
Abstract
Zinc (Zn) is an essential trace element in living organisms and plays a vital role in the regulation of both microbial virulence and host immune responses. A growing number of studies have shown that zinc deficiency or the internal Zn concentration does not meet the needs of animals and microbes, leading to an imbalance in zinc homeostasis and intracellular signalling pathway dysregulation. Competition for zinc ions (Zn2+) between microbes and the host exists in the use of Zn2+ to maintain cell structure and physiological functions. It also affects the interplay between microbial virulence factors and their specific receptors in the host. This review will focus on the role of Zn in the crosstalk between the host and microbe, especially for changes in microbial pathogenesis and nociceptive neuron-immune interactions, as it may lead to new ways to prevent or treat microbial infections.
Similar content being viewed by others


1 Introduction
Zinc (Zn) is the second most abundant trace metal in living organisms and is involved in numerous aspects of life, including but not limited to DNA replication, transcription, protein synthesis, cell proliferation, apoptosis, and signal transduction. Both animals and microbes cannot naturally produce Zn, and the strategy for withholding and using Zn is vital to their survival and development [1, 2]. Within the expected concentration range, animals and microbes can regulate their internal Zn concentrations to maintain physiological metabolism [3, 4]. However, excess amounts of zinc ions (Zn2+) will lead to some cytotoxic effects, while zinc deficiency results in disruption of normal biological activities [3, 5].
Numerous studies have shown that zinc deficiency in animals leads to retarded growth, impaired immunity, and severe pathological changes in the body [4]. However, the host uses a similar strategy to reduce the zinc concentration in a single location against bacterial infection, which is called “nutritional immunity”, limiting the growth and virulence of pathogenic bacteria [6]. During the infectious process, some zinc-sequestering proteins, such as calprotectin, are expressed and recruited to the infection site to limit zinc acquisition by bacteria [7]. To survive in the host and succeed in competing with commensal microbes, pathogenic bacteria must maintain a steady state of zinc usage by controlling zinc distribution with zinc transporters [2, 3, 8]. The presence of zinc transporters in pathogenic bacteria also contributes to maintaining zinc homeostasis and virulence [2, 9]. Moreover, zinc acts as a direct or indirect regulator to affect the communication between nociceptor neurons and immune systems in the host, modulating the inflammatory response and host defence against bacterial infection [10,11,12].
Although nutritional immunity is termed a direct and effective antibacterial immune response, these mechanisms are not well described in the case of virus infection; for example, calprotectin did not to have a proven antiviral effect in published papers. However, zinc has been noted as a direct antiviral drug, as well as an antiviral immune stimulator and an indispensable component for the replication of many viruses [13,14,15]. Thus, zinc is likely to be a potential inter-kingdom signal between the host and microbes. In this review, we describe recent advances in understanding the role of zinc, similar to host hormones and quorum sensing (QS) of bacteria [16, 17], in modulating the communication between pathogenic microbes and their hosts.
2 Zinc homeostasis in bacteria
In addition to being a cofactor for some bacterial proteins, zinc ions are required for DNA repair, enzymatic reactions, responses to oxidative stress, and regulatory roles in other physiological processes in bacteria [2, 18]. Studies have shown that at least four kinds of zinc transport systems in bacteria, including two uptake transporters, ZnuACB and ZupT, and two export proteins, ZntA and ZitB, maintain zinc concentrations in the cell [2, 19]. As shown in Figure 1, under the pressure of a high Zn2+ concentration, the protein expression of P-type ATPase, cation diffusion facilitator (CDF) family of membrane transport, and resistance nodulation division (RND)-type efflux pumps are induced and protect bacteria from zinc poisoning [20, 21]. When zinc supplementation is sufficient for basic needs, bacteria synthesize low-affinity absorption systems to control zinc transport, such as the inorganic phosphate transport (Pit) system, which catalyses the rapid exchange of zinc via chemical osmotics [21]. Under zinc-limiting conditions, bacteria absorb and transport Zn2+ against zinc deficiency via a high-affinity ZnuACB uptake transporter [22, 23].

Zinc transporter systems in bacteria [2, 9, 19,20,21,22]. Under zinc-replete conditions, membrane transport based on the P-type ATPase, CDF-type and RND-type families participates in zinc distribution. They work together to maintain zinc homeostasis in the cell. With sufficient zinc supplementation, bacteria control the cellular zinc concentration by a low-affinity absorption system, such as Pit and ZupT. Under zinc-limiting conditions, transporters of the high-affinity absorption system are induced and used to recruit Zn2+ against zinc deficiency, including ZnuACB, ZitB, ZnuD, etc.
The ZnuACB transporter usually consists of three parts, including the periplasmic Zn2+-binding protein ZnuA, the inner membrane channel protein ZnuB and the ATP enzyme protein ZnuC, which is responsible for providing energy for Zn2+ transport [21]. ZinT and ZnuA have been shown to cooperate in periplasmic zinc recruitment, and the role of ZinT in the zinc uptake process is dependent on the presence of ZnuA [23, 24]. These genes are regulated by Zur, which not only blocks the binding of RNA polymerase and inhibits the expression of ZnuACB by Zn2Zur (or Zn4Zur) dimer protein but also regulates the mobilization of zinc through ribosomal proteins and leads to ZupT transporting Zn2+ rather than ZnuACB [18, 20]. Both ZupT and ZnuACB are necessary for bacteria to grow under zinc limitation; although ZnuACB usually obscures the role of ZupT, it also contributes to zinc uptake by Escherichia coli (E. coli) and Salmonella at a zinc concentration of 10 mM [25, 26].
In recent reports, the ZnuACB transport system is the most important zinc transport system in many bacteria [2, 27, 28]. znuA gene deletion can significantly reduce the adhesion of E. coli O157:H7 and Campylobacter jejuni to host cells [24, 29] and attenuate the pathogenicity of Brucella abortus, Salmonella enteritidis, and Pasteurella multocida to susceptible animals [27, 30]. The motility and biofilm formation of uropathogenic E. coli (UPEC) CFT073△znuB decreased compared with that of the wild-type strain [28]. After the znuACB gene was deleted, the virulence of Salmonella Typhimurium (STm) was weakened, but the existence of a ZnuACB transport system allowed the bacteria to resist calprotectin-mediated Zn2+ chelation and benefit from competition with host microflora [8, 27]. Our study also found that the ZnuACB system of enterotoxigenic E. coli (ETEC) F4ac plays an important role in maintaining the formation of biofilms and adherence to porcine small intestinal epithelial cells in vitro under zinc deficiency [31]. Although deletion of the zur gene does not affect the virulence of Staphylococcus aureus (S. aureus) and Mycobacterium tuberculosis (M. tuberculosis), it decreases the mortality of mice infected with STm [32,33,34]. Under zinc-limiting conditions, the growth of E. coli Nissle 1917 did not weaken due to the deletion of zinc transporter genes, and the bacteria utilize yersiniabactin to scavenge zinc to resist calprotectin-mediated zinc sequestration in the inflamed gut [35]. The newly discovered TonB-dependent outer membrane protein receptor ZnuD in Neisseria meningitidis regulates the absorption of zinc and haem and can induce antibodies combined with vesicles to infect guinea pigs and activate complement-mediated cytotoxicity to kill bacteria [36, 37]. Thus, in addition to regulating zinc concentrations in the cell, zinc transporters can also affect the expression of virulence factors and the pathogenicity of bacteria. It is likely to be an effective target for the development of new antibacterial drugs or vaccines.
3 Zinc is an essential micronutrient for the host
Zinc sufficiency is crucial for the maintenance of zinc homeostasis and health in the host, while severe zinc deficiency induces apoptosis via the activation of the caspase pathway [38]. During this process, mitochondrial p53 mediates the nuclear translocation of apoptosis-inducing factor and endonuclease G and triggers subsequent cell death [38, 39]. Moreover, within the normal range, zinc can directly interfere with caspase and endonuclease activity and thereby suppress cell apoptosis [40]. In addition, it can effectively inhibit the accumulation of reactive oxygen species (ROS) in the body, reduce oxidative damage to proteins, and maintain cell membrane structure [41, 42].
Indeed, in the early stage of zinc insufficiency, cells upregulate the mRNA expression of the zinc-iron regulatory protein ZIP1-ZIP14 (Zrt, Irt-like protein/SLC39) and mediate zinc influx and intracellular redistribution to increase zinc uptake [43, 44]. Later, the mRNA expression of divalent metal transporter 1 (DMT1) is upregulated, and DMT1 cooperates with ZIP to maintain the stability of Zn2+ concentrations [45, 46]. On the other hand, with an excessive intake of zinc, cells increase zinc binding and zinc efflux by upregulating the mRNA expression of metallothionein (MT)-1 and the zinc transporter ZnT/solute-linked carrier 30 (ZIP/SLC30) family proteins ZnT1-ZnT10 [44, 47]. Thus, zinc transporters are critical to maintaining intracellular Zn2+ concentrations and are closely involved in zinc uptake and zinc efflux.
The absorption of Zn2+ by mammals mostly occurs in the distal small intestine, and the absorption process is mainly divided into four stages: intestinal cell uptake, mucosal cell transport, portal vein circulation, and endogenous zinc secreted back to intestinal cells [48]. Intestinal cells regulate intracellular Zn2+ concentrations mainly through exogenous zinc absorption and the efflux of endogenous zinc. Zinc transporters are also reported to be related to zinc homeostasis regulation and dynamic changes in intestinal epithelial cells [44, 47]. For instance, ZIP4 contributes to the differentiation and maintenance of Paneth cells, ZnT2 regulates antibacterial substance secretion in Paneth cells, and ZIP8 negatively regulates pro-inflammatory responses by promoting zinc efflux out of the organelles or extracellular zinc uptake into monocytes, macrophages, and pulmonary epithelial cells [47, 49]. ZIP10 inhibits apoptosis but has been implicated in gastric and colon cancer, while the ZIP6/ZIP10 heterodimer was required for zinc influx into cells to trigger the onset of mitosis and influence cell division progression [49, 50]. The loss of ZIP4 in the intestinal epithelium led to changes in mammalian rapamycin target protein (mTOR) signal transduction in villi and crypts and resulted in Paneth cell viability and functional disorders [51]. Moreover, an imbalance in Zn2+ homeostasis occurred in the intestine of ZIP7- and ZIP14-deficient mice, ZIP7 promoted the proliferation of crypt cells, and ZIP14 protected the structure of intestinal epithelial cells by maintaining stable expression and modification in tight junctions (TJs), such as phosphorylation of occludin proteins. The deletion of ZIP7 triggered endoplasmic reticulum (ER) stress in proliferative progenitor cells, resulting in a large amount of cell death, and ZIP14 deletion impaired the integrity of the intestinal epithelial barrier by reducing the expression of the zonula occludens (ZO)-1 and claudin-1 proteins [49, 52, 53].
The stability and catalytic activity of proteins is mostly promoted by metal binding, whereas incorrect metallization causes protein denaturation, enzyme inactivation, and even cell death. To avoid damage caused by insufficient zinc binding in proteins, cells have to control the expression or activity of zinc transporters to maintain a steady cellular zinc balance [49, 54]. Intracellular labile zinc is mostly harvested in organelles, including mitochondria, the ER, and the Golgi apparatus [55]. Even the accumulation of zinc ions leads to ER stress and sometimes reduces cell viability. These ions are essential for maintaining the activation of specific proteins and zinc homeostasis in life [42, 56]. For example, zinc ions stored in the ER of Arabidopsis thaliana will be triggered by the appropriate signal and released into the cytoplasm to cope with the redistribution of Zn2+ under conditions of zinc deficiency [57]. Yeast can effectively regulate intracellular zinc concentrations by changing the zinc proteome and the expression of zinc transporters at the transcriptional and posttranslational levels, especially in zinc-deficient environments [56].
Zinc transporter activity is also related to the zinc-sensing receptor protein ZnR/GPR39 (G Protein-Coupled Receptor 39) in intestinal epithelial cells [58]. The ZnR/GPR39 protein belongs to the G protein-coupled receptor (GPCR) family and mediates Zn2+-dependent signal transduction in keratinocytes, pancreatic cells, prostate cancer cells, salivary gland cells, neurons and bone cells [58, 59]. The GPR39 protein binds to Zn2+ through two histidine residues, His17 and His19, and the aspartic acid residue Asp313. It senses changes in Zn2+ concentrations but is not activated by Mn2+, Cu2+ or Fe2+ [58]. After activation by Zn2+, ZnR/GPR39 controls the absorption of Cl− and reduces fluid loss during diarrhoea by upregulating potassium-chlorine cotransporter 1 in the basolateral colon [60]. It also mediates Gq-dependent downstream signal transduction, inducing increased expression of Na+/H+ exchanger (NHE), which is attributed to the recovery of the pH in the intestine [58, 60]. An acidic pH of 6.5 can have a profound effect on ZnR/GPR39 activity, leading to a loss of stimulation of the extracellular protein kinase (ERK1/2) or protein kinase B (Akt) pathway and reduced NHE activation [58]. Asp313 was proven to be the key residue for ZnR/GPR39 to sense extracellular pH changes; therefore, for survival, ZnR/GPR39 must maintain pH homeostasis in the intestine [61]. In human colon epithelial cancer cells (Caco-2), through the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways, ZnR/GPR39 activated Zn2+-dependent mTOR/p70 ribosomal S6 protein kinase (p70S6K) and protein kinase C-ζ (PKCz), enhanced cell proliferation and differentiation, and promoted the abundant expression of ZO-1, occludin and E-cadherin to attenuate the colitis and intestinal damage caused by STm infections [59, 62].
4 Zinc plays an important role in the host-pathogenic bacteria interaction
In normal organisms, to prevent the proliferation and infection of pathogens, host cells limit the concentration of intracellular free Zn2+ by zinc binding and efflux, while bacteria rely on an efficient zinc transport system to maintain zinc supplementation for proliferation and disruption of the host defence system to establish infection [9, 18, 27]. Therefore, there exists competition in zinc acquisition between the host and pathogenic bacteria, and gut health seems to be a good target for studying this competitive relationship.
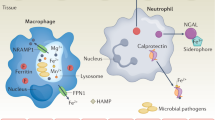
The intestinal mucosal barrier is the first line of defence against pathogens and other stimulators in the gut (Figure 2) [44, 63]. The protective effect is determined by the integrity of intestinal epithelial cells, the stability of the mucus layer, and normal expression or distribution of the E-cadherin, occludin, claudins, and ZO proteins [47, 64]. It has been reported that E. coli infections can change the expression of TJ proteins in the host and induce cytoplasmic distribution of claudin-2, resulting in an increase in intestinal epithelial permeability and a rapid decrease in transepithelial electrical resistance [65]. Upregulation of claudin-2 is induced by interleukin (IL)-22, resulting in enhancement of epithelial permeability and water efflux, which facilitates Citrobacter rodentium (C. rodentium) infection clearance through diarrhoea [65, 66]. In fact, intestinal mucosal immune responses in the host are activated shortly after an infection, and during this process, a number of pro-inflammatory cytokines and chemokines are expressed and released, including monocyte chemoattractant protein-1, IL-8/CXCL8, and tumour necrosis factor (TNF)-β [67]. According to recent reports, IL-17 exerts anti-inflammatory effects in the intestine by regulating the secretion of secretory immunoglobulin A (sIgA). IL-17 promotes the secretion of chemokines, such as CXCL1, CXCL2, and CCL20, in a positive feedback way to recruit neutrophils to enhance inflammatory responses [68, 69].

Regulation of mucosal immune homeostasis in the intestine [7, 71, 78, 108, 123, 133]. The dynamic change in competition in zinc acquisition between the host and pathogens is complicated. In addition to playing a role in enzymes and structural cofactors for the survival of all organisms, zinc also affects the activity of some transmembrane receptor proteins, including TLRs, which are responsible for the recognition of microbes or antigen molecules and the development and modulation of immune responses. Upon pathogen attack, some zinc-sequestering proteins are expressed and recruited to the infected site to chelate zinc ions and limit the growth and virulence of pathogenic bacteria, such as calprotectin, also known as the S100A8/S100A9 heterodimer, which is vital for strategic nutritional immunity. During the infectious process, the host also releases other S100 proteins, and then the binding of S100 proteins to cell surface receptors, such as TLR4, RAGE, and GPCRs, plays an important role in the regulation of inflammatory signal transduction. In turn, by regulating downstream signalling pathways, zinc can enhance the integrity of the intestinal mucosal barrier and reduce inflammation and diarrhoea caused by pathogenic infection.
As part of the innate defence against intestinal inflammation, neutrophils are essential for controlling bacterial diseases. Neutrophils engulf and kill bacteria and release an abundant amount of ROS to form a special structure called neutrophil extracellular traps, which are known as critical factors that contribute to resisting bacterial infection [70]. Another aspect of the antibacterial strategy in neutrophils is termed nutritional immunity. The mechanism inhibits the growth of pathogens and reduces the activity of bacterial virulence factors by limiting nutritional metal availability to bacteria, including that of zinc, manganese, iron, and copper [6, 71]. These metals are necessary in enzymes and structural cofactors for the survival of all organisms. Among them, the difficult competition between bacteria and the host over zinc has recently been considered a possible target for new antimicrobial therapies [18, 72, 73].
Upon pathogen attack, the S100A8/S100A9 heterodimer, also known as calprotectin, as one of the most abundant antibacterial proteins in neutrophils, is recruited to the infected site to chelate zinc ions and limit their access to invasive pathogens, further inactivating the superoxide defence of bacteria and inhibiting the growth of E. coli, S. aureus, Salmonella and other microbes [6, 71, 74, 75]. S100A7 (psoriasin) is expressed and acts as an effective chemical barrier in epithelial cells by zinc chelation to inhibit microbial growth [76, 77]. The zinc-binding protein S100A12 (calgranulin C) mostly participates in superoxide formation to exert its antibacterial activity. It also affects ubiquitin and beta-catenin degradation in host cells by interacting with S100A9 (also known as calgranulin B or MRP14) or directly with calcyclin-binding protein and Siah-1-interacting protein [71, 78]. During inflammation, both IL-17 and IL-22 can upregulate the gene expression of S100A8 (calgranulin A, MRP8), S100A9 and S100A12 [79]. When these proteins are released from the cell, they can bind and activate cell surface receptors (Figure 2), such as Toll-like receptor 4 (TLR4), receptor for advanced glycation end-products (RAGE), GPCRs, etc.; initiate intracellular inflammatory signal transduction; and play an important role in the regulation of immune and inflammatory responses [7, 78]. Although S100A9 activates TLR4 to induce inflammation that does not depend on zinc ions, it still acts as a zinc chelator and results in decreased intracellular free zinc levels in the cell [80]. The apoptosis-inducing activity of S100A8/S100A9 is also dependent on both receptor-mediated and zinc exclusion-modulated pathways [81, 82]. Therefore, zinc nutritional immunity is an excellent strategy to fight against bacterial infection in the host.
Recent studies have shown that nociceptors play a role in host defence against STm infection, especially those nociceptors with transient receptor potential cation channel V1 (TRPV1) and tetrodotoxin-resistant voltage-gated sodium channel NaV1.8 expression, decreasing the number of M cells in ileal Peyer’s patches and increasing the colonization of segmental filamentous bacteria, resulting in a reduction in Salmonella invasion [11]. This is not a commonly known antimicrobial defence mechanism. As reported, TRPV1-expressing nociceptors can sense bacterial infections, including STm, C. rodentium and ETEC, and release calcitonin gene-related peptide (CGRP) or substance P (SP) within the ileum to promote host defence [11, 12, 83, 84]. Elucidation of the complex interactions between intestinal immune cells, neurons, and microorganisms (the “microbe-gut-brain axis”) in mammals will further enhance our understanding of intestinal immunity and host defence [12, 85].
In fact, there are abundant neurons in the gastrointestinal tract. They can detect specific metabolites, cell wall components and toxins of pathogens and play an important role in regulating inflammation, bowel movements and intestinal secretion [12, 85]. Simultaneously, the innate and acquired immune responses of the gastrointestinal tract can be activated or inhibited via the varied stimulation of SP, CGRP, vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide released from the enteric nervous system [12]. Nociceptors are the main sensory receptors that respond to gastrointestinal disorders and potentially harmful stimuli, including pathogens and their components, extreme temperature, toxic chemicals, inflammatory mediators, and mechanical and tissue damage, evoking the sensation of pain and improving the body’s resistance during periods of infection [85,86,87]. The endogenous transient receptor potential (TRP) superfamily of ion channels responds to agonists, stimulates the activity of nociceptive sensory neurons and results in neurogenic inflammation and pain [10]. Intracellular zinc (10 nM) is available for TRP ankyrin subtype 1 protein activation and the subsequent release of inflammatory neuropeptides [10, 88]. Recently, neuropeptides were also reported to be used as a substitute for antibiotics in diarrhoea treatment. For example, VIP treatment can reduce the incidence of diarrhoea and increase the growth rate in newborn weaned piglets. In addition, the increase in inflammatory mediators induced by ETEC infection, including IL-2, IL-12, interferon (IFN)-γ, and TNF-α, and enterotoxin-induced jejunal fluid secretion can be reversed in a dose-dependent manner by VIP treatment [89].
5 Zinc affects the activity of host receptors and immune responses
In a recent study, zinc was found to affect the biological activity of host receptors and influence the interaction between pathogenic microbes and the host. For example, porcine aminopeptidase N (APN) is a Zn2+-dependent membrane-bound exopeptidase that is widely expressed in various tissues and cells, especially in the small intestine mucosa [90]. It has been proven to be a fimbrial receptor protein for ETEC F4 and mediates the endocytosis of F4 bacteria in the host [91]. In our previous study, we observed that APN directly interacted with the major FaeG subunit of F4 fimbriae and affected F4 bacterial adherence to host cells [92]. The binding determinants of the APN-FaeG interaction contain residues important for zinc binding, and in a certain concentration range, the change in Zn2+ concentrations affects the biological activity of the APN protein as well as the adherence between E. coli F4 and host intestinal epithelial cells [90, 93]. In addition to cleaving N-terminal amino acids from small oligopeptides on the apical surface of intestinal epithelial cells, APN participates in regulating the MAPK/ERK1/2 signalling pathway in monocytes, while its zinc-binding site is blocked by inhibitory antibodies [91, 94]. APN has also been described as a cancer-specific biomarker and reported to be involved in the progression of tumour metastasis, angiogenesis and prognosis, as well as protein overexpression in cancer cells, while tissue invasion and metastasis in human prostate cancer could be attenuated by using Zn2+ to inhibit APN biological activity in a dose-dependent manner [95,96,97].
Zinc is also closely related to some transmembrane receptor proteins, including B cell receptors (BCRs), TLRs and nucleotide-binding oligomerization domain (NOD)-like receptors, which are responsible for the recognition of microbes or antigen molecules and involved in the development and modulation of intestinal homeostasis [72, 98, 99]. ZIP9 increased the intracellular zinc level and enhanced Akt and Erk phosphorylation in response to BCR activation [100]. ZIP7 plays an important role in B cell development and positively regulates pre-BCR and/or BCR signalling, while ZIP10 deficiency leads to impairment of BCR signalling in immune responses [99, 101, 102]. In addition, zinc induces inflammatory responses via the TLR/nuclear factor kappa-B (NF-κB) pathway and indirectly regulates TLR signalling via zinc transporters [103, 104].
Interestingly, the antimicrobial roles of macrophages vary in defending against different pathogens. Macrophages deploy both zinc starvation and/or zinc toxicity as an antimicrobial strategy [72, 98, 105]. The clearance of Streptococcus in macrophages is promoted by both calprotectin-mediated zinc starvation and intracellular zinc toxicity [106], whereas zinc starvation is utilized as a strategy against Histoplasma capsulatum infection, and excessive zinc poisoning within the macrophage phagolysosome exerts direct bactericidal effects on M. tuberculosis and UPEC [98, 107, 108]. In addition, continuous stimulation of TLRs and NOD2 alters the expression of MT and increases the level of intracellular zinc, leading to increased autophagy and enhanced bacterial clearance of S. typhimurium, S. aureus, and adherent invasive E. coli in intestinal macrophages [98, 109,110,111].
Zinc also causes macrophage malfunction, further triggering an abnormal inflammatory response, and impairs the innate immune system [111, 112]. For example, IRF/IL-23-mediated M1 macrophage activation and GATA3/IL-4-mediated M2 macrophage inhibition are both aggravated by zinc deficiency, resulting in severe intestinal inflammation and nitrogen metabolism disorder in patients with liver cirrhosis, whereas zinc supplementation can improve ammonia and protein metabolism and regulate the activation of M1/M2 macrophages [113]. Alternative activation of macrophages does not contribute to defending against intracellular pathogens in some cases. IL-4 induces macrophage polarization to the M2 phenotype and increases the expression of MT3 and ZnT4 but not MT1 and MT2. In addition to promoting zinc uptake by intracellular pathogens, MT3 and ZnT4 augments intracellular labile Zn2+ and contributes to pathogen persistence in M2 macrophages [114].
Moreover, zinc itself has a regulatory role in intestinal immune responses and the healthy balance of the intestinal flora [3, 108]. After antigen stimulation, zinc attenuates the intestinal stress response and promotes the secretion of proteins and pro-inflammatory cytokines, such as sIgA, antimicrobial peptides, IL-1β, and IL-6 [54]. By regulating the MAPK and Akt signalling pathways, zinc can enhance intestinal mucosal barrier integrity and reduce inflammation and diarrhoea caused by bacterial infections [115]. Zinc also increases the expression of zinc finger protein A20 (also known as tumour necrosis factor alpha-induced protein 3) in intestinal epithelial cells to inhibit the inflammatory response and apoptosis mediated by some signal transduction pathways, such as NF-κB, and subsequent TNF-α production [116, 117]. However, A20 is a potent inhibitor of NF-κB-, TLR3-, and retinoic acid-inducible gene I-mediated IFN induction; thus, along with increased activation of NF-κB, interferon regulatory factor (IRF)-3, and IRF7, viral clearance was improved in A20-deficient cells, and this deficiency may protect the host against viral infection [118, 119].
As reported, zinc could optimize anti-tumour effects by inhibiting LPS-, ROS- or other immune factor-induced oncogenic signalling pathways, such as NF-κB, activator protein-1, Janus tyrosine kinase/signal transducer and activator of transcription (JAK/STAT), and PI3K/Akt; decrease oxidative stress and inflammatory responses induced by chemo- and radiotherapy; promote the development and differentiation of T and B lymphocytes; and improve the tumour microenvironment to reduce the risk of prostate, oesophageal, lung, and oral cancers [120,121,122]. High dietary zinc treatment has a beneficial effect on the modulation of mucosal structure and immune responses in the gut. It blocked TNF-α-mediated degradation of IκBα, decreased the mRNA expression of IFN-γ and IL-8, and upregulated the mRNA expression of ZO-1, occludin and transforming growth factor-β, resulting in an improvement in the antioxidant capacity and prevention of postweaning diarrhoea in piglets [63, 123].
Additionally, zinc is also reported to influence the modulation of biological rhythm and activity. By effectively stabilizing the binding of period proteins and cryptochrome proteins, zinc ions help to sustain the rhythmicity of the mammalian circadian clock in resisting the interference of negative factors, such as improper diet and drug abuse [124]. Previous studies have shown that zinc can regulate the absorption of water and electrolytes in the intestine and has a general inhibitory effect on voltage-gated ion channels, i.e., those for K+, Na+, Ca2+, and Cl− [125]. A new type of selective chloride channel regulated by Zn2+ in enteroendocrine cells of Drosophila larvae’s digestive tract, called Hodor, was found to act as a zinc ion sensor in the intestine and regulate individual feeding behaviour [126, 127]. Zn2+ is also used to promote the efficiency of antibiotics, such as vancomycin, quinolones, aminoglycosides, tetracycline, and macrolides [128]. For example, in gram-negative bacteria, the presence of zinc ions leads to an increase in the absorption of quinolones by changing the permeability of the bacterial outer membrane porin OmpF protein, while Zn2+ can effectively inhibit the acetylation of aminoglycosides catalysed by aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] and enhance the sensitivity of bacteria, especially for some aminoglycoside-resistant bacteria [128, 129].
When zinc deficiency occurs, the cytotoxicity of natural killer cells, the phagocytic activity of neutrophils and the chemotaxis of monocytes are significantly decreased, but the phagocytosis and oxidative burst of monocytes are not affected. Zinc shortage has an influence on only IL-6 and TNF-α production in monocytes [54, 112]. In addition, the loss of copper-zinc superoxide dismutase activities results in damage to the stability of the cell membrane structure and dysfunction in host defensive systems, such as the regulation of neutrophil apoptosis and neutrophil-mediated tissue injury [117]. Because of this dysfunction, zinc deficiency enhances the severity of infectious diseases and illness in animals, whereas probiotics were reported to overcome the host defence defect in zinc-deficient cells by abolishing the activation of pro-inflammatory signalling via the ERK and p38 pathways [130].
6 Zinc is vital for viral infections
It is known that the host is more vulnerable to enterovirus or toxigenic bacterial infections in zinc-limited conditions, while those pathogens both stimulate chloride secretion by activating intracellular guanosine monophosphate and adenylate cyclase [131]. During this period, poor nutritional absorption and diarrhoea further aggravate the compromised mineral state, and zinc supplementation can attenuate this damage [132, 133]. Thus, zinc plays an important role in both bacterial and viral infections.
Indeed, zinc was previously considered a potential agent for the prevention of H1N1 influenza (“swine flu”) and has direct antiviral properties [134]. Studies have shown that the replication of severe acute respiratory syndrome coronavirus (SARS-CoV) and hepatitis C virus (HCV) was inhibited by zinc oxide or zinc salt, while zinc could be deposited onto the surfaces of herpes simplex virus (HSV) virions, and the viral ubiquitin–proteasome pathway could be targeted with the zinc ionophore pyrithione to inhibit HSV-1 and HSV-2 infections [14, 135,136,137]. Upon exposure to low endosomal pH, zinc binding with a specific histidine residue on the viral E1 protein can effectively inhibit membrane fusion and block particle release of respiratory syncytial virus, HSV, Semliki Forest virus, and sindbis viruses [138, 139]. Moreover, zinc concentration is positively correlated with MT synthesis [54], which is an abundant zinc-binding protein and has a selective antiviral effect against viruses. MT acts as a zinc chaperone, promotes antiviral signal transduction and interferes with a potent antiviral response by modulating zinc influx or redistribution. Influenza, human immunodeficiency virus (HIV), HCV, measles virus, human cytomegalovirus and coxsackievirus infections are associated with upregulated MT expression, leading to a higher NF-κB DNA binding affinity [139, 140]. In the case of HIV, a significant increase in intracellular zinc and MT expression contributed to the inhibition of HIV-infected monocyte apoptosis by suppressing caspase-3 activation, which is conducive to the persistence of HIV infection [141, 142].
Thus, as a recent review summarized, the antiviral effects of zinc can be separated into two major categories. First, virus replication or infection-related symptoms are specifically inhibited or attenuated by zinc treatment. Second, zinc supplementation has protective effects against viruses by boosting the antiviral and systemic immunity of patients with zinc deficiency [108, 139]. In addition, zinc is an integral structural element of many viral proteins, including enzymes, proteases, and polymerase, and is thus crucial for virus transmission, as well as innate and adaptive antiviral responses [139, 143]. The protective role of zinc against coronavirus disease 2019 (COVID-19, caused by SARS-CoV-2) might be a good example to understand this effect. From the outset, zinc directly inhibits RNA-dependent RNA polymerase activity to block viral replication, such as that of SARS-CoV, equine arteritis virus (EAV), hepatitis E virus, etc. Both chloroquine (CQ) and its derivative hydroxychloroquine depend on this strategy to prevent the replication of SARS-CoV-2 in host cells [13, 144, 145]. CQ also induces the uptake of zinc, which results in an increase in cellular zinc accumulation and a lower risk of secondary infection, especially for patients with viral pneumonia coinfected with Streptococcus pneumoniae (S. pneumoniae) since zinc inhibits the growth of S. pneumoniae by regulating bacterial Mn(II) homeostasis [13, 144, 146].
Apart from the direct antiviral and antibacterial effects mentioned above, zinc is essential to ameliorate SARS-CoV-2-induced lung injury and other worse outcomes. For instance, zinc represents a promising agent to preserve the barrier function of the respiratory epithelium; it functions as a potent antioxidant and promotes ciliary beat frequency, mucociliary clearance, and ZO-1 and claudin-1 expression [13, 147, 148]. In a recent study, zinc appeared to affect the entry of SARS-CoV-2 into cells by decreasing the activity of angiotensin-converting enzyme 2, which is a receptor that binds primarily to the spike protein on the surface of coronaviruses [13, 149]. Zinc also has a critical role in resisting COVID-19. It degrades viral RNA and inhibits gene translation by various antiviral mediators, such as ribonuclease L and RNA-dependent protein kinase, which is induced by IFN-α via the JAK/STAT pathway [144, 150]. In view of the fact that severe COVID-19 frequently occurs along with pulmonary fibrosis and septic shock, a deficiency in zinc intake under the condition of sepsis may further increase the incidence and severity of COVID-19, and zinc supplements may stave off LPS-induced neutrophil recruitment into the lung and thus alleviate lung injury in systemic inflammation [13, 147]. In addition, zinc also minimizes the risk of excessive release of inflammatory cytokines (“cytokine storm”) by inhibiting IKK activity and subsequent NF-κB signal transduction, as well as regulatory T cell functions [146, 151].
7 Conclusions
In recent decades, host hormones and QS of bacteria were found to be effective regulators of crosstalk between bacteria and their host [16, 17]. The inter-kingdom signalling between them provides a new way to understand microbial infections and host immunity. Likewise, zinc serves as an important agent for the development of cell structure and functions. It may promote the survival and virulence of bacteria, viruses, or other microbes and participate in host defence by modulating various modes of signal transduction and inflammation in a concentration-dependent way [5, 22, 72, 98].
Both zinc deficiency and excess zinc toxicity can be used as a strategy to resist microbial infections. Upon stimulation, due to the variety of zinc sensors and channels distributed in different regions or tissues in the body, zinc may have an adverse effect on host immune regulation in different cells. Thus, zinc homeostasis in cells must be strictly regulated. In this review, we also discussed a recent study on zinc competition between hosts and microbes, inflammation and the immune response modulated by zinc-mediated nociceptive sensory neuron-immune interactions, and the change in host receptors with zinc treatment during infection, which provides a basis to clarify pathogenesis, representing a profound impact on our understanding of host defence and a promising target for new antimicrobial strategies. However, the molecular mechanism of zinc in the interplay between the host and microbes still needs to be elucidated.
Availability of data and materials
Not applicable.
Abbreviations
- APN:
-
Aminopeptidase N
- CDF:
-
Cation diffusion facilitator
- CGRP:
-
Calcitonin gene-related peptide
- COVID-19:
-
Coronavirus disease 2019
- C. rodentium :
-
Citrobacter rodentium
- DMT1:
-
Divalent metal transporter 1
- EAV:
-
Equine arteritis virus
- ER:
-
Endoplasmic reticulum
- ETEC:
-
Enterotoxigenic E. coli
- UPEC:
-
Uropathogenic E. coli
- GPCRs:
-
G protein-coupled receptors
- GPR39:
-
G protein-coupled receptor 39
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- HSV:
-
Herpes simplex virus
- IL:
-
Interleukin
- IFN:
-
Interferon
- IRF:
-
Interferon regulatory factor
- MAPK:
-
Mitogen-activated protein kinase
- MT:
-
Metallothionein
- mTOR:
-
Mammalian rapamycin target protein
- M. tuberculosis :
-
Mycobacterium tuberculosis
- NHE:
-
Na+/H+ exchanger
- NOD:
-
Nucleotide-binding oligomerization domain
- QS:
-
Quorum sensing
- RAGE:
-
Receptor for advanced glycation end-products
- RND:
-
Resistance nodulation division
- Pit:
-
Inorganic phosphate transport
- PI3K:
-
Phosphatidylinositol 3-kinase
- p70S6K:
-
P70 ribosomal S6 protein kinase
- PKCz:
-
Protein kinase C-ζ
- RdRp:
-
RNA-dependent RNA polymerase
- ROS:
-
Reactive oxygen species
- S. aureus :
-
Staphylococcus aureus
- SARS-CoV:
-
Severe acute respiratory syndrome coronavirus
- sIgA:
-
Secretory immunoglobulin A
- SP:
-
Substance P
- S. pneumoniae :
-
Streptococcus pneumoniae
- STm:
-
Salmonella Typhimurium
- TJ:
-
Tight junction
- TLR4:
-
Toll-like receptor 4
- TNF-β:
-
Tumour necrosis factor β
- TRPV1:
-
Transient receptor potential cation channel V1
- VIP:
-
Vasoactive intestinal peptide
- ZO-1:
-
Zonula occludens-1
References
Prasad AS (1995) Zinc: an overview. Nutrition 11(1 Suppl):93–99
Hantke K (2005) Bacterial zinc uptake and regulators. Curr Opin Microbiol 8:196–202. https://doi.org/10.1016/j.mib.2005.02.001
Gielda LM, DiRita VJ (2012) Zinc competition among the intestinal microbiota. mBio 3:e00171-00112. https://doi.org/10.1128/mBio.00171-12
Lichten LA, Cousins RJ (2009) Mammalian zinc transporters: nutritional and physiologic regulation. Annu Rev Nutr 29:153–176. https://doi.org/10.1146/annurev-nutr-033009-083312
Murakami M, Hirano T (2008) Intracellular zinc homeostasis and zinc signaling. Cancer Sci 99:1515–1522. https://doi.org/10.1111/j.1349-7006.2008.00854.x
Hood MI, Skaar EP (2012) Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol 10:525–537. https://doi.org/10.1038/nrmicro2836
Gonzalez LL, Garrie K, Turner MD (2020) Role of S100 proteins in health and disease. Biochim Biophys Acta Mol Cell Res 1867:118677. https://doi.org/10.1016/j.bbamcr.2020.118677
Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE, Restrepo NA, Hosking MP, Edwards RA, Battistoni A, Pasquali P, Lane TE, Chazin WJ, Vogl T, Roth J, Skaar EP, Raffatellu M (2012) Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe 11:227–239. https://doi.org/10.1016/j.chom.2012.01.017
Cerasi M, Ammendola S, Battistoni A (2013) Competition for zinc binding in the host-pathogen interaction. Front Cell Infect Microbiol 3:108. https://doi.org/10.3389/fcimb.2013.00108
Hu H, Bandell M, Petrus MJ, Zhu MX, Patapoutian A (2009) Zinc activates damage-sensing TRPA1 ion channels. Nat Chem Biol 5:183–190. https://doi.org/10.1038/nchembio.146
Lai NY, Musser MA, Pinho-Ribeiro FA, Baral P, Jacobson A, Ma P, Potts DE, Chen Z, Paik D, Soualhi S, Yan Y, Misra A, Goldstein K, Lagomarsino VN, Nordstrom A, Sivanathan KN, Wallrapp A, Kuchroo VK, Nowarski R, Starnbach MN, Shi H, Surana NK, An D, Wu C, Huh JR, Rao M, Chiu IM (2020) Gut-innervating nociceptor neurons regulate Peyer’s patch microfold cells and SFB levels to mediate Salmonella host defense. Cell 180:33-49.e22. https://doi.org/10.1016/j.cell.2019.11.014
Lai NY, Mills K, Chiu IM (2017) Sensory neuron regulation of gastrointestinal inflammation and bacterial host defence. J Intern Med 282:5–23. https://doi.org/10.1111/joim.12591
Skalny AV, Rink L, Ajsuvakova OP, Aschner M, Gritsenko VA, Alekseenko SI, Svistunov AA, Petrakis D, Spandidos DA, Aaseth J, Tsatsakis A, Tinkov AA (2020) Zinc and respiratory tract infections: perspectives for COVID-19. Int J Mol Med 46:17–26. https://doi.org/10.3892/ijmm.2020.4575
Shittu MO, Afolami OI (2020) Improving the efficacy of Chloroquine and Hydroxychloroquine against SARS-CoV-2 may require Zinc additives - A better synergy for future COVID-19 clinical trials. Infez Med 28:192–197
Kar M, Khan NA, Panwar A, Bais SS, Basak S, Goel R, Sopory S, Medigeshi GR (2019) Zinc chelation specifically inhibits early stages of dengue virus replication by activation of NF-κB and induction of antiviral response in epithelial cells. Front Immunol 10:2347. https://doi.org/10.3389/fimmu.2019.02347
Hughes DT, Sperandio V (2008) Inter-kingdom signalling: communication between bacteria and their hosts. Nat Rev Microbiol 6:111–120. https://doi.org/10.1038/nrmicro1836
Li Q, Ren Y, Fu X (2019) Inter-kingdom signaling between gut microbiota and their host. Cell Mol Life Sci 76:2383–2389. https://doi.org/10.1007/s00018-019-03076-7
Capdevila DA, Wang J, Giedroc DP (2016) Bacterial strategies to maintain zinc metallostasis at the host-pathogen interface. J Biol Chem 291:20858–20868. https://doi.org/10.1074/jbc.R116.742023
Graham AI, Hunt S, Stokes SL, Bramall N, Bunch J, Cox AG, McLeod CW, Poole RK (2009) Severe zinc depletion of Escherichia coli: roles for high affinity zinc binding by ZinT, zinc transport and zinc-independent proteins. J Biol Chem 284:18377–18389. https://doi.org/10.1074/jbc.M109.001503
Gilston BA, Wang S, Marcus MD, Canalizo-Hernández MA, Swindell EP, Xue Y, Mondragón A, O’Halloran TV (2014) Structural and mechanistic basis of zinc regulation across the E. coli Zur regulon. PLoS Biol 12:e1001987. https://doi.org/10.1371/journal.pbio.1001987
Hantke K (2001) Bacterial zinc transporters and regulators. Biometals 14:239–249. https://doi.org/10.1023/A:1012984713391
Sheng Y, Fan F, Jensen O, Zhong Z, Kan B, Wang H, Zhu J (2015) Dual zinc transporter systems in Vibrio cholerae promote competitive advantages over gut microbiome. Infect Immun 83:3902–3908. https://doi.org/10.1128/iai.00447-15
Petrarca P, Ammendola S, Pasquali P, Battistoni A (2010) The Zur-regulated ZinT protein is an auxiliary component of the high-affinity ZnuABC zinc transporter that facilitates metal recruitment during severe zinc shortage. J Bacteriol 192:1553–1564. https://doi.org/10.1128/jb.01310-09
Gabbianelli R, Scotti R, Ammendola S, Petrarca P, Nicolini L, Battistoni A (2011) Role of ZnuABC and ZinT in Escherichia coli O157:H7 zinc acquisition and interaction with epithelial cells. BMC Microbiol 11:36. https://doi.org/10.1186/1471-2180-11-36
Grass G, Franke S, Taudte N, Nies DH, Kucharski LM, Maguire ME, Rensing C (2005) The metal permease ZupT from Escherichia coli is a transporter with a broad substrate spectrum. J Bacteriol 187:1604–1611. https://doi.org/10.1128/jb.187.5.1604-1611.2005
Karlinsey JE, Maguire ME, Becker LA, Crouch ML, Fang FC (2010) The phage shock protein PspA facilitates divalent metal transport and is required for virulence of Salmonella enterica sv. Typhimurium Mol Microbiol 78:669–685. https://doi.org/10.1111/j.1365-2958.2010.07357.x
Ammendola S, Pasquali P, Pistoia C, Petrucci P, Petrarca P, Rotilio G, Battistoni A (2007) High-affinity Zn2+ uptake system ZnuABC is required for bacterial zinc homeostasis in intracellular environments and contributes to the virulence of Salmonella enterica. Infect Immun 75:5867–5876. https://doi.org/10.1128/iai.00559-07
Gunasekera TS, Herre AH, Crowder MW (2009) Absence of ZnuABC-mediated zinc uptake affects virulence-associated phenotypes of uropathogenic Escherichia coli CFT073 under Zn (II)-depleted conditions. FEMS Microbiol Lett 300:36–41. https://doi.org/10.1111/j.1574-6968.2009.01762.x
Davis LM, Kakuda T, DiRita VJ (2009) A Campylobacter jejuni znuA orthologue is essential for growth in low-zinc environments and chick colonization. J Bacteriol 191:1631–1640. https://doi.org/10.1128/jb.01394-08
Yang X, Becker T, Walters N, Pascual DW (2006) Deletion of znuA virulence factor attenuates Brucella abortus and confers protection against wild-type challenge. Infect Immun 74:3874–3879. https://doi.org/10.1128/iai.01957-05
Quan G, Xia P, Lian S, Wu Y, Zhu G (2020) Zinc uptake system ZnuACB is essential for maintaining pathogenic phenotype of F4ac (+) enterotoxigenic E. coli (ETEC) under a zinc restricted environment. Vet Res 51:127. https://doi.org/10.1186/s13567-020-00854-1
Campoy S, Jara M, Busquets N, Pérez de Rozas AM, Badiola I, Barbé J (2002) Role of the high-affinity zinc uptake znuABC system in Salmonella enterica Serovar typhimurium virulence. Infect Immun 70:4721–4725. https://doi.org/10.1128/iai.70.8.4721-4725.2002
Lindsay JA, Foster SJ (2001) zur: a Zn2+-responsive regulatory element of Staphylococcus aureus. Microbiology 147:1259–1266. https://doi.org/10.1099/00221287-147-5-1259
Maciąg A, Dainese E, Rodriguez GM, Milano A, Provvedi R, Pasca MR, Smith I, Palù G, Riccardi G, Manganelli R (2007) Global analysis of the Mycobacterium tuberculosis Zur (FurB) regulon. J Bacteriol 189:730–740. https://doi.org/10.1128/jb.01190-06
Zhi H, Behnsen J, Aron A, Subramanian V, Liu JZ, Gerner RR, Petras D, Green KD, Price SL, Camacho J, Hillman H, Tjokrosurjo J, Montaldo NP, Hoover E, Treacy-Abarca S, Gilston BA, Skaar EP, Chazin WJ, Garneau-Tsodikova S, Lawrenz MB, Perry RD, Nuccio S-P, Dorrestein PC, Raffatellu M (2020) Siderophore-mediated zinc acquisition enhances enterobacterial colonization of the inflamed gut. In press, bioRxiv:2020.2007.2020.212498. doi:https://doi.org/10.1101/2020.07.20.212498
Hubert K, Devos N, Mordhorst I, Tans C, Baudoux G, Feron C, Goraj K, Tommassen J, Vogel U, Poolman JT, Weynants V (2013) ZnuD, a potential candidate for a simple and universal Neisseria meningitidis vaccine. Infect Immun 81:1915–1927. https://doi.org/10.1128/iai.01312-12
Stork M, Bos MP, Jongerius I, de Kok N, Schilders I, Weynants VE, Poolman JT, Tommassen J (2010) An outer membrane receptor of Neisseria meningitidis involved in zinc acquisition with vaccine potential. PLoS Pathog 6:e1000969. https://doi.org/10.1371/journal.ppat.1000969
Seth R, Corniola RS, Gower-Winter SD, Morgan TJ Jr, Bishop B, Levenson CW (2015) Zinc deficiency induces apoptosis via mitochondrial p53- and caspase-dependent pathways in human neuronal precursor cells. J Trace Elem Med Biol 30:59–65. https://doi.org/10.1016/j.jtemb.2014.10.010
Le Bras M, Rouy I, Brenner C (2006) The modulation of inter-organelle cross-talk to control apoptosis. Med Chem 2:1–12. https://doi.org/10.2174/157340606775197787
Truong-Tran AQ, Carter J, Ruffin RE, Zalewski PD (2001) The role of zinc in caspase activation and apoptotic cell death. Biometals 14:315–330. https://doi.org/10.1023/a:1012993017026
Choi S, Liu X, Pan Z (2018) Zinc deficiency and cellular oxidative stress: prognostic implications in cardiovascular diseases. Acta Pharmacol Sin 39:1120–1132. https://doi.org/10.1038/aps.2018.25
Lee SR (2018) Critical role of zinc as either an antioxidant or a prooxidant in cellular systems. Oxid Med Cell Longev 2018:9156285. https://doi.org/10.1155/2018/9156285
Wang X, Zhou B (2010) Dietary zinc absorption: a play of Zips and ZnTs in the gut. IUBMB Life 62:176–182. https://doi.org/10.1002/iub.291
Ohashi W, Hara T, Takagishi T, Hase K, Fukada T (2019) Maintenance of intestinal epithelial homeostasis by zinc transporters. Dig Dis Sci 64:2404–2415. https://doi.org/10.1007/s10620-019-05561-2
Huang D, Zhuo Z, Fang S, Yue M, Feng J (2016) Different zinc sources have diverse impacts on gene expression of zinc absorption related transporters in intestinal porcine epithelial cells. Biol Trace Elem Res 173:325–332. https://doi.org/10.1007/s12011-016-0655-x
Colvin RA, Fontaine CP, Laskowski M, Thomas D (2003) Zn2+ transporters and Zn2+ homeostasis in neurons. Eur J Pharmacol 479:171–185. https://doi.org/10.1016/j.ejphar.2003.08.067
Ohashi W, Fukada T (2019) contribution of zinc and zinc transporters in the pathogenesis of inflammatory bowel diseases. J Immunol Res 2019:8396878. https://doi.org/10.1155/2019/8396878
Gefeller EM, Martens H, Aschenbach JR, Klingspor S, Twardziok S, Wrede P, Pieper R, Lodemann U (2015) Effects of age and zinc supplementation on transport properties in the jejunum of piglets. J Anim Physiol Anim Nutr 99:542–552. https://doi.org/10.1111/jpn.12232
Hojyo S, Fukada T (2016) Zinc transporters and signaling in physiology and pathogenesis. Arch Biochem Biophys 611:43–50. https://doi.org/10.1016/j.abb.2016.06.020
Nimmanon T, Ziliotto S, Ogle O, Burt A, Gee J, Andrews G, Kille P, Hogstrand C, Maret W, Taylor K (2021) The ZIP6/ZIP10 heteromer is essential for the zinc-mediated trigger of mitosis. Cell Mol Life Sci 78:1781-1798. https://doi.org/10.1007/s00018-020-03616-6
Geiser J, Venken KJ, De Lisle RC, Andrews GK (2012) A mouse model of acrodermatitis enteropathica: loss of intestine zinc transporter ZIP4 (Slc39a4) disrupts the stem cell niche and intestine integrity. PLoS Genet 8:e1002766. https://doi.org/10.1371/journal.pgen.1002766
Ohashi W, Kimura S, Iwanaga T, Furusawa Y, Irié T, Izumi H, Watanabe T, Hijikata A, Hara T, Ohara O, Koseki H, Sato T, Robine S, Mori H, Hattori Y, Watarai H, Mishima K, Ohno H, Hase K, Fukada T (2016) Zinc transporter SLC39A7/ZIP7 promotes intestinal epithelial self-renewal by resolving ER Stress. PLoS Genet 12:e1006349. https://doi.org/10.1371/journal.pgen.1006349
Guthrie GJ, Aydemir TB, Troche C, Martin AB, Chang SM, Cousins RJ (2015) Influence of ZIP14 (slc39A14) on intestinal zinc processing and barrier function. Am J Physiol Gastrointest Liver Physiol 308:G171-178. https://doi.org/10.1152/ajpgi.00021.2014
Olechnowicz J, Tinkov A, Skalny A, Suliburska J (2018) Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J Physiol Sci 68:19–31. https://doi.org/10.1007/s12576-017-0571-7
Lu Q, Haragopal H, Slepchenko KG, Stork C, Li YV (2016) Intracellular zinc distribution in mitochondria, ER and the Golgi apparatus. Int J Physiol Pathophysiol Pharmacol 8:35–43
Bird AJ, Wilson S (2020) Zinc homeostasis in the secretory pathway in yeast. Curr Opin Chem Biol 55:145–150. https://doi.org/10.1016/j.cbpa.2020.01.011
Sinclair SA, Senger T, Talke IN, Cobbett CS, Haydon MJ, Krämer U (2018) Systemic upregulation of MTP2- and HMA2-mediated Zn partitioning to the shoot supplements local Zn deficiency responses. Plant Cell 30:2463–2479. https://doi.org/10.1105/tpc.18.00207
Hershfinkel M (2018) The Zinc sensing receptor, ZnR/GPR39, in health and disease. Int J Mol Sci 19:439. https://doi.org/10.3390/ijms19020439
Cohen L, Sekler I, Hershfinkel M (2014) The zinc sensing receptor, ZnR/GPR39, controls proliferation and differentiation of colonocytes and thereby tight junction formation in the colon. Cell Death Dis 5:e1307. https://doi.org/10.1038/cddis.2014.262
Sunuwar L, Asraf H, Donowitz M, Sekler I, Hershfinkel M (2017) The Zn2+-sensing receptor, ZnR/GPR39, upregulates colonocytic Cl– absorption, via basolateral KCC1, and reduces fluid loss. Biochim Biophys Acta Mol Basis Dis 1863:947–960. https://doi.org/10.1016/j.bbadis.2017.01.009
Cohen L, Asraf H, Sekler I, Hershfinkel M (2012) Extracellular pH regulates zinc signaling via an Asp residue of the zinc-sensing receptor (ZnR/GPR39). J Biol Chem 287:33339–33350. https://doi.org/10.1074/jbc.M112.372441
Shao YX, Lei Z, Wolf PG, Gao Y, Guo YM, Zhang BK (2017) Zinc supplementation, via GPR39, upregulates PKCζ to protect intestinal barrier integrity in Caco-2 cells challenged by Salmonella enterica serovar Typhimurium. J Nutr 147:1282–1289. https://doi.org/10.3945/jn.116.243238
Sargeant HR, Miller HM, Shaw MA (2011) Inflammatory response of porcine epithelial IPEC J2 cells to enterotoxigenic E. coli infection is modulated by zinc supplementation. Mol Immunol 48:2113–2121. https://doi.org/10.1016/j.molimm.2011.07.002
Deng Z, Luo XM, Liu J, Wang H (2020) Quorum sensing, biofilm, and intestinal mucosal barrier: involvement the role of probiotic. Front Cell Infect Microbiol 10:538077. https://doi.org/10.3389/fcimb.2020.538077
Alvarez C-S, Giménez R, Cañas M-A, Vera R, Díaz-Garrido N, Badia J, Baldomà L (2019) Extracellular vesicles and soluble factors secreted by Escherichia coli Nissle 1917 and ECOR63 protect against enteropathogenic E. coli-induced intestinal epithelial barrier dysfunction. BMC Microbiol 19:166. https://doi.org/10.1186/s12866-019-1534-3
Tsai PY, Zhang B, He WQ, Zha JM, Odenwald MA, Singh G, Tamura A, Shen L, Sailer A, Yeruva S, Kuo WT, Fu YX, Tsukita S, Turner JR (2017) IL-22 upregulates epithelial claudin-2 to drive diarrhea and enteric pathogen clearance. Cell Host Microbe 21:671-681.e674. https://doi.org/10.1016/j.chom.2017.05.009
Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, Kagnoff MF (1995) A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest 95:55–65. https://doi.org/10.1172/jci117676
Luo Y, Xu J, Zhang C, Jiang C, Ma Y, He H, Wu Y, Devriendt B, Cox E, Zhang H (2019) Toll-like receptor 5-mediated IL-17C expression in intestinal epithelial cells enhances epithelial host defense against F4+ ETEC infection. Vet Res 50:48. https://doi.org/10.1186/s13567-019-0665-8
Luo Y, Van Nguyen U, de la Fe Rodriguez PY, Devriendt B, Cox E (2015) F4+ ETEC infection and oral immunization with F4 fimbriae elicits an IL-17-dominated immune response. Vet Res 46:121. https://doi.org/10.1186/s13567-015-0264-2
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. https://doi.org/10.1126/science.1092385
Damo SM, Kehl-Fie TE, Sugitani N, Holt ME, Rathi S, Murphy WJ, Zhang Y, Betz C, Hench L, Fritz G, Skaar EP, Chazin WJ (2013) Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc Natl Acad Sci U S A 110:3841–3846. https://doi.org/10.1073/pnas.1220341110
Wątły J, Potocki S, Rowińska-Żyrek M (2016) Zinc homeostasis at the bacteria/host interface-from coordination chemistry to nutritional immunity. Chemistry 22:15992–16010. https://doi.org/10.1002/chem.201602376
Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP (2008) Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319:962–965. https://doi.org/10.1126/science.1152449
Abtin A, Eckhart L, Gläser R, Gmeiner R, Mildner M, Tschachler E (2010) The antimicrobial heterodimer S100A8/S100A9 (calprotectin) is upregulated by bacterial flagellin in human epidermal keratinocytes. J Invest Dermatol 130:2423–2430. https://doi.org/10.1038/jid.2010.158
Kehl-Fie Thomas E, Chitayat S, Hood MI, Damo S, Restrepo N, Garcia C, Munro Kim A, Chazin Walter J, Skaar Eric P (2011) Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 10:158–164. https://doi.org/10.1016/j.chom.2011.07.004
Gläser R, Meyer-Hoffert U, Harder J, Cordes J, Wittersheim M, Kobliakova J, Fölster-Holst R, Proksch E, Schröder JM, Schwarz T (2009) The antimicrobial protein psoriasin (S100A7) is upregulated in atopic dermatitis and after experimental skin barrier disruption. J Invest Dermatol 129:641–649. https://doi.org/10.1038/jid.2008.268
Hattori F, Kiatsurayanon C, Okumura K, Ogawa H, Ikeda S, Okamoto K, Niyonsaba F (2014) The antimicrobial protein S100A7/psoriasin enhances the expression of keratinocyte differentiation markers and strengthens the skin’s tight junction barrier. Br J Dermatol 171:742–753. https://doi.org/10.1111/bjd.13125
Carvalho A, Lu J, Francis JD, Moore RE, Haley KP, Doster RS, Townsend SD, Johnson JG, Damo SM, Gaddy JA (2020) S100A12 in digestive diseases and health: a scoping review. Gastroenterol Res Pract 2020:2868373. https://doi.org/10.1155/2020/2868373
Chiricozzi A, Nograles KE, Johnson-Huang LM, Fuentes-Duculan J, Cardinale I, Bonifacio KM, Gulati N, Mitsui H, Guttman-Yassky E, Suárez-Fariñas M, Krueger JG (2014) IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS One 9:e90284. https://doi.org/10.1371/journal.pone.0090284
Loes AN, Shi R, Harms MJ (2019) Zinc-independent activation of Toll-like receptor 4 by S100A9. bioRxiv 796:219. https://doi.org/10.1101/796219
Hashemi M, Chitayat S, Ande SR, Chazin W (2010) Apoptosis-inducing activity of the S100A8/A9 heterodimer. Antiinflamm Antiallergy Agents Med Chem. https://doi.org/10.2174/187152309789838993
Nakatani Y, Yamazaki M, Chazin WJ, Yui S (2005) Regulation of S100A8/A9 (calprotectin) binding to tumor cells by zinc ion and its implication for apoptosis-inducing activity. Mediators Inflamm 2005:280–292. https://doi.org/10.1155/mi.2005.280
Lapointe TK, Basso L, Iftinca MC, Flynn R, Chapman K, Dietrich G, Vergnolle N, Altier C (2015) TRPV1 sensitization mediates postinflammatory visceral pain following acute colitis. Am J Physiol Gastrointest Liver Physiol 309:G87-99. https://doi.org/10.1152/ajpgi.00421.2014
Brierley SM (2020) Gut nociceptors: sentinels promoting host defense. Cell Res 30:279–280. https://doi.org/10.1038/s41422-020-0278-9
Ghaisas S, Maher J, Kanthasamy A (2016) Gut microbiome in health and disease: linking the microbiome-gut-brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol Ther 158:52–62. https://doi.org/10.1016/j.pharmthera.2015.11.012
Gerner RR, Raffatellu M (2020) Gut pain sensors help to combat infection. Nature 580:594–595. https://doi.org/10.1038/d41586-020-01105-1
Steensels S, Depoortere I (2018) Chemoreceptors in the Gut. Annu Rev Physiol 80:117–141. https://doi.org/10.1146/annurev-physiol-021317-121332
Andersson DA, Gentry C, Moss S, Bevan S (2009) Clioquinol and pyrithione activate TRPA1 by increasing intracellular Zn2+. Proc Natl Acad Sci U S A 106:8374–8379. https://doi.org/10.1073/pnas.0812675106
Xu C, Wang Y, Sun R, Qiao X, Shang X, Niu W (2014) Modulatory effects of vasoactive intestinal peptide on intestinal mucosal immunity and microbial community of weaned piglets challenged by an enterotoxigenic Escherichia coli (K88). PLoS One 9:e104183. https://doi.org/10.1371/journal.pone.0104183
Chen L, Lin YL, Peng G, Li F (2012) Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc Natl Acad Sci USA 109:17966–17971. https://doi.org/10.1073/pnas.1210123109
Melkebeek V, Rasschaert K, Bellot P, Tilleman K, Favoreel H, Deforce D, De Geest BG, Goddeeris BM, Cox E (2012) Targeting aminopeptidase N, a newly identified receptor for F4ac fimbriae, enhances the intestinal mucosal immune response. Mucosal Immunol 5:635–645. https://doi.org/10.1038/mi.2012.37
Xia P, Wang Y, Zhu C, Zou Y, Yang Y, Liu W, Hardwidge PR, Zhu G (2016) Porcine aminopeptidase N binds to F4+ enterotoxigenic Escherichia coli fimbriae. Vet Res 47:24. https://doi.org/10.1186/s13567-016-0313-5
Xia P, Quan G, Yang Y, Zhao J, Wang Y, Zhou M, Hardwidge PR, Zhu J, Liu S, Zhu G (2018) Binding determinants in the interplay between porcine aminopeptidase N and enterotoxigenic Escherichia coli F4 fimbriae. Vet Res 49:23. https://doi.org/10.1186/s13567-018-0519-9
Santos AN, Langner J, Herrmann M, Riemann D (2000) Aminopeptidase N/CD13 is directly linked to signal transduction pathways in monocytes. Cell Immunol 201:22–32. https://doi.org/10.1006/cimm.2000.1629
Ishii K, Usui S, Sugimura Y, Yamamoto H, Yoshikawa K, Hirano K (2001) Inhibition of aminopeptidase N (AP-N) and urokinase-type plasminogen activator (uPA) by zinc suppresses the invasion activity in human urological cancer cells. Biol Pharm Bull 24:226–230. https://doi.org/10.1248/bpb.24.226
Ishii K, Usui S, Sugimura Y, Yoshida S, Hioki T, Tatematsu M, Yamamoto H, Hirano K (2001) Aminopeptidase N regulated by zinc in human prostate participates in tumor cell invasion. Int J Cancer 92:49–54
John E, Laskow TC, Buchser WJ, Pitt BR, Basse PH, Butterfield LH, Kalinski P, Lotze MT (2010) Zinc in innate and adaptive tumor immunity. J Transl Med 8:118. https://doi.org/10.1186/1479-5876-8-118
von Pein JB, Stocks CJ, Schembri MA, Kapetanovic R, Sweet MJ (2021) An alloy of zinc and innate immunity: galvanising host defence against infection. Cell Microbiol 23:e13268. https://doi.org/10.1111/cmi.13268
Fukada T, Hojyo S, Hara T, Takagishi T (2019) Revisiting the old and learning the new of zinc in immunity. Nat Immunol 20:248–250. https://doi.org/10.1038/s41590-019-0319-z
Taniguchi M, Fukunaka A, Hagihara M, Watanabe K, Kamino S, Kambe T, Enomoto S, Hiromura M (2013) Essential role of the zinc transporter ZIP9/SLC39A9 in regulating the activations of Akt and Erk in B-cell receptor signaling pathway in DT40 cells. PLoS One 8:e58022. https://doi.org/10.1371/journal.pone.0058022
Anzilotti C, Swan D, Boisson B, Deobagkar M, Oliveira C, Chabosseau P, Engelhardt K, Xu X, Chen R, Alvarez L, Berlinguer Palmini R, Bull K, Cawthorne E, Cribbs A, Crockford T, Dang T, Fearn A, Fenech E, Jong S, Hambleton S (2019) An essential role for the Zn2+ transporter ZIP7 in B cell development. Nat Immunol 20:350–361. https://doi.org/10.1038/s41590-018-0295-8
Hojyo S, Miyai T, Fujishiro H, Kawamura M, Yasuda T, Hijikata A, Bin BH, Irié T, Tanaka J, Atsumi T, Murakami M, Nakayama M, Ohara O, Himeno S, Yoshida H, Koseki H, Ikawa T, Mishima K, Fukada T (2014) Zinc transporter SLC39A10/ZIP10 controls humoral immunity by modulating B-cell receptor signal strength. Proc Natl Acad Sci USA 111:11786–11791. https://doi.org/10.1073/pnas.1323557111
Faghfouri AH, Zarrin R, Maleki V, Payahoo L, Khajebishak Y (2020) A comprehensive mechanistic review insight into the effects of micronutrients on toll-like receptors functions. Pharmacol Res 152:104619. https://doi.org/10.1016/j.phrs.2019.104619
Tsou TC, Liou SH, Yeh SC, Tsai FY, Chao HR (2013) Crucial role of Toll-like receptors in the zinc/nickel-induced inflammatory response in vascular endothelial cells. Toxicol Appl Pharmacol 273:492–499. https://doi.org/10.1016/j.taap.2013.09.014
Botella H, Peyron P, Levillain F, Poincloux R, Poquet Y, Brandli I, Wang C, Tailleux L, Tilleul S, Charrière GM, Waddell SJ, Foti M, Lugo-Villarino G, Gao Q, Maridonneau-Parini I, Butcher PD, Castagnoli PR, Gicquel B, de Chastellier C, Neyrolles O (2011) Mycobacterial p (1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe 10:248–259. https://doi.org/10.1016/j.chom.2011.08.006
Ong CY, Berking O, Walker MJ, McEwan AG (2018) new insights into the role of zinc acquisition and zinc tolerance in group A streptococcal infection. Infect Immun 86:e00048-e118. https://doi.org/10.1128/iai.00048-18
Stocks CJ, Phan MD, Achard MES, Nhu NTK, Condon ND, Gawthorne JA, Lo AW, Peters KM, McEwan AG, Kapetanovic R, Schembri MA, Sweet MJ (2019) Uropathogenic Escherichia coli employs both evasion and resistance to subvert innate immune-mediated zinc toxicity for dissemination. Proc Natl Acad Sci USA 116:6341–6350. https://doi.org/10.1073/pnas.1820870116
Subramanian Vignesh K, Deepe GS Jr (2016) Immunological orchestration of zinc homeostasis: the battle between host mechanisms and pathogen defenses. Arch Biochem Biophys 611:66–78. https://doi.org/10.1016/j.abb.2016.02.020
Lahiri A, Abraham C (2014) Activation of pattern recognition receptors up-regulates metallothioneins, thereby increasing intracellular accumulation of zinc, autophagy, and bacterial clearance by macrophages. Gastroenterology 147:835–846. https://doi.org/10.1053/j.gastro.2014.06.024
Stocks C, Schembri M, Sweet M, Kapetanovic R (2018) For when bacterial infections persist: toll-like receptor-inducible direct antimicrobial pathways in macrophages. J Leukoc Biol 103:35–51. https://doi.org/10.1002/JLB.4RI0917-358R
Gao H, Dai W, Zhao L, Min J, Wang F (2018) The role of zinc and zinc homeostasis in macrophage function. J Immunol Res 2018:6872621. https://doi.org/10.1155/2018/6872621
Bonaventura P, Benedetti G, Albarède F, Miossec P (2015) Zinc and its role in immunity and inflammation. Autoimmun Rev 14:277–285. https://doi.org/10.1016/j.autrev.2014.11.008
Kodama H, Tanaka M, Naito Y, Katayama K, Moriyama M (2020) Japan’s practical guidelines for zinc deficiency with a particular focus on taste disorders, inflammatory bowel disease, and liver cirrhosis. Int J Mol Sci 21:2941. https://doi.org/10.3390/ijms21082941
Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Divanovic S, Caruso JA, Deepe GS (2016) IL-4 induces metallothionein 3- and SLC30A4-dependent increase in intracellular Zn2+ that promotes pathogen persistence in macrophages. Cell Rep 16:3232–3246. https://doi.org/10.1016/j.celrep.2016.08.057
Shao Y, Wolf PG, Guo S, Guo Y, Gaskins HR, Zhang B (2017) Zinc enhances intestinal epithelial barrier function through the PI3K/AKT/mTOR signaling pathway in Caco-2 cells. J Nutr Biochem 43:18–26. https://doi.org/10.1016/j.jnutbio.2017.01.013
Oshima N, Ishihara S, Rumi MA, Aziz MM, Mishima Y, Kadota C, Moriyama I, Ishimura N, Amano Y, Kinoshita Y (2010) A20 is an early responding negative regulator of Toll-like receptor 5 signalling in intestinal epithelial cells during inflammation. Clin Exp Immunol 159:185–198. https://doi.org/10.1111/j.1365-2249.2009.04048.x
Jarosz M, Olbert M, Wyszogrodzka G, Młyniec K, Librowski T (2017) Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 25:11–24. https://doi.org/10.1007/s10787-017-0309-4
Shembade N, Ma A, Harhaj EW (2010) Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327:1135–1139. https://doi.org/10.1126/science.1182364
Maelfait J, Roose K, Bogaert P, Sze M, Saelens X, Pasparakis M, Carpentier I, van Loo G, Beyaert R (2012) A20 (Tnfaip3) deficiency in myeloid cells protects against influenza A virus infection. PLoS Pathog 8:e1002570. https://doi.org/10.1371/journal.ppat.1002570
Bao B, Thakur A, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, Ali S, Lum LG, Sarkar FH (2012) The immunological contribution of NF-kappaB within the tumor microenvironment: a potential protective role of zinc as an anti-tumor agent. Biochim Biophys Acta 1825:160–172. https://doi.org/10.1016/j.bbcan.2011.11.002
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49:1603–1616. https://doi.org/10.1016/j.freeradbiomed.2010.09.006
Sun R, Wang J, Young KH (2017) Oncogenic signaling pathways and pathway-based therapeutic biomarkers in lymphoid malignancies. Crit Rev Oncog 22:527–557. https://doi.org/10.1615/CritRevOncog.2017020816
Zhu C, Lv H, Chen Z, Wang L, Wu X, Chen Z, Zhang W, Liang R, Jiang Z (2016) Dietary zinc oxide modulates antioxidant capacity, small intestine development, and jejunal gene expression in weaned piglets. Biol Trace Elem Res 175:331–338. https://doi.org/10.1007/s12011-016-0767-3
Schmalen I, Reischl S, Wallach T, Klemz R, Grudziecki A, Prabu JR, Benda C, Kramer A, Wolf E (2014) Interaction of circadian clock proteins CRY1 and PER2 is modulated by zinc binding and disulfide bond formation. Cell 157:1203–1215. https://doi.org/10.1016/j.cell.2014.03.057
Peralta FA, Huidobro-Toro JP (2016) Zinc as allosteric ion channel modulator: ionotropic receptors as metalloproteins. Int J Mol Sci 17:1059. https://doi.org/10.3390/ijms17071059
Redhai S, Pilgrim C, Gaspar P, Giesen LV, Lopes T, Riabinina O, Grenier T, Milona A, Chanana B, Swadling JB, Wang YF, Dahalan F, Yuan M, Wilsch-Brauninger M, Lin WH, Dennison N, Capriotti P, Lawniczak MKN, Baines RA, Warnecke T, Windbichler N, Leulier F, Bellono NW, Miguel-Aliaga I (2020) An intestinal zinc sensor regulates food intake and developmental growth. Nature 580:263–268. https://doi.org/10.1038/s41586-020-2111-5
Remnant EJ, Williams A, Lumb C, Yang YT, Chan J, Duchêne S, Daborn PJ, Batterham P, Perry T (2016) Evolution, expression, and function of nonneuronal ligand-gated chloride channels in drosophila melanogaster. G3 6:2003–2012. https://doi.org/10.1534/g3.116.029546
Ye Q, Chen W, Huang H, Tang Y, Wang W, Meng F, Wang H, Zheng Y (2020) Iron and zinc ions, potent weapons against multidrug-resistant bacteria. Appl Microbiol Biotechnol 104:5213–5227. https://doi.org/10.1007/s00253-020-10600-4
Lin DL, Tran T, Alam JY, Herron SR, Ramirez MS, Tolmasky ME (2014) Inhibition of aminoglycoside 6’-N-acetyltransferase type Ib by zinc: reversal of amikacin resistance in Acinetobacter baumannii and Escherichia coli by a zinc ionophore. Antimicrob Agents Chemother 58:4238–4241. https://doi.org/10.1128/aac.00129-14
Broekaert IJ, Nanthakumar NN, Walker WA (2007) Secreted probiotic factors ameliorate enteropathogenic infection in zinc-deficient human Caco-2 and T84 cell lines. Pediatr Res 62:139–144. https://doi.org/10.1203/PDR.0b013e31809fd85e
Berni Canani R, Buccigrossi V, Passariello A (2011) Mechanisms of action of zinc in acute diarrhea. Curr Opin Gastroenterol 27:8–12. https://doi.org/10.1097/MOG.0b013e32833fd48a
Santosham M, Chandran A, Fitzwater S, Fischer-Walker C, Baqui AH, Black R (2010) Progress and barriers for the control of diarrhoeal disease. Lancet 376:63–67. https://doi.org/10.1016/s0140-6736(10)60356-x
Wessels I, Maywald M, Rink L (2017) Zinc as a gatekeeper of immune function. Nutrients 9:1286. https://doi.org/10.3390/nu9121286
Ghaffari H, Tavakoli A, Moradi A, Tabarraei A, Bokharaei-Salim F, Zahmatkeshan M, Farahmand M, Javanmard D, Kiani SJ, Esghaei M, Pirhajati-Mahabadi V, Monavari SH, Ataei-Pirkooh A (2019) Inhibition of H1N1 influenza virus infection by zinc oxide nanoparticles: another emerging application of nanomedicine. J Biomed Sci 26:70. https://doi.org/10.1186/s12929-019-0563-4
Kümel G, Schrader S, Zentgraf H, Daus H, Brendel M (1990) The mechanism of the antiherpetic activity of zinc sulphate. J Gen Virol 71:2989–2997. https://doi.org/10.1099/0022-1317-71-12-2989
Krenn BM, Gaudernak E, Holzer B, Lanke K, Van Kuppeveld FJ, Seipelt J (2009) Antiviral activity of the zinc ionophores pyrithione and hinokitiol against picornavirus infections. J Virol 83:58–64. https://doi.org/10.1128/jvi.01543-08
Qiu M, Chen Y, Chu Y, Song S, Yang N, Gao J, Wu Z (2013) Zinc ionophores pyrithione inhibits herpes simplex virus replication through interfering with proteasome function and NF-κB activation. Antiviral Res 100:44–53. https://doi.org/10.1016/j.antiviral.2013.07.001
Liu CY, Kielian M (2012) Identification of a specific region in the e1 fusion protein involved in zinc inhibition of semliki forest virus fusion. J Virol 86:3588–3594. https://doi.org/10.1128/jvi.07115-11
Read SA, Obeid S, Ahlenstiel C, Ahlenstiel G (2019) The role of zinc in antiviral immunity. Adv Nutr 10:696–710. https://doi.org/10.1093/advances/nmz013
Kim CH, Kim JH, Lee J, Ahn YS (2003) Zinc-induced NF-kappaB inhibition can be modulated by changes in the intracellular metallothionein level. Toxicol Appl Pharmacol 190:189–196. https://doi.org/10.1016/s0041-008x(03)00167-4
Raymond AD, Gekonge B, Giri MS, Hancock A, Papasavvas E, Chehimi J, Kossenkov AV, Nicols C, Yousef M, Mounzer K, Shull J, Kostman J, Showe L, Montaner LJ (2010) Increased metallothionein gene expression, zinc, and zinc-dependent resistance to apoptosis in circulating monocytes during HIV viremia. J Leukoc Biol 88:589–596. https://doi.org/10.1189/jlb.0110051
Costello LC, Guan Z, Franklin RB, Feng P (2004) Metallothionein can function as a chaperone for zinc uptake transport into prostate and liver mitochondria. J Inorg Biochem 98:664–666. https://doi.org/10.1016/j.jinorgbio.2004.02.005
Chaturvedi UC, Shrivastava R (2005) Interaction of viral proteins with metal ions: role in maintaining the structure and functions of viruses. FEMS Immunol Med Microbiol 43:105–114. https://doi.org/10.1016/j.femsim.2004.11.004
Wessels I, Rolles B, Rink L (2020) The potential impact of zinc supplementation on COVID-19 pathogenesis. Front Immunol 11:1712. https://doi.org/10.3389/fimmu.2020.01712
Kaushik N, Subramani C, Anang S, Muthumohan R, Shalimar NB, Ranjith-Kumar CT, Surjit M (2017) Zinc salts block hepatitis E virus replication by inhibiting the activity of viral RNA-dependent RNA polymerase. J Virol 91:e00754-e817. https://doi.org/10.1128/jvi.00754-17
Eijkelkamp BA, Morey JR, Neville SL, Tan A, Pederick VG, Cole N, Singh PP, Ong CY, Gonzalez de Vega R, Clases D, Cunningham BA, Hughes CE, Comerford I, Brazel EB, Whittall JJ, Plumptre CD, McColl SR, Paton JC, McEwan AG, Doble PA, McDevitt CA (2019) Dietary zinc and the control of Streptococcus pneumoniae infection. PLoS Pathog 15:e1007957. https://doi.org/10.1371/journal.ppat.1007957
St Croix CM, Leelavaninchkul K, Watkins SC, Kagan VE, Pitt BR (2005) Nitric oxide and zinc homeostasis in acute lung injury. Proc Am Thorac Soc 2:236–242. https://doi.org/10.1513/pats.200501-007AC
Woodworth BA, Zhang S, Tamashiro E, Bhargave G, Palmer JN, Cohen NA (2010) Zinc increases ciliary beat frequency in a calcium-dependent manner. Am J Rhinol Allergy 24:6–10. https://doi.org/10.2500/ajra.2010.24.3379
Imai Y, Kuba K, Ohto-Nakanishi T, Penninger JM (2010) Angiotensin-converting enzyme 2 (ACE2) in disease pathogenesis. Circ J 74:405–410. https://doi.org/10.1253/circj.cj-10-0045
Fleming SB (2016) Viral inhibition of the IFN-induced JAK/STAT signalling pathway: development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines (Basel) 4:23. https://doi.org/10.3390/vaccines4030023
Liu MJ, Bao S, Napolitano JR, Burris DL, Yu L, Tridandapani S, Knoell DL (2014) Zinc regulates the acute phase response and serum amyloid A production in response to sepsis through JAK-STAT3 signaling. PLoS One 9:e94934. https://doi.org/10.1371/journal.pone.0094934
Acknowledgements
The authors gratefully acknowledge the funding support.
Funding
This study was supported by Chinese National Science Foundation grants (Nos. 32072820, 31702242, 31972651, and 31800121), a grant from Jiangsu Government Scholarship for Overseas Studies (JS20190246), and a project funded by the Priority Academic Program of Development Jiangsu High Education Institution and High-level Talents of Yangzhou University Scientific Research Foundation.
Author information
Authors and Affiliations
Contributions
PX and GZ participated in the structural design of this paper. PX prepared the figures and drafted the manuscript. SL, YW, GQ and GZ critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
All the authors approved and gave explicit consent for publication of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xia, P., Lian, S., Wu, Y. et al. Zinc is an important inter-kingdom signal between the host and microbe. Vet Res 52, 39 (2021). https://doi.org/10.1186/s13567-021-00913-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-021-00913-1