppA plasmid, necessitated 5 μg/mL of puromycin. Escherichia coli DH5α and BL21(DE3) strains were employed for gene cloning and protein expression, respectively, with E. coli BL21 (DE3) recognized as a prevalent T7 expression host suitable for transformation and protein synthesis.
Antibodies and reagents
Reagents such as mouse anti-fibronectin, donkey anti-mouse Alexa Fluor 488, and donkey anti-rabbit Alexa Fluor 647 were sourced from Thermo Fisher Scientific, USA. Rabbit anti-plasminogen was procured from AssayPro, USA. Rabbit antibodies against collagen IV, laminin, tPA, and vitronectin were supplied by Abcam, USA. Rabbit anti-HA, anti-ANXA2, and mouse anti-Flag antibodies were obtained from Proteintech Group, Inc., China. Biochemicals including bovine-derived fibronectin and plasminogen, as well as human-derived tPA, were purchased from Thermo Fisher Scientific, Cell Sciences, and Solarbio Science & Technology Co., Ltd., China, respectively. Human-derived collagen IV and laminin were acquired from Sigma-Aldrich, USA.
DNA constructs and transfection
The complete sequence of M. bovis LppA (GenBank accession no. CP002188.1, MBOVPG45_0682) was optimized for E. coli-preferred codons (see Supplementary material) and synthesized by Sangon Biotech. Plasmids pIRR45-LppA, pCAGGS-FLAG-N-LppA, pCAGGS-HA-N-ANXA2, and pET30a-LppA were constructed using an In-Fusion kit (TaKaRa). Specifically, to complement the mutant, we developed a complementation plasmid, pIRR45, employing primer pairs pIRR45-TfibZT-F/R and pIRR45-LppA-F/R (Additional file 1) to amplify the LppA gene (inclusive of its presumed promoter) and subcloned it into the pIRR45 plasmid. The resulting pIRR45-LppA recombinant plasmid was introduced into the ΔLppA mutant strain via PEG 8000, generating the complemented ΔLppA:LppA strain. Plasmids pCAGGS-FLAG-LppA were created using primer pairs pCAGGS-FLAG-ZT-F/R and pCAGGS-FLAG-LppA-F/R (Additional file 1). The cattle ANXA2 gene was cloned into pCAGGS-HA using primer pairs pCAGGS-FLAG-ZT-F/R and pCAGGS-HA-ANXA2-F/R (Additional file 1), yielding pCAGGS-HA-ANXA2 plasmids. Both pCAGGS-FLAG-LppA and pCAGGS-HA-ANXA2 plasmids were co-transfected into 293T cells using Lipofectamine 3000 (Invitrogen, USA), following the manufacturer’s guidelines. The coding region of the M. bovis LppA gene underwent double digestion with EcoRI and XhoI restriction enzymes and was then ligated into the similarly digested pET30a-LppA plasmid. The derivative plasmid, pET30a-LppA, was introduced into E. coli strain BL21(DE3) through heat shock.
Preparation of polyclonal antibodies against LppA
Polyclonal antibodies against M. bovis LppA were produced by immunizing 6-week-old Kunming mice with rLppA protein. Purified rLppA was emulsified with complete and incomplete Freund’s adjuvant (1:1, v/v), and the mice were immunized subcutaneously on four occasions at two-week intervals. Antisera were collected 2 weeks after the fourth immunization and titrated via enzyme-linked immunosorbent assay (ELISA). In brief, 96-well plates were coated with 10 μg/mL rLppA protein overnight at 4 °C. Following a block with 5% bovine serum albumin (BSA), double serial dilutions of both anti-LppA serum and pre-immune serum were applied to the wells and incubated at 37 °C for 30 min, succeeded by the addition of 100 µL of goat anti-mouse IgG-horseradish peroxidase (HRP) at a 1:10 000 dilution. After a 1-h incubation at room temperature, 3,3′,5,5′-tetramethylbenzidine (TMB) was added, and the reaction was terminated with H2SO4. Optical density (OD) readings were taken at 450 nm using an iMark microplate reader (Bio-Rad). The highest antibody dilution that met the criteria (OD value > 0.4) was recorded as the antibody titer. The specificity of the resulting polyclonal antibodies was confirmed through western blot analysis.
Bioinformatics analysis
The NCBI database (Accessed on June 13, 2023) was utilized to search for M. bovis isolates from locations worldwide, yielding 262 isolates (Additional file 2). A comprehensive set of 150 572 proteome sequences was compiled into a self-constructed M. bovis protein database (MB-DATABASE). Similarly, the NCBI database (Accessed on June 13, 2023) was explored for Mycoplasma isolates from global locations, uncovering 89 distinct Mycoplasma isolates (Additional file 2). In total, 71 084 proteome sequences were assembled into a Mycoplasma self-constructed protein database (MY-DATABASE). An LppA Hidden Markov Model (LppA-HMM) was utilized through the hmmbuild function of the HMMER 3.0 software [33]. Upon retrieving the MB-DATABASE or MY-DATABASE using LppA-HMM, the screening criteria were established at an e-value < 10−5 to identify sequences similar to LppA. NCBI BLAST (blastp program) searches were executed with the acquired amino acid sequences. Amino acid sequences labeled as “LppA protein” and “Mycoplas_LppA” in NCBI annotation or domain annotation were chosen. Subsequent multiple sequence alignments were conducted employing MAFFT software [34], and a neighbor-joining phylogenetic tree was reconstituted with 1000 bootstraps using MEGA 11 software [35]. The LppA domain structure was examined using the SMART databases [36], and the structural characteristic model of LppA was formulated in AlphaFold v2.0 [37]. The 1–350 amino acid sequence of LppA was scrutinized for antigenic epitope regions using Protean software (Lasergene; DNAStar 6.0).
Mycoplasma adhesion assay
EBL cells were allocated into a 12-well plate (1 × 105 cells per well) and incubated in 5% CO2 at 37 °C overnight. M. bovis was introduced to the EBL cells at a multiplicity of infection of 1000 and incubated for 1.5 h, after which the cells were rinsed thrice with 1 × phosphate-buffered saline. The cells were subsequently digested with TrypLE™ Express (Gibco), and dilutions were distributed on PPLO agar plates. Viable Mycoplasma cells were quantified by counting colonies following 3–5 days of cultivation.
Confocal microscopy and immunoelectron microscopy
EBL/293T cells were stabilized with 2% paraformaldehyde, permeabilized with 0.1% Triton X-100, and subsequently incubated with the specified antibodies. Images were captured using a Leica laser scanning confocal microscope and processed with LAS-AF-Lite 3.3.0 software. Immunogold electron microscopy was employed to delineate the subcellular localization of LppA. In brief, washed M. bovis cells were secured in 2% paraformaldehyde and 0.5% glutaraldehyde and processed at the Lilai Biomedicine Experiment Center (Chengdu, China). For immunogold labeling, mouse anti-LppA serum served as the primary antibody, while goat anti-mouse IgG linked to 10-nm colloidal gold particles (Beijing Biosynthesis Biotechnology) was utilized as the secondary antibody. The ultimately desiccated samples were examined using transmission electron microscopy (JEM-1400FLASH).
Immunoprecipitation (IP)
IP-mass spectrometry (IP-MS)
rLppA protein (10 μg) was incubated with EBL whole-cell lysates overnight at 4 °C. Anti-LppA serum was introduced to the mixture and rotated at 4 °C for 8 h. Immunoprecipitation experiments were conducted using protein A/G magnetic beads following the manufacturer’s instructions (Invitrogen), and the results were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) with silver staining and mass spectrometry. Briefly, 25 µL (0.25 mg) of Pierce Protein A/G Magnetic Beads were placed in a 1.5 mL microcentrifuge tube. 175 µL of 1 × TBS containing 0.05% Tween-20 (TBST) was added, and the tube was gently shaken for mixing. The tube was then placed in a magnetic stand to gather the beads, and the supernatant was discarded following a 1-min wash with 1 mL of TBST. The beads were collected with a magnetic stand, and the supernatant was removed. The mixture of Anti-LppA serum and rLppA protein was added to the tube containing pre-washed magnetic beads and incubated at room temperature for 1 h with agitation. The beads were subsequently immobilized using a magnetic stand, and the supernatant was reserved for analysis. The magnetic beads underwent two washes with 500 µL of TBST, followed by two washes with 500 µL of purified water. After each wash, the beads were collected using a magnetic stand, and the supernatant was discarded. After removal from the magnetic stand, 100 µL of 1 × SDS-PAGE loading buffer was added to the tube, thoroughly mixed, and heated at 95 °C for 10 min. Finally, the supernatant containing the target antigen was magnetically separated from the beads, and the supernatant was retained. Silver staining was executed using the Fast Silver Stain kit (Beyotime) per the manufacturer’s guidelines. Mass spectrometry analysis was conducted by Bioprofile (Shanghai) Technology Co., Ltd.
Co-IP
To investigate the LppA protein’s interaction with ANXA2, 293T cells were seeded into 6-well culture plates and transfected with pCAGGS-FLAG-N-LppA and pCAGGS-HA-N-ANXA2 plasmids. Transfected cells were harvested at 24 h post-transfection and lysed in NP-40 Lysis Buffer containing 1 mM protease inhibitor. The lysates were centrifuged at 12 000 × g for 10 min, and the supernatants, containing 500 μg of total protein, were incubated overnight with 10 μg of mouse monoclonal antibody against Flag, with gentle agitation at 4 °C. Immunoprecipitation experiments were conducted with Protein A/G magnetic beads as previously detailed. Western blot detection was carried out using anti-HA rabbit polyclonal antibody.
Western blot and dot blot analyses
Western blot
Protein samples were separated on a 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Amersham). The membranes were blocked with 5% BSA for 1 hat room temperature, followed by overnight incubation with primary antibodies at 4 °C. After three washes with TBST), the membranes were exposed to HRP-coupled goat anti-mouse antibody (Abcam, 1:5000 dilution) or HRP-coupled goat anti-rabbit antibody (Abcam, 1:5000 dilution) for 1 h at room temperature. Detection was performed using ECL (Thermo Fisher Scientific), and the signal was captured with the ChemiDoc imaging system (Bio-Rad).
Dot blot
Serial twofold dilutions of rLppA protein and 6 × His peptides (5.0–0.3125 μg) were applied onto nitrocellulose (NC) membranes (Solarbio, China). After drying at room temperature for 1 h, the NC membranes were blocked with 5% BSA for 1 h at room temperature. They were then incubated with 10 μg of fibronectin, collagen IV, laminin, vitronectin, tPA, or plasminogen, respectively, on a rocking platform overnight at 4 °C. The membranes were washed three times for 10 min each with 1 × TBST. Primary antibodies such as anti-fibronectin, anti-collagen IV, anti-laminin, anti-vitronectin, anti-tPA, or anti-plasminogen IgG were added and incubated for 1 h at room temperature. Subsequently, Goat anti-mouse HRP-IgG or goat anti-rabbit HRP-IgG was applied as secondary antibodies for 1 h at room temperature, followed by ECL detection. Signals were captured using the ChemiDoc imaging system.
Enzyme-linked immunosorbent assay
ECM components (fibronectin, collagen IV, laminin, and vitronectin; 300 ng/well) or EBL cell membrane extracts (200 ng per well) were coated onto a 96-well plate and incubated at 4 ℃ overnight. For the cell membrane microplate binding assay, various dilutions (200–0.8 ng) of rLppA protein were introduced into ELISA wells previously coated with EBL cell membrane extracts. For the ECM microplate binding assay, rLppA protein was serially diluted (250–1.95 ng) and applied to ELISA wells coated with ECM proteins. In the adhesion inhibition test, different dilutions of anti-LppA mouse serum (1:10–1:1280 dilution) were initially combined with M. bovis PG45 and incubated at 37 °C for 1 h; thereafter, the mixture was introduced to ELISA wells coated with EBL cell membrane extracts. Anti-LppA serum and goat anti-mouse HRP-IgG served as primary and secondary antibodies, respectively. The chromogenic substrate used was TMB, and the OD was measured at 405 nm using an iMark microplate reader. BSA treatment served as a negative control.
Plasminogen activity assay
The rLppA protein (1 μg) was incubated with plasminogen (1 μg) at 37 °C for 1 h, followed by its addition to a 96-well plate. Subsequently, tPA (50 ng) or the lysine analog aminoacetic acid ε-ACA (80 mM) was introduced and the mixture was incubated for 15 min at 37 ℃. Absorbance changes at 405 nm were monitored at 15-min intervals using an iMark microplate reader, following the addition of 0.5 mM of the specific substrate D-Val-Leu-Lys p-nitroaniline dihydrochloride.
ANXA2 translocation assay
The pCAGGS-FLAG-N and pCAGGS-FLAG-N-LppA plasmids were transfected into EBL cells for 24 h, respectively. The membrane and cytoplasm of EBL cells, either transfected with plasmid or infected with M. bovis, were separately extracted using a commercial Membrane Protein Extraction Kit (Thermo Fisher Scientific). Changes in the expression of ANXA2 in the cell membrane and cytoplasm were detected using western blot using an anti-ANXA2 antibody. α-Tubulin and Na+/K+-ATPase were employed as marker proteins for the cytoplasm and membrane, respectively.
Statistical analysis
Each experiment was independently replicated three times, and each sample was analyzed in triplicate. Statistical analyses were conducted using unpaired t-tests and one-way/two-way analysis of variance with GraphPad Prism 6.0 software. Significant differences were indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. The term “ns” denotes a lack of statistical significance.
Results
Sequence and immunogenicity analyses of LppA
Genome analysis has revealed that the LppA gene is universally present in M. bovis isolates, establishing it as a core gene of this species. M. bovis LppA exhibits highly conserved amino acid sequences within the 1–340 amino acid (aa) region, particularly in the 16–59 aa and 207–340 aa regions, which are completely consistent across all M. bovis strains (Figure 1A). These regions may represent the core functional areas of M. bovis’s LppA. Throughout its evolution, LppA from different M. bovis isolates has experienced deletions in various regions, most notably, significant losses of amino acid segments within the 65–202 aa or 341–617 aa regions (Figure 1A). Phylogenetic analysis, based on LppA’s amino acid sequence in Mycoplasma, demonstrated that LppA divides into five main groups, with M. bovis and Mycoplasma agalactiae clustering closely together (Additional file 3A). Further multiple sequence alignment analysis revealed that LppA is highly conserved among Mycoplasma species, with numerous conserved sites suggesting similar functions across different Mycoplasma species (Additional file 3B). Functional domain prediction analysis of LppA, using SMART software, identified a signal peptide at the N-terminal 1 to 21 aa. Analysis of LppA’s tertiary structure with AlphaFold 2 indicated that the 207–340 aa region contains multiple helical structures (Figure 1B).

Sequence characterization of LppA in M. bovis. A Multiple conserved sites of LppA in M. bovis. Highly conserved residues are shown in red. B Protein structures of LppA were modeled using AlphaFold2; red indicates the most conserved region. C Multiple epitopes of 1–340 amino acids of M. bovis LppA conserved regions predicted using DNAstar 6.0 software.
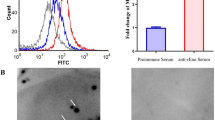
To determine whether LppA serves as a potential antigenic target for M. bovis, we identified several antigenic sites within LppA’s highly conserved amino acid region through bioinformatics analysis (Figures 1A and C). Anti-LppA mouse serum was produced following procedures for prokaryotic expression, recombinant protein induction, and polyclonal antibody generation (Additional file 4). We observed that rLppA reacted with bovine serum from natural and experimental M. bovis infections, as well as with anti-LppA mouse serum, but not with M. bovis-negative bovine serum (Additional file 5A). LppA from M. bovis was successfully detected in all six clinical isolates tested (Additional file 5B). These results suggest that LppA is highly immunogenic and could serve as a vaccine candidate for M. bovis.
Surface localization of M. bovis LppA
To ascertain LppA’s localization, we separately extracted the membrane proteins and cytoplasmic proteins of M. bovis (Figure 2A). LppA was primarily located in the cell membrane of M. bovis, with lesser quantities found in the cytoplasm. This membrane-centric localization of LppA was further confirmed through an immunogold labeling assay (Figure 2B).

Surface localization of LppA in M. bovis. A Distribution of LppA in M. bovis by immunoblotting assay. The 180-kDa protein marker (M) is labeled. Total protein (lane 1), cell membrane fraction (lane 2), and cytoplasmic fraction (lane 3) of M. bovis were subjected to western blotting with anti-LppA serum. B Immunoelectron microscopy localization of LppA in M. bovis. (Left) Anti-LppA mouse serum was used as the primary antibody. Black arrows indicate colloidal gold particles. (Right) Negative control using pre-immune mouse serum. Scale bar for transmission electron microscopy image: × 150 000 magnification, 100 nm.
LppA as a novel adhesin in M. bovis
The interaction between rLppA and membrane extracts from EBL cells was analyzed using ELISA. rLppA was found to bind to the EBL cell membrane in a dose-dependent manner within the range of 12.5–200 ng, compared with the control group (Figure 3A). This binding was inhibited by anti-LppA serum at a dilution of 1:640 (p < 0.0001) (Figure 3B). These data indicate that LppA directly interacts with EBL cells, identifying it as a novel cytoadhesin of M. bovis.

Binding of M. bovis LppA to EBL cell membrane extracts. A rLppA binds to embryonic bovine lung (EBL) cell membrane extracts in a dose-dependent manner. Different concentrations of rLppA (200–0.8 ng) were added to the wells of plates coated with EBL cell membrane extracts (200 ng) and were then incubated with anti-LppA serum for 1 h, followed by incubation with horseradish peroxidase–conjugated goat anti-mouse secondary antibody. Subsequently, after 10 min of incubation with tetramethylbenzidine, bound rLppA was detected by measuring the optical density of the wells at 405 nm using a microtiter plate reader. Bovine serum albumin served as the negative control. B Inhibition of rLppA binding to EBL cell membranes by anti-LppA serum. rLppA (200 ng) was pre-incubated with various concentrations of anti-LppA serum for 1 h and added to wells coated with EBL cell membrane extracts (200 ng). Pre-immune serum was used as the negative control. Values are presented as mean ± standard error of the mean of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
LppA binds host ECM components
To further investigate LppA’s adhesion to host cells, we evaluated its potential interaction with ECM components. Both dot blot analysis and ELISA were utilized to assess rLppA’s binding affinity for fibronectin, collagen IV, laminin, and vitronectin, using 6 × His peptides as a negative control. The results indicated that rLppA could bind to fibronectin, collagen IV, laminin, and vitronectin in a dose-dependent manner (Figure 4).

Binding of M. bovis LppA to host ECM components. A–D Binding ability of M. bovis LppA to host extracellular matrix components (fibronectin, collagen IV, laminin, and vitronectin) was tested by dot blot, with 6 × His peptides as the negative control. E–H LppA binds to fibronectin, collagen IV, laminin, and vitronectin in a dose-dependent manner. Different concentrations of rLppA (250–1.95 ng) were added to wells coated with fibronectin, collagen IV, laminin, or vitronectin (200 ng). The optical density was measured at 405 nm.
LppA promotes plasminogen activity by binding host plasminogen and tPA
rLppA’s ability to bind plasminogen in a dose-dependent manner was demonstrated through a dot blot test (Figure 5A). The plasmin-specific chromogenic substrate d-Val-Leu-Lys 4-nitroanilide dihydrochloride was employed to detect the activation of plasminogen bound to rLppA by the activator tPA. Wells containing rLppA protein, plasminogen, and tPA exhibited higher OD values at 405 nm compared to control wells with 6 × His peptides (Figure 5C). This suggests that rLppA enhances the efficiency of plasminogen conversion to active plasmin. The trend of tPA-mediated plasminogen activation was observed both in the presence and absence of rLppA. The inclusion of rLppA significantly accelerated the rate of plasminogen activation from 15 to 120 min compared to wells containing only plasminogen and tPA (p < 0.0001; Figure 5C). However, no increase in OD values was noted in wells containing only plasminogen and rLppA or rLppA and tPA (Figure 5C), indicating that rLppA solely enhanced tPA’s activation of plasminogen and did not possess the functionalities of tPA or plasmin. The OD values in wells with rLppA, tPA, and plasminogen significantly decreased upon incubation with the lysine analog ε-ACA (Figure 5C), suggesting ε-ACA also affects rLppA’s activation capacity. Additionally, rLppA was observed to bind to tPA (Figure 5B).

Activation of plasminogen bound to LppA. A, B Binding ability of M. bovis LppA to plasminogen and tPA was detected by dot blot, with 6 × His peptides acting as the negative control. C Determination of kinetic curves of LppA-promoted plasminogen activation by tPA. Plasminogen and rLppA were incubated on a microtiter plate for 1 h, and then tPA and the chromogenic substrate d-Val-Leu-Lys p-nitroaniline dihydrochloride were added. Plasmin activity was detected photometrically at 405 nm for every 15 min for 135 min. Wells without plasminogen or tPA were used as negative controls. rLppA-bound plasminogen was converted to plasmin, which cleaved the substrate d-Val-Leu-Lys p-nitroaniline dihydrochloride in a time-dependent manner. ε-ACA inhibited d-Val-Leu-Lys p-nitroaniline dihydrochloride cleavage. The absorbance at 405 nm of the 6 × His peptide control was significantly lower than that of the rLppA-added wells.
Annexin A2 is a cellular receptor of LppA
To discern additional host proteins interacting with M. bovis LppA, we employed IP-MS and identified several potential LppA-interacting proteins (Figure 6A). LppA’s interaction with ANXA2 was substantiated through Co-IP (Figure 6B), and immunofluorescence microscopy revealed co-localization of LppA and ANXA2 in 293T cells (Figure 6C). These findings confirm that LppA interacts with the ANXA2 protein.

Annexin A2 is a cellular receptor of M. bovis LppA. A LppA-immunoprecipitated proteins identified by immunoprecipitation-mass spectrometry (IP-MS) assays. Yellow arrowheads indicate LppA; red arrowheads indicate host proteins that interact with LppA. B Co-immunoprecipitation analysis. Lysates from 293T cells (+) transfected with pCAGGS-FLAG-N-LppA and pCAGGS-HA-N-ANXA2 plasmids were immunoprecipitated using an anti-Flag antibody and analyzed using western blotting with antibodies to FLAG and HA. Lysates from 293T cells (−) not transfected with pCAGGS-FLAG-N-LppA or pCAGGS-HA-N-ANXA2 plasmids were used as the control group. Whole-cell lysates (WCL) of 293T were used as the input. C Co-localization of the LppA and ANXA2 proteins was analyzed using confocal microscopy. 293T cells were co-transfected with pCAGGS-FLAG-N-LppA and pCAGGS-HA-N-ANXA2 plasmids and labeled with mouse anti-HA and rabbit anti-Flag, followed by Alexa-Fluor-conjugated donkey anti-mouse (488) and donkey anti-rabbit (647) secondary antibodies. The cells were then observed using a confocal microscope. Green represents LppA, red represents ANXA2, and blue represents the nuclear stain 4′,6-diamidino-2-phenylindole (DAPI); co-localization assay suggests that LppA is almost completely coincident with ANXA2.
LppA promotes the enrichment of ANXA2 protein on the cell membrane
The expression levels of ANXA2 in the cytoplasm and cell membrane of EBL cells were assessed through Western blot analysis, with α-tubulin and Na+/K+-ATPase serving as markers for cytoplasmic and membrane proteins, respectively. ANXA2 expression levels were similar in the cytoplasmic and membrane fractions of either uninfected EBL cells or cells transfected with the pCAGGS-FLAG-N plasmid (Figure 7A and Additional file 6A). However, in cells infected with M. bovis or transfected with the pCAGGS-FLAG-N-LppA plasmid, there was a notable enrichment of ANXA2 in the membrane, suggesting an increased presence of ANXA2 on the cell surface (Figure 7A and Additional file 6A). This observation was corroborated by confocal microscopy, which showed a substantial increase in the ANXA2 fluorescence signal on the surface of cells either transfected with the pCAGGS-FLAG-N-LppA plasmid or infected with M. bovis PG45, compared to control cells transfected with the pCAGGS-FLAG-N plasmid or uninfected cells (Figures 7B, C, D and Additional files 6B, C, D). These findings imply that LppA might assist M. bovis in promoting the accumulation of ANXA2 on the cell membrane.

LppA promotes the enrichment of ANXA2 on the cell membrane. The pCAGGS-FLAG-N and pCAGGS-FLAG-N-LppA plasmids were separately transfected into embryonic bovine lung (EBL) cells for 24 h. A Expression changes of ANXA2 in the cell membrane and cytoplasm were detected using an anti-ANXA2 antibody. α-Tubulin and Na+/K+-ATPase were used as marker proteins for the cytoplasm and cell membrane, respectively. B Relative ANXA2 expression in the cell membrane and cytoplasm. C EBL cells transfected with the pCAGGS-FLAG-N plasmid. D EBL cells transfected with the pCAGGS-FLAG-N-LppA plasmid. Blue represents DAPI, red represents ANXA2, and green represents rLppA. The white arrow indicates the enrichment of the ANXA2 protein on the cell membrane.
Disruption of the LppA gene markedly reduces the adhesion of M. bovis to cells in vitro
The LppA-deficient M. bovis mutant (ΔLppA) was generated using the transposon mutagenesis method (Additional file 7). The ΔLppA:LppA complementation strain was subsequently created by transforming ΔLppA mutants with the pIRR45-LppA plasmid. Polymerase chain reaction and Sanger sequencing, utilizing specific primers (Additional file 1), confirmed the insertion of the transposon into 47.9% of the full coding sequence of the LppA gene downstream of the start codon in the ΔLppA mutants (spanning nucleotides 880–881). The expression levels of LppA in the ΔLppA and ΔLppA:LppA strains were validated through Western blot analysis using anti-LppA serum (Figure 8A). Growth curves for M. bovis wild-type, ΔLppA, and ΔLppA:LppA strains revealed no significant differences, suggesting that LppA’s absence does not impede M. bovis growth (Figure 8B).

LppA is a novel adhesion protein of M. bovis. A Presence of LppA in M. bovis wild-type, M. bovis ΔLppA, and M. bovis ΔLppA:LppA was examined using anti-LppA mouse serum. B Comparative growth curves of M. bovis wild-type, M. bovis ΔLppA, and M. bovis ΔLppA:LppA. C M. bovis wild-type, M. bovis ΔLppA, and M. bovis ΔLppA:LppA were used to infect embryonic bovine lung (EBL) cells for 1.5 h. Subsequently, the cells were washed three times with phosphate-buffered saline, digested with trypsin, and the adherent mycoplasmas were counted. D Adhesion inhibition assay of anti-LppA serum to M. bovis. M. bovis was pre-incubated with various concentrations of anti-LppA serum for 1 h and then used to infect EBL cells. Pre-immune serum was used as the control. Values are presented as mean ± standard error of the mean of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001). E M. bovis wild-type, ΔLppA, and ΔLppA:LppA were stained with CFDA-SE for 30 min and then used to infect EBL cells. The nuclei of the cells were stained with DAPI and the cytoskeleton was stained with rhodamine phalloidin. Green represents M. bovis, red represents the cytoskeleton, and blue (DAPI) represents the nucleus.
The adhesion capability of M. bovis wild-type, ΔLppA, and ΔLppA:LppA strains to EBL cells was then examined. The ΔLppA mutant displayed a significantly diminished adhesion to EBL cells compared with the parental strain PG45, but this adhesion trait was partially reinstated in the complemented ΔLppA:LppA strain, as further evidenced by confocal microscopy analysis (Figure 8C, E). Pre-incubating M. bovis with anti-LppA serum before infecting EBL cells also markedly reduced M. bovis’s adhesion to EBL cells (Figure 8D). This adhesion inhibition was dose-dependent, with a 1/10 dilution of anti-LppA serum resulting in more pronounced inhibition compared to a 1/100 dilution (p < 0.0001). Additionally, a 1/100 dilution of anti-LppA serum significantly curtailed M. bovis’s adhesion capacity compared to pre-immune mouse serum (p < 0.0001). These outcomes highlight LppA’s crucial role in facilitating the adhesion of M. bovis to EBL cells.
Discussion
Adhesion to and colonization of host cells are crucial biological processes for Mycoplasma to infect the host. However, the specific virulence factors utilized by M. bovis for adhesion, invasion, and immune evasion remain unidentified. A significant obstacle impeding the functional annotation of Mycoplasma genes is their low sequence similarity to those of other bacteria, compounded by the scarcity of effective genetic manipulation tools for Mycoplasma [1]. Several proteins, including enolase, TrmFo, Mbov_0503, and mbfN, have been previously identified as adhesion-related proteins in M. bovis [11,12,13,14,15,16,17,18,19,20,21]. Nevertheless, only a few proteins have been validated as cytoadhesion-related proteins at the bacterial level. Consequently, research into the molecular mechanisms of Mycoplasma pathogenicity trails significantly behind that of other pathogenic bacteria. In this study, we evaluated the adhesion of M. bovis PG45, ΔLppA, and ΔLppA:LppA to EBL cells. The adhesion of ΔLppA to EBL cells was notably reduced compared to that of the parental strain and ΔLppA:LppA. Treating M. bovis PG45 with a specific anti-LppA serum led to a substantial decrease in adhesion to EBL cells. Together, these findings suggest that LppA is a novel adhesion-related protein in M. bovis.
Surface-localized adhesion-associated proteins in M. bovis present potential targets for developing drugs, vaccines, and immunodiagnostic assays against this economically significant ruminant pathogen. In this research, immunoelectron microscopy and Western blot analysis demonstrated that LppA is primarily located on the cell membrane surface of M. bovis. LppA is immunogenic and recognized by the host to trigger an immune response. Recent studies have shown that the LppA of Mycoplasma mycoides, as a primary antigen, can provoke a robust immune reaction in the host, such as a T-cell response, during the initial infection stages [24, 38]. In other pathogenic bacteria such as Salmonella enterica serovar Typhimurium and Mycoplasma mycoides subspecies mycoides, LppA also serves as a powerful inducer of host inflammatory and immune responses [25, 39]. In recent years, several immunogenic surface adhesion proteins of M. bovis, such as Vsp [8, 21], have been uncovered. However, the expression of the Vsp family of proteins is variable. In different M. bovis isolates, the expression patterns of the Vsp family differ, contributing significantly to the immune evasion tactics of M. bovis [40]. LppA-related proteins are commonly found in M. bovis strains, and the 1–340 amino acid region is highly conserved, containing numerous antigenic epitopes. Hence, the newly characterized adhesion protein LppA of M. bovis highlighted in this study emerges as a potential candidate target for drugs, vaccines, and immunodiagnostic development. Future work will explore the role of LppA in the pathogenesis of M. bovis infections in cattle.
Phylogenetic and sequence analyses showed that LppA-related proteins presented five main groups in Mycoplasma, among which M. bovis and Mycoplasma agalactiae clustered together. Multiple sequence alignment analysis has shown that LppA possesses numerous identical conserved sites across Mycoplasma species, and these sites maintain similar spatial conformations in the protein’s tertiary structure. Therefore, these domains might be critical to LppA’s biological function. Interestingly, some M. bovis strains exhibit a partial absence of the amino acid sequence (amino acids 65–202 or 341–617) at the carboxy-terminus of LppA. LppA is not unique in this aspect; similar fragment deletions have been observed in MbfN, a leucine-rich repeat lipoprotein of M. bovis [16]. The absence of these protein fragments might induce functional alterations in M. bovis. Nevertheless, the reasons for these deletions and the functions of the missing amino acid sequences remain to be explored. Genetic closeness between M. bovis/Mycoplasma agalactiae and the Mycoplasma mycoides cluster was noted in phylogenetic and sequence analyses, suggesting the possibility of lateral gene transfer among them. The presence of LppA-like sequences in other bacteria and their evolutionary relationships warrant further investigation.
The host ECM is central to numerous biological processes, including cell adhesion, differentiation, growth, and migration. However, components of the host ECM also serve as common facilitators in pathogen–host cell interactions [41, 42]. Pathogens initiate contact with a host through the interaction of their virulence factors and ECM components, a critical step in the infection process [16]. Leptospira is known to bind to ECM components such as collagen, fibronectin, and laminin [43, 44], with certain bacterial proteins identified as mediating these interactions [44]. For instance, the LigA and LigB proteins of Leptospira interrogans serovar Copenhageni facilitate the bacterium’s binding to fibronectin, laminin, type IV collagen, and the plasma protein fibrinogen [43]. Similarly, the ability of S. aureus to bind to fibronectin constitutes a significant virulence trait, indicative of the bacterium’s invasive capacity [45]. Previous research indicated that M. bovis secures and perpetuates its infection in host cells by adhering to multiple ECM receptors [6, 16]. In this study, we discovered that rLppA binds to fibronectin, collagen IV, laminin, and vitronectin in a dose-dependent fashion, underscoring LppA’s pivotal role in M. bovis adhesion to host cells through interactions with ECM components.
Plasminogen and ECM proteins share a close relationship within the host [46]. Plasminogen, a vital source of plasmin, is central to the fibrinolytic system. This extensive proteolytic system plays a crucial role in various physiological processes, such as the breakdown of fibrin clots and the degradation of various ECM and connective tissue components [47]. Numerous pathogenic microorganisms express factors that can bind to and enhance plasminogen activity [48,49,50], allowing pathogens to co-opt the host’s fibrinolytic system to aid their invasion and colonization [47, 49, 51]. For instance, GAPDH and enolase in Mycoplasma hyorhinis serve as plasminogen receptors (PlgRs), binding and activating plasminogen to assist the pathogen in degrading the host’s ECM [27,28,29]. This interaction may be a potential mechanism through which M. hyorhinis breaches tissue barriers in vivo, leading to serous fibrinous inflammation in piglets’ body cavities and joints [29]. This study investigated the role of M. bovis’s LppA in plasminogen hijacking and identified an interaction between plasminogen and rLppA. rLppA-bound plasminogen, once activated by tPA, forms plasmin, degrading specific substrates. The lysine analog ε-ACA impeded the activation of LppA-bound plasminogen, indicating that lysine residues are vital functional sites at which LppA promotes the conversion of plasminogen to plasmin. These findings suggest LppA is a novel PlgR in M. bovis, potentially aiding the pathogen’s tissue dissemination.
ANXA2, a member of the extensive annexin family of Ca2+-regulatory and phospholipid-binding proteins, participates in several cellular physiological processes, such as membrane dynamics, cell–cell interactions, and cell adhesion [52, 53]. Although primarily found in the cytoplasm, ANXA2 can traverse the cell membrane to perform various functions, including enhancing plasmin activities [52,53,54,55]. Several studies have documented microorganisms’ interactions with ANXA2. It has been identified as a novel receptor for the adhesion of spotted fever group Rickettsia and Staphylococcus aureus to vascular endothelial cells [56] and is known to interact with the CARDS toxin of Mycoplasma pneumoniae, promoting host cell vacuolization [57], and heat shock protein GroEL of Mycoplasma gallisepticum interacts with ANXA2, facilitating host infection [58]. ANXA2 also engages with M. hyorhinis p37, activating the NF-κB pathway to mediate infection [59]. Recent research indicated that M. bovis can utilize ANXA2 for adhesion, invasion, and the downregulation of proteins essential for the IL-17 signaling pathway [60]. However, the specific M. bovis factors that interact with host ANXA2 were previously unidentified. In this study, we discovered an interaction between LppA and ANXA2 through IP-MS and Co-IP analyses, noting that LppA fosters the enrichment of ANXA2 on the EBL cell membrane. This suggests LppA assists M. bovis in adhering to the host through interactions with the host’s ECM and ANXA2.
In conclusion, this study identified LppA as a multifunctional surface protein on M. bovis, capable of adhering to host cells. LppA interacts with multiple ECM components, host plasminogen, and ANXA2, promoting M. bovis colonization and spread during infection. These insights enhance our understanding of M. bovis pathogenesis and offer new perspectives for preventing and controlling M. bovis infections.
Availability of data and materials
The datasets analysed in the current study are available from the corresponding author on reasonable request.
References
Zhu X, Baranowski E, Dong Y, Li X, Hao Z, Zhao G, Zhang H, Lu D, Rasheed MA, Chen Y, Hu C, Chen H, Sagne E, Citti C, Guo A (2020) An emerging role for cyclic dinucleotide phosphodiesterase and nanoRNase activities in Mycoplasma bovis: securing survival in cell culture. PLoS Pathog 16:e1008661
Caswell JL, Bateman KG, Cai HY, Castillo-Alcala F (2010) Mycoplasma bovis in respiratory disease of feedlot cattle. Vet Clin North Am Food Anim Pract 26:365–379
Josi C, Burki S, Stojiljkovic A, Wellnitz O, Stoffel MH, Pilo P (2018) Bovine epithelial in vitro infection models for Mycoplasma bovis. Front Cell Infect Microbiol 8:329
Burki S, Gaschen V, Stoffel MH, Stojiljkovic A, Frey J, Kuehni-Boghenbor K, Pilo P (2015) Invasion and persistence of Mycoplasma bovis in embryonic calf turbinate cells. Vet Res 46:53
Burki S, Frey J, Pilo P (2015) Virulence, persistence and dissemination of Mycoplasma bovis. Vet Microbiol 179:15–22
Guo M, Wang G, Lv T, Song X, Wang T, Xie G, Cao Y, Zhang N, Cao R (2014) Endometrial inflammation and abnormal expression of extracellular matrix proteins induced by Mycoplasma bovis in dairy cows. Theriogenology 81:669–674
Brank M, Le Grand D, Poumarat F, Bezille P, Rosengarten R, Citti C (1999) Development of a recombinant antigen for antibody-based diagnosis of Mycoplasma bovis infection in cattle. Clin Diagn Lab Immunol 6:861–867
Sachse K, Helbig JH, Lysnyansky I, Grajetzki C, Muller W, Jacobs E, Yogev D (2000) Epitope mapping of immunogenic and adhesive structures in repetitive domains of Mycoplasma bovis variable surface lipoproteins. Infect Immun 68:680–687
Le Grand D, Solsona M, Rosengarten R, Poumarat F (1996) Adaptive surface antigen variation in Mycoplasma bovis to the host immune response. FEMS Microbiol Lett 144:267–275
Nussbaum S, Lysnyansky I, Sachse K, Levisohn S, Yogev D (2002) Extended repertoire of genes encoding variable surface lipoproteins in Mycoplasma bovis strains. Infect Immun 70:2220–2225
Song Z, Li Y, Liu Y, Xin J, Zou X, Sun W (2012) Alpha-Enolase, an adhesion-related factor of Mycoplasma bovis. PLoS One 7:e38836
Gao X, Bao S, Xing X, Fu X, Zhang Y, Xue H, Wen F, Wei Y (2018) Fructose-1,6-bisphosphate aldolase of Mycoplasma bovis is a plasminogen-binding adhesin. Microb Pathog 124:230–237
Zhu X, Dong Y, Baranowski E, Li X, Zhao G, Hao Z, Zhang H, Chen Y, Hu C, Chen H, Citti C, Guo A (2020) Mbov_0503 encodes a novel cytoadhesin that facilitates Mycoplasma bovis interaction with tight junctions. Microorganisms 8:164
Sachse K, Grajetzki C, Rosengarten R, Hanel I, Heller M, Pfutzner H (1996) Mechanisms and factors involved in Mycoplasma bovis adhesion to host cells. Zentralbl Bakteriol 284:80–92
Xu QY, Pan Q, Wu Q, Xin JQ (2022) Mycoplasma bovis adhesins and their target proteins. Front Immunol 13:1016641
Adamu JY, Mitiku F, Hartley CA, Sansom FM, Marenda MS, Markham PF, Browning GF, Tivendale KA (2020) Mycoplasma bovis mbfN encodes a novel LRR lipoprotein that undergoes proteolytic processing and binds host extracellular matrix components. J Bacteriol 203:e00154-20
Adamu JY, Wawegama NK, Kanci Condello A, Marenda MS, Markham PF, Browning GF, Tivendale KA (2020) Mycoplasma bovis membrane protein MilA is a multifunctional lipase with movel lipid and glycosaminoglycan binding activity. Infect Immun 88:e00945
Zhao G, Zhang H, Chen X, Zhu X, Guo Y, He C, Anwar Khan F, Chen Y, Hu C, Chen H, Guo A (2017) Mycoplasma bovis NADH oxidase functions as both a NADH oxidizing and O(2) reducing enzyme and an adhesin. Sci Rep 7:44
Chen X, Huang J, Zhu H, Guo Y, Khan FA, Menghwar H, Zhao G, Guo A (2018) P27 (MBOV_RS03440) is a novel fibronectin binding adhesin of Mycoplasma bovis. Int J Med Microbiol 308:848–857
Guo Y, Zhu H, Wang J, Huang J, Khan FA, Zhang J, Guo A, Chen X (2017) TrmFO, a fibronectin-binding adhesin of Mycoplasma bovis. Int J Mol Sci 18:1732
Zou X, Li Y, Wang Y, Zhou Y, Liu Y, Xin J (2013) Molecular cloning and characterization of a surface-localized adhesion protein in Mycoplasma bovis Hubei-1 strain. PLoS ONE 8:e69644
Browning GF, Marenda MS, Noormohammadi AH, Markham PF (2011) The central role of lipoproteins in the pathogenesis of mycoplasmoses. Vet Microbiol 153:44–50
Rottem S (2003) Interaction of mycoplasmas with host cells. Physiol Rev 83:417–432
Dedieu L, Totte P, Rodrigues V, Vilei EM, Frey J (2010) Comparative analysis of four lipoproteins from Mycoplasma mycoides subsp. mycoides small colony identifies LppA as a major T-cell antigen. Comp Immunol Microbiol Infect Dis 33:279–290
Carozza M, Rodrigues V, Unterfinger Y, Galea S, Coulpier M, Klonjkowski B, Thiaucourt F, Totte P, Richardson J (2015) An adenoviral vector expressing lipoprotein A, a major antigen of Mycoplasma mycoides subspecies mycoides, elicits robust immune responses in mice. Vaccine 33:141–148
Lemichez E, Lecuit M, Nassif X, Bourdoulous S (2010) Breaking the wall: targeting of the endothelium by pathogenic bacteria. Nat Rev Microbiol 8:93–104
Wang J, Li Y, Pan L, Li J, Yu Y, Liu B, Zubair M, Wei Y, Pillay B, Olaniran AO, Chiliza TE, Shao G, Feng Z, Xiong Q (2021) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) moonlights as an adhesin in Mycoplasma hyorhinis adhesion to epithelial cells as well as a plasminogen receptor mediating extracellular matrix degradation. Vet Res 52:80
Wang J, Li S, Chen J, Gan L, Wang J, Xiong Q, Feng Z, Li Q, Deng Z, Yuan X, Yu Y (2023) Hijacking of host plasminogen by Mesomycoplasma (Mycoplasma) hyopneumoniae via GAPDH: an important virulence mechanism to promote adhesion and extracellular matrix degradation. Microbiol Spectr 11:e0021823
Wang J, Yu Y, Li Y, Li S, Wang L, Wei Y, Wu Y, Pillay B, Olaniran AO, Chiliza TE, Shao G, Feng Z, Xiong Q (2022) A multifunctional enolase mediates cytoadhesion and interaction with host plasminogen and fibronectin in Mycoplasma hyorhinis. Vet Res 53:26
Li M, Ma Y, Lian X, Lu Y, Li Y, Xi Y, Sun X (2022) Study on the biological effects of ZnO nanosheets on EBL cells. Front Bioeng Biotechnol 10:915749
DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP (1987) Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol 7:379–387
Chen S, Hao H, Zhao P, Ji W, Li M, Liu Y, Chu Y (2018) Differential immunoreactivity to bovine convalescent serum between Mycoplasma bovis biofilms and planktonic cells revealed by comparative immunoproteomic analysis. Front Microbiol 9:379
Eddy SR (2011) Accelerated profile HMM searches. PLoS Comput Biol 7:e1002195
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027
Letunic I, Khedkar S, Bork P (2021) SMART: recent updates, new developments and status in 2020. Nucleic Acids Res 49:D458–D460
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589
Monnerat MP, Thiaucourt F, Poveda JB, Nicolet J, Frey J (1999) Genetic and serological analysis of lipoprotein LppA in Mycoplasma mycoides subsp. mycoides LC and Mycoplasma mycoides subsp. capri. Clin Diagn Lab Immunol 6:224–230
Fadl AA, Sha J, Klimpel GR, Olano JP, Niesel DW, Chopra AK (2005) Murein lipoprotein is a critical outer membrane component involved in Salmonella enterica serovar typhimurium systemic infection. Infect Immun 73:1081–1096
Rosengarten R, Behrens A, Stetefeld A, Heller M, Ahrens M, Sachse K, Yogev D, Kirchhoff H (1994) Antigen heterogeneity among isolates of Mycoplasma bovis is generated by high-frequency variation of diverse membrane surface proteins. Infect Immun 62:5066–5074
Henderson B, Nair S, Pallas J, Williams MA (2011) Fibronectin: a multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS Microbiol Rev 35:147–200
Schwarz-Linek U, Hook M, Potts JR (2004) The molecular basis of fibronectin-mediated bacterial adherence to host cells. Mol Microbiol 52:631–641
Choy HA, Kelley MM, Chen TL, Moller AK, Matsunaga J, Haake DA (2007) Physiological osmotic induction of Leptospira interrogans adhesion: LigA and LigB bind extracellular matrix proteins and fibrinogen. Infect Immun 75:2441–2450
Breiner DD, Fahey M, Salvador R, Novakova J, Coburn J (2009) Leptospira interrogans binds to human cell surface receptors including proteoglycans. Infect Immun 77:5528–5536
Hauck CR, Ohlsen K (2006) Sticky connections: extracellular matrix protein recognition and integrin-mediated cellular invasion by Staphylococcus aureus. Curr Opin Microbiol 9:5–11
Franco-Serrano L, Sanchez-Redondo D, Najar-Garcia A, Hernandez S, Amela I, Perez-Pons JA, Pinol J, Mozo-Villarias A, Cedano J, Querol E (2021) Pathogen moonlighting proteins: from ancestral key metabolic enzymes to virulence factors. Microorganisms 9:1300
Law RH, Abu-Ssaydeh D, Whisstock JC (2013) New insights into the structure and function of the plasminogen/plasmin system. Curr Opin Struct Biol 23:836–841
Dasari P, Koleci N, Shopova IA, Wartenberg D, Beyersdorf N, Dietrich S, Sahagun-Ruiz A, Figge MT, Skerka C, Brakhage AA, Zipfel PF (2019) Enolase from aspergillus fumigatus is a moonlighting protein that binds the human plasma complement proteins factor H, FHL-1, C4BP, and plasminogen. Front Immunol 10:2573
Hu Z, Li H, Zhao Y, Wang G, Shang Y, Chen Y, Wang S, Tian M, Qi J, Yu S (2022) NADH oxidase of Mycoplasma synoviae is a potential diagnostic antigen, plasminogen/fibronectin binding protein and a putative adhesin. BMC Vet Res 18:455
Castiblanco-Valencia MM, Fraga TR, Pagotto AH, Serrano SM, Abreu PA, Barbosa AS, Isaac L (2016) Plasmin cleaves fibrinogen and the human complement proteins C3b and C5 in the presence of Leptospira interrogans proteins: a new role of LigA and LigB in invasion and complement immune evasion. Immunobiology 221:679–689
Ikeda R, Ichikawa T (2014) Interaction of surface molecules on Cryptococcus neoformans with plasminogen. FEMS Yeast Res 14:445–450
Su Z, Chang Q, Drelich A, Shelite T, Judy B, Liu Y, Xiao J, Zhou C, He X, Jin Y, Saito T, Tang S, Soong L, Wakamiya M, Fang X, Bukreyev A, Ksiazek T, Russell WK, Gong B (2020) Annexin A2 depletion exacerbates the intracerebral microhemorrhage induced by acute rickettsia and Ebola virus infections. PLoS Negl Trop Dis 14:e0007960
Bharadwaj A, Bydoun M, Holloway R, Waisman D (2013) Annexin A2 heterotetramer: structure and function. Int J Mol Sci 14:6259–6305
Luo M, Hajjar KA (2013) Annexin A2 system in human biology: cell surface and beyond. Semin Thromb Hemost 39:338–346
Hajjar KA (2015) The biology of annexin A2: from vascular fibrinolysis to innate immunity. Trans Am Clin Climatol Assoc 126:144–155
He X, Zhang W, Chang Q, Su Z, Gong D, Zhou Y, Xiao J, Drelich A, Liu Y, Popov V, Zhao X, Wakamiya M, Gaitas A, Lu F, Gong B (2019) A new role for host annexin A2 in establishing bacterial adhesion to vascular endothelial cells: lines of evidence from atomic force microscopy and an in vivo study. Lab Invest 99:1650–1660
Somarajan SR, Al-Asadi F, Ramasamy K, Pandranki L, Baseman JB, Kannan TR (2014) Annexin A2 mediates Mycoplasma pneumoniae community-acquired respiratory distress syndrome toxin binding to eukaryotic cells. MBio 5:e01497-14
Yu Y, Zhang L, Chen Y, Li Y, Wang Z, Li G, Wang G, Xin J (2019) GroEL protein (heat shock protein 60) of Mycoplasma gallisepticum induces apoptosis in host cells by interacting with Annexin A2. Infect Immun 87:e00248-19
Duan H, Chen L, Qu L, Yang H, Song SW, Han Y, Ye M, Chen W, He X, Shou C (2014) Mycoplasma hyorhinis infection promotes NF-kappaB-dependent migration of gastric cancer cells. Cancer Res 74:5782–5794
Zhang H, Lu D, Zhang Y, Zhao G, Raheem A, Chen Y, Chen X, Hu C, Chen H, Yang L, Guo A (2022) Annexin A2 regulates Mycoplasma bovis adhesion and invasion to embryo bovine lung cells affecting molecular expression essential to inflammatory response. Front Immunol 13:974006
Acknowledgements
We thank Francois Thiaucourt for kindly providing pMT/mNeonGreen plasmid. We thank Professor Glenn Browning (the University of Melbourn) for kindly providing M. bovis oriC plasmid pIRR45. We also thank Dr Xiaochun Wu (Gansu Agricultural University) for kindly providing EBL cells.
Funding
This research was supported by grants from the National Key Research and Development Program of China (2022YFD1800704), the Science and Technology Major-Project of Gansu Province (22ZD6NA001), the Key Talents Program of Gansu Province (2021RCXM047), the LVRI Young Elite Project (NKLS2020-119, NKLS2022-12), the Chinese Academy of Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2021-LVRI) and Key Project (CAAS-ASTIP-JBGS-20210701). The funders had no role in study design, data collection, analysis, publishing decisions, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
YC and SC conceived and designed the study. SL and ZL drafted the manuscript. S Liu, ZL, SC, S Lan, HH, and AAB critically revised manuscript. S Lan, HH, S Liu, SC, XJ, JL conducted experimental work. S Lan, ZL and SC analyzed and interpreted data. XY and PG contributed to reagents and materials. YC and SC contributed to the fund acquisition the study and supervised the work. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments were performed following the requirements of the Animal Ethics Procedures and Guidelines of the People’s Republic of China. The animal experiments were approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (LVRIAEC2021-028).
Competing interests
The authors declare that they have no competing interests.
Additional information
Handling editor: Freddy Haesebrouck.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Plasmids, oligonucleotides, cell lines and strains used in this study.
List of plasmids, primers, M. bovis strains, and cell line used in this study.
Additional file 2. The strains information of MB-DATABASE and MY-DATABASE.
NCBI accession numbers of strains used to construct the MB-DATABASE and MY-DATABASE databases. Genetic analysis of LppA sequences in Mycoplasmas and bacteria.
Additional file 3. Sequence characterization of LppA in different species of the genus Mycoplasma.
A. Phylogenetic tree constructed based on LppA sequences was divided into five groups based on the topological structure. Groups 1 to 5 are marked with yellow, blue, purple, pink, and green, respectively. The LppA of M. bovis and M. agalactiae clustered in Group 1. B. Multiple conserved sites of LppA in Mycoplasma were revealed. Highly conserved residues are shown in red. C. Protein structures of M. bovis LppA were modeled using AlphaFold2; red indicates the most conserved region.
Additional file 4. Recombinant LppA expression and polyclonal antibody preparation.
A. Purification of the rLppA protein washed with different concentrations of imidazole solution and analyzed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis. B. LppA of M. bovis PG45 was examined by western blot using anti-LppA mouse serum and negative serum.
Additional file 5. Immunogenicity analysis of LppA in
M. bovis. A. rLppA protein was examined using western blot using different sera. M. bovis-negative bovine serum was used as the negative control. B. Presence of LppA was examined in six strains of M. bovis by using anti-LppA serum.
Additional file 6.
M. bovis infection promotes ANXA2 translocation from the cytoplasm to the membrane. The embryonic bovine lung (EBL) cells were infected with M. bovis PG45 for 24 h. A. Expression changes of ANXA2 in the cell membrane and cytoplasm were detected by using an anti-ANXA2 antibody. α-Tubulin and Na + /K + -ATPase were used as marker proteins for the cell cytoplasm and membrane, respectively. B. Relative ANXA2 expression in the cell membrane and cytoplasm. C. EBL cells uninfected with the M. bovis PG45. D. EBL cells infected with the M. bovis PG45. Blue represents DAPI, red represents ANXA2, and green represents M. bovis. The white arrow indicates ANXA2 translocation from the cytoplasm to the membrane.
Additional file 7. The methods of construction and identification of
M. bovis PG45 mutant strains, expression and purification of the recombinant protein, extraction of the membrane and the cytoplasm, and immunogenicity analysis of LppA protein. The sequence of Tn inserts sites of M. bovisΔLppA strain and the sequence of M. bovis LppA optimized with E. coli-preferred codons.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, S., Li, Z., Lan, S. et al. LppA is a novel plasminogen receptor of Mycoplasma bovis that contributes to adhesion by binding the host extracellular matrix and Annexin A2. Vet Res 54, 107 (2023). https://doi.org/10.1186/s13567-023-01242-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-023-01242-1