
Abstract
Background
The cause of immune-mediated myositis (IMM), characterized by recurrent, rapid-onset muscle atrophy in Quarter Horses (QH), is unknown. The histopathologic hallmark of IMM is lymphocytic infiltration of myofibers. The purpose of this study was to identify putative functional variants associated with equine IMM.
Methods
A genome-wide association (GWA) study was performed on 36 IMM QHs and 54 breed matched unaffected QHs from the same environment using the Equine SNP50 and SNP70 genotyping arrays.
Results
A mixed model analysis identified nine SNPs within a ~ 2.87 Mb region on chr11 that were significantly (Punadjusted < 1.4 × 10− 6) associated with the IMM phenotype. Associated haplotypes within this region encompassed 38 annotated genes, including four myosin genes (MYH1, MYH2, MYH3, and MYH13). Whole genome sequencing of four IMM and four unaffected QHs identified a single segregating nonsynonymous E321G mutation in MYH1 encoding myosin heavy chain 2X. Genotyping of additional 35 IMM and 22 unaffected QHs confirmed an association (P = 2.9 × 10− 5), and the putative mutation was absent in 175 horses from 21 non-QH breeds. Lymphocytic infiltrates occurred in type 2X myofibers and the proportion of 2X fibers was decreased in the presence of inflammation. Protein modeling and contact/stability analysis identified 14 residues affected by the mutation which significantly decreased stability.
Conclusions
We conclude that a mutation in MYH1 is highly associated with susceptibility to the IMM phenotype in QH-related breeds. This is the first report of a mutation in MYH1 and the first link between a skeletal muscle myosin mutation and autoimmune disease.
Similar content being viewed by others

Background
Inflammatory myopathies are infectious or immune-mediated disorders that are characterized by the presence of lymphocytes in the skeletal muscle. Immune-mediated myositides (IMMs) are an important cause of morbidity and, in some cases, mortality in several species including humans [1], dogs [2], and horses [3, 4]. Common clinical features include malaise, muscle atrophy, and weakness with a histopathologic hallmark of inflammatory infiltrates, particularly lymphocytes, surrounding blood vessels, and within myocytes [5, 6]. There are several different IMM subtypes including inclusion body myositis in humans [7], polymyositis and dermatomyositis in dogs and humans [5], canine masticatory myositis [8], and equine IMM [3, 4]. Equine IMM is characterized by CD4+, CD8+, and CD20+ lymphocytic infiltrates surrounding blood vessels and infiltrating myofibers without evidence of rimmed vacuoles [3, 6]. Similar to human IMM, equine IMM has a bimodal age distribution affecting young horses (< 8 years of age) or older horses (> 17 years of age) [3, 9].
Causes of autoimmune diseases such as IMM are not well understood, but environmental stimuli, combined with a genetic predilection, appear to be important initiating factors [10, 11].The precise environmental trigger for equine IMM is not clear, but 39% of horses with IMM are reported to have a recent history of infection, particularly with Streptococci spp., or vaccination with influenza, herpes virus-1, or Streptococcus equi subsp. equi 3 to 4 weeks prior to onset [3, 4]. While recurrence of muscle wasting is reported with equine IMM, an improvement in clinical signs is often noted following treatment with corticosteroids. Full muscle mass is typically regained in 1–10 weeks, with corticosteroid treatment decreasing the time to full recovery [3].
Genetic associations with IMM have been found with various major histocompatibility complex loci in humans and dogs [10, 12]. Because the majority of horses affected by IMM are of Quarter Horse (QH)-related breeds and since certain stallions appear to be overrepresented in the genetic lineage of QHs with IMM, we hypothesized that there is an underlying genetic variant that causes susceptibility to IMM in QHs [3, 13]. The first objective of this study was to identify associated genomic regions underlying risk for developing equine IMM by performing a genome-wide association (GWA) study of QHs and related breeds with and without IMM that were housed in the same environment and therefore exposed to the same risk factors that may result in the IMM phenotype. The second objective was to evaluate the region of association from the GWA using whole genome sequencing to identify a putative functional variant associated with the equine IMM phenotype. The third objective was to determine if the alteration encoded by the putative functional variant altered protein structure and was targeted by inflammation.
Methods
All blood and muscle samples were collected with approval from the Animal Care and Use Committee at the University of Minnesota and University of California, Davis.
IMM case and control selection
GWA cohort
IMM-affected QHs (n = 36; 11 geldings, 10 stallions, and 15 mares) were selected based on a history of muscle atrophy (particularly of the epaxial or gluteal muscles, Fig. 1) and the presence of lymphocytes invading myofibers or cuffing blood vessels in a formalin-fixed or fresh muscle biopsy as previously described [3]. Horses with type 1 polysaccharide storage myopathy based on amylase-resistant polysaccharide in myofibers or the presence of the H309A GYS1 mutation [14] were excluded. The mean ± SEM age at the time of biopsy was 4.6 ± 0.8 years (range 0.1–19 years) for IMM-affected QHs. Due to the importance of environmental triggers, unaffected QHs (n = 54; 5.6 ± 0.6 years range 1–17; 17 geldings, 3 stallions and 34 mares) were selected from two herds that had active IMM cases. Unaffected horses had no history of muscle atrophy or stiffness consistent with IMM. Horses were selected such that they were not related at least within one generation. Of the horses used for the GWA, 1/36 affected and 41/54 unaffected horses were used in a previous genetic study [15].

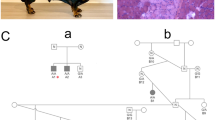
a Normal muscle mass in an MYH1 E321G homozygote prior to developing IMM. b The same horse 4 months after an episode of IMM. The spine is prominent due to loss of epaxial muscles (arrow). c Atrophy of middle and superficial gluteal muscles (arrow) is present
Whole-genome sequencing
From the GWA cohort, four of the most severely affected IMM QHs (1 gelding, 1 stallion, and 2 mares) and four unaffected QHs (2 geldings and 2 mares) were selected for whole-genome sequencing.
Follow-up cohort
The follow-up cohort included an additional 35 IMM QHs (13 geldings, 11 stallions, 11 mares; mean age at biopsy 3.4 ± 0.8 years [range 0.5–18 years]), phenotyped clinically by muscle atrophy (Fig. 1) (n = 25/35) and stiffness (n = 4/35) with mild lymphocytic infiltrates in myocytes or vascular cuffing in muscle samples. Unaffected QHs (n = 22; 5 geldings, 2 stallions, 15 mares, > 2 years) in this follow-up cohort were housed on the same property as IMM-affected cases and therefore exposed to the same environmental risk factors to develop IMM. While these horses may have gone on to develop myositis later in life, all had no history of disease at the time of sampling. All horses in this cohort were genotyped for the non-synonymous MYH1 E321G variant.
Random QH cohort
A cohort of 28 healthy QHs (n = 22) and a related breed, Paint horses (6), that were embryo transfer recipient horses of unknown bloodlines were genotyped for the putative variant to assess the prevalence of the MYH1 E321G variant in distantly related or unrelated cohorts. There was no history of IMM in this herd.
Across breed cohort
Genotyping of the MYH1 E321G variant was performed in a total of 64 horses across 6 breeds (Additional file 1: Table S1). Additionally, publically mapped whole-genome sequences from the Sequence Read Archive (SRA; https:/www.ncbi.nlm.nih.gov/sra) were available for 110 horses across 21 breeds.
Pedigree analysis
From the original GWA cohort, pedigrees were available for 23/36 QH. Pedigrees were analyzed using Pedigraph [16]. Following genotyping of all horses for the MYH1 E321G variant, additional pedigrees were created of individual families.
Genome-wide association (GWA)
DNA isolations were prepared from whole blood (ArchivePure™ DNA Blood Kit VWR International, Radnor, PA) or muscle (ArchivePure™ DNA Tissue Kit VWR International, Radnor, PA) samples according to the provided protocols. Genotyping of a subset of samples was performed on the Equine SNP 50K BeadChip (Illumina, San Diego, CA) (1 IMM and 41 unaffected horses), prior to the creation of the Equine SNP 70K BeadChip (Illumina, San Diego, CA). Thirty-five IMM QHs and 13 unaffected QHs were genotyped across 74,500 SNP markers with the Equine SNP 70K BeadChip (Illumina, San Diego, CA).
Statistical methods and data analysis
Datasets were merged and only SNPs that passed quality control settings (minor allele frequency > 1%, genotyping across individuals > 90%, and Hardy-Weinberg p > 0.001) were selected. A genome-wide efficient mixed model association was performed using GEMMA software using the standardized relatedness matrix option (−gk 2) [17]. Population stratification was estimated by assessing the genomic control inflation factor (λ). A Bonferroni correction for 39,589 tests (the number of useable SNPs) from the GEMMA analysis, based on a Pgenome-wide of 0.05, was determined as 1.26 × 10− 6.
Haplotype analysis
For chr11, which demonstrated the only genome-wide significant associations on the GWA, haplotypes were reconstructed on the individual chromosome using Haploview [18]. SNPs were filtered based on genotyping (> 90%) and minor allele frequency (> 1%). Association testing of both the single markers and haplotypes was performed using 1000 permutations. The adjusted haplotype-wide significance threshold was Ppermuted = 0.05.
Whole-genome sequencing
Using Illumina’s TruSeq DNA PCR-free library preparation kit (Illumina, San Diego, CA, USA) and following the manufacturer’s instructions, libraries were prepared with median insert size of 300–400 bp from the four IMM QHs and four unaffected QHs. The eight libraries were barcoded and pooled across eight lanes of a 125PE flow cell on an Illumina HiSeq2500, generating an average of 10.2× coverage per horse. Following quality trimming, reads were mapped to the EquCab2.0 reference genome using the Burrows-Wheeler Aligner (BWA) version 0.7.5a [19] using default settings. After sorting the mapped reads by the coordinates of the sequence, PCR duplicates were labeled with Picard tools (http://sourceforge.net/projects/picard/). The Genome Analysis Tool Kit (GATK version v.2.7.4) was used to perform local realignment [20]. Variant calls were made across all eight samples simultaneously using standard hard filtering parameters or variant quality score recalibration with Haplotype Caller according to GATK Best Practices Recommendations [21, 22].
Statistical methods and data analysis
SnpEFF [23] and SnpSift [24] were used to predict the functional effects of detected variants across the genome and within chr11 candidate region and filter by segregation using Fisher’s exact test. Variants were filtered by region in the entire associated haplotype block on chr11:49,915,548–56,207,873. To further refine the segregation, an unaffected Arabian horse from a previously published study [25] was included. Within this region, segregating variants were further filtered by the Fisher’s exact allelic P value (< 0.0003), allowing for one heterozygote in the IMM-affected group. Segregating variants within this region were further evaluated using all publically available mapped whole-genome sequences in the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra). Putative segregating variants were excluded if found in > 1 breed other than the QH. In addition to variant calling, visual inspection of the raw reads using the Integrated Genomics Viewer [26] within the chr11 region of association was performed. As conservation scores are not available in the EquCab2.0 genome browser within UCSC (https://genome.ucsc.edu/), scores were determined for each orthologous human variant using the 100 vertebrate score by phastCons (https://genome.ucsc.edu/). Whole genome sequences were deposited in the NCBI Sequence Read Archive (https://submit.ncbi.nlm.nih.gov/subs/sra/) (SRP119975).
Genotyping
Primer pairs were designed using Primer3Plus software [27] (F-CCCAAGATCTCAATGGCACT and R-ACCTTGTGGGAACATTCAGC) to amplify and subsequently genotype the nonsynonymous MYH1 E321G variant in an additional cohort of IMM-affected and unaffected QHs and a cohort of unaffected Arabian horses. Amplification of products was performed using endpoint PCR and visualized with the QIAxcel Advance System (QIAGEN, Valencia, CA, USA) and the QIAxcel DNA Screening Kit (QIAGEN, Valencia, CA, USA). The 20-μL PCR reactions were comprised of 2 U of Hot-start TAQ and 2.0 μL of 10× Buffer (Applied Biosystems, Foster City, CA, USA), 0.25 mM dNTPs (Thermo Fisher, Waltham, MA, USA), 0.5 μM of both forward and reverse primers (Invitrogen Life Technologies, Carlsbad, CA, USA), and 20 ng genomic DNA. Standard PCR conditions were performed as follows: hot-start TAQ activation and initial denaturation at 95 °C for 10 min; 35 cycles of 95 °C denaturation for 30 s, 60 °C annealing for 1 min, and 72 °C extension for 1 min; and final extension at 72 °C for 10 min. PCR products were purified using ExoSAP-IT® PCR Product Cleanup Kit (Affymetrix, San Diego, CA, USA). Sanger sequencing was performed using ABI 2500 automated sequencers. Resulting sequences were aligned to EquCab2.0 (http://www.ncbi.nlm.nih.gov/genome/145) and analyzed with Sequencher® software (Gene Codes Corporation, Ann Arbor, MI, USA).
Quantitative real-time PCR
Skeletal muscle MYH1 expression was assessed in 14 IMM-affected QH (11 E321G MYH1 homozygotes, 3 heterozygotes) and 11 unaffected horses across various breeds (all homozygous wild-type). Primers were designed for MYH1 (F-CACCACCAACCCGTATGACT and R-GAAGCCCAAGATCTCAATGG) and the reference gene ACTB (F-AAGGAGAAGCTCTGCTATGTCG and R-GGGCAGCTCGTAGCTCTTC), RT-qPCR was performed, and data were analyzed as previously reported [28].
Protein modeling and side chain analysis
Conformational changes caused by the identified E321G MYH1 variant were modeled using online G23D tool [29] and Homo sapiens myosin gene chain A (PDB ID: 4pa0). The amino acid in the mutated position was modeled using SCcomp [30]. Contact surface areas and solvent accessible surface areas were calculated using G23D, which applies Voronoi tessellation to allocate contact surfaces between neighboring atoms [31, 32]. Stability analysis of the E317G variant was performed using I-Mutant-2 [33], directly accessed from G23D.
Inflammation and muscle fiber type composition
Formalin-fixed or fresh skeletal muscle biopsy specimens were obtained by referring veterinarians and shipped on gel packs to the Neuromuscular Diagnostic Laboratory. Fresh samples were frozen in isopentane suspended in liquid nitrogen within 48 of the initial biopsy, and samples were stored at − 80 °C.
In a study of IMM published previously, inflammatory cell types were identified in fresh muscle samples obtained from horses of Quarter Horse-related breeds that presented with a history of gross atrophy of gluteal and epaxial muscles evident on physical examination [13]. Macrophages were identified with acid phosphatase stains, and immunohistochemical staining was used to identify CD4+, CD8+, and CD20+ [6]. Detailed methods can be found elsewhere [3, 6]. For the purposes of the present study, the type of inflammatory cells infiltrating myofibers was re-assessed in relationship to MYH1 genotype (13 horses GG, 3 horses G/A).
Adequate well-preserved frozen muscle tissue remained after examining inflammatory cell types from 10 IMM horses that were then used to evaluate fiber types. In six IMM cases, inflammation was identified in a formalin-fixed gluteal sample and fiber typing was performed in a concomitantly submitted fresh semimembranosus sample lacking substantial inflammation. Semimembranosus or gluteal muscle samples from five horses that were homozygous wild-type for the MYH1 E321G variant were used as unaffected controls. Unaffected horses had normal muscle mass as assessed by physical examination and lacked lymphocytes or macrophages in muscle biopsies. Frozen samples from semimembranosus (4 E321G MYH1 homozygotes, 3 heterozygous, 4 wild-type) and middle gluteal (3 E321G MYH1 homozygotes, 0 heterozygous, 1 wild-type) muscles were assessed.
Serial 10-μm sections stained with hematoxylin and eosin (HE) and labeled by immunofluorescence for fiber type were used to identify inflammatory infiltrates within fiber types. Type 1, 2A, 2AX, and 2X muscle fiber types were identified by multiple fluorescent labeling according to Tulloch et al. [34]. Briefly, sections were incubated with a goat polyclonal anti-collagen V IgG antibody (1350-01 Southern Biotech, Birmingham, AL) 1:100 for 1 h at room temperature. Next, three separate mouse monoclonal antibodies to detect type 1, slow myosin IgG 1:100 (MAB1628 Millipore, Burlington, MA), type 2a IgG 1:6 (A4.74 DSHB), and both type 2A and 2X IgG 1:10 (NCL-MHCf Leica Biosystems, Buffalo Grove, IL) were conjugated to fluorescent IgG1 Fab fragments using Zenon® Mouse IgG labeling kits (Life Technologies, Carlsbad, CA), Alexa Fluor® 488 (A4.74), Alexa Fluor® 594 (NCL-MHCF), and Pacific Blue™ (MAB1628). The three Zenon® labeled antibodies were mixed together, added to the tissue sections, and incubated at 4 °C overnight. A secondary antibody for Collagen V, FITC-rabbit anti-goat IgG (61-1611, Invitrogen, Carlsbad, CA) 1:500 was applied to the cryosections and incubated for 1 h at room temperature. Sections were subsequently mounted using VECTASHIELD mounting medium (H1000, Vector Labs, Burlingame, CA) and examined using a fluorescence microscope (Olympus, Tokyo, Japan) with filters designed for each of the different emitting wavelengths. Images were captured and pseudo-colored composites generated.
Statistical methods and data analysis
The total number of type 1, 2A, 2AX, and 2X muscle fibers was determined for the entire muscle section, and fiber type compositions were determined by dividing the total number of fibers of each type by the total number of muscle fibers counted (range 447 to 3244 muscle fibers/sample). The fiber type composition of IMM samples with inflammation and wild-type samples were compared by genotype and disease status using a two-way ANOVA.
Results
Pedigree analysis
From the original GWA cohort, pedigrees were available for 23/36 QH. All affected horses could be traced back to a common sire within eight generations (Additional file 2: Figure S1). Pedigree analysis supported either an autosomal dominant or autosomal recessive mode of inheritance.
Genome-wide association study
Following quality control of the 73,706 SNPs available on the array, 39,589 SNPs remained (1601 excluded for minor allele frequency < 1%, 32,439 excluded for genotyping < 90%, and 77 excluded for failing hardy Weinberg equilibrium [P < 0.001]). Genomic inflation (λ) was estimated at 1.98, indicative of population stratification. Due to the elevated genomic inflation, a mixed model analysis was performed utilizing GEMMA with the same filters [17]. Using the GEMMA relationship matrix, genomic inflation was controlled for in the population (λ = 1.02). Nine SNPs on chromosome 11 reached genome-wide significance (Table 1 and Fig. 2a).

a Manhattan plot and (top right insert) QQ-plot demonstrating a genome-wide significant association with the IMM phenotype using GEMMA analysis [17] on chr11. Minimal genomic inflation was present. b Genotypes for the MYH1 E321G variant across IMM-affected horses (n = 71 GWA and follow-up cohorts combined), at risk horses (n = 75, housed on the same farms as IMM horses), a cohort of random QH (n = 28), and 21 other breeds (n = 179)
Haplotype analysis and candidate region
Haplotype analysis of 1109 SNPs on chr11 that passed quality control (838 removed for genotyping < 90% and 64 removed for minor allele frequency < 1%) using Haploview [18] identified 46 significantly associated haplotype blocks (Ppermuted < 0.05), spanning ~ 6.3 Mb from chr11: 49,915,548–56,207,873. Of these, four highly significant haplotype blocks (Ppermuted < 0.001) spanning ~ 3.1 Mb from chr11:52379156-55487290 were identified. This region overlapped the ~ 2.87 Mb region flanked by eight of the nine genome-wide significant SNPs from the GWA. Therefore, while the entire 6.3 Mb region was evaluated, the 3.1 Mb region was prioritized.
The 6.3 Mb region identified through GWA and subsequent haplotype analysis encompassed 148 Ensembl annotated genes, with 277 annotated transcripts in equine skeletal muscle [35]. Within the 3.1 Mb prioritized region, 38 Ensembl annotated genes and 48 annotated skeletal muscle transcripts were identified [35]. Of these 38 genes, four myosin genes (MYH1, MYH2, MYH3, and MYH13) and myocardin (MYOCD) were located within the associated region. MYH1 and MYH2 are expressed in adult equine skeletal muscle while MYH3 is expressed at the embryonic stage and MYH13 is expressed primarily in ocular skeletal muscle [36].
Whole-genome sequencing
Whole-genome sequencing was performed on four IMM and four unaffected QHs at ~ 10× coverage. Visual inspection of the raw reads using the Integrated Genomics Viewer [26] within the GWA region did not identify any structural variants across the eight horses. Using Haplotype Caller according to GATK Best Practices Recommendations [21, 22], an average of 5,107,127 single nucleotide variants (SNVs) and 655,690 insertions/deletions were identified across all eight horses. Variants were filtered based on a region of interest from the GWA and haplotype analysis, spanning the entire 6.3 Mb associated region (chr11:49,915,548–56,207,873), and 628 variants within this filtered region were significantly associated with the IMM phenotype (Fisher’s exact test, Punadjusted < 0.00003). These 628 variants were examined in all publically available mapped whole-genome sequences (n = 110 across 21 breeds) from the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra). Only 15 of these variants were unique to the QH. As IMM has not been reported in a breed other than QH-related breeds, the other 613 variants were excluded from further analysis. The remaining 15 breed-specific variants were prioritized for further evaluation (Table 2). Two of these variants were classified as MODERATE missense variants while other variants were classified as MODIFIERS (n = 12) or a LOW synonymous variant (n = 1) (Table 2).
To prioritize these 15 variants for further genotyping, the custom equine transcriptome recently published by our laboratory was utilized [35]. These RNA-Seq datasets included skeletal muscle and embryo (both inner cellular mass and trophoectoderm). Within all available sets, the MYH3 variant was non-coding (Table 2). Therefore, the MYH1 variant was initially prioritized for genotyping of a larger group of horses.
Genotyping of putative variants
DNA from 46/48 horses used in the GWA study was available for genotyping the MYH1 E321G variant and a significant association was identified (P = 6.76 × 10− 15). Within the follow-up cohort of 35 mildly affected IMM horses, a significant allelic association was validated (P = 2.95 × 10− 5) with the MYH1 E321G variant. In the two IMM cohorts combined, 87.7% of horses were homozygous or heterozygous for the variant (Fig. 2b). In the random cohort of QHs without a history of IMM, three heterozygotes (11%) were identified. Genotyping of 175 horses across 21 non-QH breeds did not identify any other breeds with the MYH1 E321G variant (Fig. 2b).
To further exclude the possibility that additional segregating variants were responsible for IMM, we genotyped a subset of horses for chr11:52,944,928 and chr11:53,105,764 in horses that lacked the MYH1 E321G variant but were classified as IMM based on lymphocytic infiltrates (n = 2 in GWA and n = 7 in follow-up cohort). The subset of horses genotyped included the n = 9 IMM-affected horses that genotyped A/A at chr11:52,993,878, IMM-affected horses that genotyped G/G (n = 11), and unaffected horses that genotyped A/A (n = 11). The nine IMM-affected horses that had genotyped A/A at chr11:52,993,878 also genotyped for the reference allele at the two other segregating variants, resembling the unaffected horse genotype. These nine horses with an inflammatory myopathy therefore appeared to be phenocopies. In the other IMM-affected horses, the two other variants are genotyped as alternate allele/alternate allele. Because these variants lacked a strong functional effect, were non-coding in adult equine skeletal muscle, and lacked any known connection to immune or muscle function, the MYH1 E321G variant was identified as the putative functional variant for equine IMM (Table 2).
Pedigree evaluation based on MYH1 E321G genotype
Available pedigrees were re-evaluated on the 23 IMM-affected horses based on the MYH1 E321G genotype and could be distinctly summarized as linking to four founder stallions (Fig. 3). Two of these founder stallions were traced back to the stallion in family A within one to three generations. For family D, the dam line traced back to the stallion in family A within four generations.

a–d Pedigrees from four subsets of the larger pedigree (Additional file 2: Figure S1) demonstrating a founder effect of four stallions for the IMM phenotype. Circles = females, squares = males. Open circles = phenotype unknown. Colored circles = IMM-affected with red = G/G genotype, blue = G/A genotype, and gray = A/A phenotype. Two of these founder stallions were traced back to the stallion in family A within one to three generations. For family D, the dam line traced back to the stallion in family A within four generations (Additional file 2: Figure S1)
RT-qPCR
There was no significant difference in MYH1 skeletal muscle expression between IMM-affected horses and controls (data not shown).
Protein modeling, contact area, and stability analysis
The missense mutation identified in MYH1 is located in a highly conserved region of the myosin globular head in subfragment-1 between the helix loop-helix region of Helix J and Helix K (Fig. 4a, b). The helix loops lay between SWITCH1 and SWITCH2 motifs that have been identified as RAS GTP proteins, and play a role in the binding of ATP [29]. The MYH1 E321G mutation substitutes a negatively charged glutamic acid (E) for a non-polar glycine (G) that lacks side chains necessary for hydrogen bond formation. Contact area and solute accessibility analyses showed that 14 residues were directly affected by the MYH1 E321G mutation (Fig. 4c) with the largest reduction of contact between the SWITCH1 and helix 1 domains of the myosin 2X globular head (Fig. 4d, e). Furthermore, this single mutation of E317G is responsible for significantly decreasing the stability of the protein at physiological conditions with a RI score of 8 (Fig. 4f).

Schematic representation of MYH1 (MyHC 2X) with wild type (E317E, wt) and MYH1 mutation (E317G, mut). a Wild-type (wt) variant is shown as light green (red arrow) and E317G mutation (mut) is shown on the adjacent panel as teal (red arrow). In both panels, helices are represented as blue and additionally loop, SWITCH regions and actin binding sites are labeled. b Detailed alignments of the query protein (upper sequence line) and structure hits showing the protein sequence (PDB SEQRES record) and the resolved coordinates (PDB ATOM record). The input variant is indicated by green arrows (below and above the alignment) and highlighted amino acids represent residues that appear in dbSNP as well as common associated somatic mutation in cancer (COSMIC) variants. c Side chain and stability analysis of MYH1 E317G mutation showing contact surface areas between atoms and solvent accessible surfaces. Table includes contact surface area (Å2) and minimal atomic distance (Å) between two atoms of the two residues for the 15 amino acid residues that were determined to be impacted by the mutation. d A pictorial presentation of contact/residues affected by the MYH1 E317G mutation and its association to SWITCH1, helix I and helix J regions of MYH1. The residues affected are represented with an asterisk (*). Helix K is partially represented. The input variant is indicated by green arrows (below and above the alignment) and highlighted amino acids represent residues that appear in dbSNP as well as common associated somatic mutation in cancer (COSMIC) variants. e G23D model displaying a close up of combined MYH1 mutation (teal only) and wild type (green). Blue represent the helices and shows the proximity of the MYH1 mutation to the SWITCH 1 region and to the G677 and P710 residues found between Loop2 and SH2 helix domain of MYH1. f Stability analysis of the E317G mutation using I-mutant-2 at physiological pH, which predicts decreased stability of the mutant protein
Inflammation and muscle fiber type composition
CD4+ and CD8+ lymphocytes and macrophages were identified within and surrounding myofibers in all horses homozygous and heterozygous for the E321G MYH1 variant (Fig. 5a). Inflammatory cells were present in type 2X fibers which contain myosin heavy chain 2X that is encoded by MYH1 (Fig. 5b, c). Significantly fewer type 2X fibers (mean 27%; range 0–50%) were found in E321G MYH1 homozygous and heterozygous samples with inflammation compared to controls (mean 60; range 43–87%; P < 0.0001) (Fig. 5d, e) Small numbers of inflammatory cells, atrophic type 2X fibers, and a predominance of type 2A and 2AX fibers over 2X fibers were identified in the semimembranosus muscle of an IMM homozygote with a 4-month long history of IMM (Fig. 5f–h). In contrast, in the complete absence of inflammation, the proportion of type 1, 2A, 2AX, and 2X fibers of control horses did not differ significantly from IMM muscle samples (Fig. 5i–k). The mean percentage of type 2X fibers in IMM E321G horse muscle without inflammatory infiltrates (4 semimembranosus, 2 gluteal) was 45%, ranging from 30 to 56%, as compared to control samples (3 semimembranosus, 2 gluteal), where the mean percentage of type 2X fibers was 60%, ranging from 43–87%.

a Marked CD4+ lymphocytic infiltrates (red) within myofibers of horse 1, an E321G MYH1 homozygote. IHC × 20. b Serial section to c demonstrating inflammatory cell infiltrates (arrows) in horse 1. HE × 20. c Serial section to b demonstrating that fibers infiltrated by lymphocytes (vertical arrows) are 2X fibers (red) and that some fibers with cellular infiltrates (horizontal arrows) do not have any remaining myosin staining. IF × 20. d Serial section to e from horse 2 showing inflammatory cells (arrows). HE × 10. e Serial section to D demonstrating a complete absence of 2X fibers. Dark regions with no staining (arrows) correspond to regions of inflammation in D. IF × 10. f Normal fiber type distribution and fiber sizes in another homozygous wild-type horse for comparison. IF × 20. g Section of muscle from horse 3 that had a 4-month history of IMM showing a small amount of inflammatory cell infiltrates. HE × 10. h Another section of horse 3 demonstrating atrophic type 2X fibers (brown). IF × 10. i Preponderance of type 2A (yellow) or 2AX fibers (intermediate) over type 2X fibers (brown) in another region of the sample from horse 3. IF × 10. j Normal fiber type distribution of muscle from a wild-type horse (type 1 blue, type 2A yellow, type 2X brown) showing type 2X fibers that are larger than type 2A fibers. IF × 10. k Normal fiber type distribution and fiber sizes in the semimembranosus muscle that lacks inflammation from horse 4 that was homozygous for the E321G variant. IF × 20. Bars in all figures represents 100 μm
Discussion
This is the first report of a mutation in MYH1 gene associated with susceptibility to a specific myopathy. A GWA study initially identified a ~ 6.3 Mb region on chr11 associated with IMM, and haplotype analyses narrowed this region to ~ 3.1 Mb that encompassed two myosin genes expressed in adult equine muscle, namely MYH1, encoding type 2X myosin heavy chain (MyHC), and MYH2, encoding MyHC 2A. Type 2X fibers comprise 40–80% of all fibers and type 2A comprise 30-40% of all fibers present in muscles typically affected with IMM [37]. One nonsynonymous variant was identified in MYH1 that resulted in substitution of a charged glutamic acid for a non-polar amino acid, glycine, at 321 in the MyHC globular head. Credence for the significance of the variant was provided by the fact that glutamic acid is highly conserved (Table 2), present in this position in seven other species (Sus, Bos, Homo, Oryctol, Rat, Canis) and in the corresponding position of MyHC 2A, perinatal, extraocular, embryonic, and cardiac/slow MyHC [38]. We propose that the other segregating variants with no known functional effect were in linkage disequilibrium with this MYH1 functional variant, although they cannot be conclusively excluded as putative variants for IMM. The MYH1 E321G variant is all the more significant, however, because of a strong selection pressure against mutations in skeletal muscle. MyHC appears to exist based on the remarkable orthology of myosin heavy chain genes across species [38, 39]. Thus, the E321G variant identified in MYH1 in IMM horses is both novel and strongly associated with susceptibility to develop equine IMM.
The MYH1 E321G variant appears to be variably penetrant, conferring susceptibility to disease, potentially dependent upon whether other factors needed to trigger an immune-mediated disease are present. A significant proportion of both IMM and in contact unaffected horses were heterozygous for the MYH1 E321G variant. Heterozygosity, however, was much less in the unrelated group of QH and absent in the Arabian breed of horse as well as in the publically available mapped NCBI SRA database of 21 breeds. The high degree of heterozygosity in the original cohort of horses can be explained by the fact that the group of horses in contact with IMM-affected horses were closely related to the affected horses. The unadjusted genomic inflation factor for the initial GWA was 1.98. To control for environmental exposure to factors such as other respiratory bacteria, viruses, and vaccinations that can trigger IMM, we selected horses in the same environment, which was a breeding farm in one case and a private farm with preference for certain bloodlines in another [4]. The M protein of S. equi in particular shows considerable similarity in amino acid sequence with MyHC 2X (encoded by MYH1; Additional file 3: Figure S2) and is a common trigger of IMM [3]. Lack of IMM onset in some heterozygotes could be the result of differential expression of the affected allele, subthreshold environmental triggering of autoimmunity, or a lack of initial priming of the immune system from a first exposure to mutant MyHC 2X. It is also possible that some of the in contact horses developed IMM at a later stage. Together, our results suggest that rather than consistently causing a myopathy, homozygosity and, in some cases, heterozygosity for the MYH1 variant predisposed horses to a myopathy under certain environmental triggers.
Nine of 71 horses diagnosed with IMM and genotyped for MYH1 E321G were homozygous reference. Of these, two were from the initial GWA cohort, where horses were phenotyped more stringently (i.e., moderate to severe lymphocytic infiltrate on muscle biopsy), and seven were from the follow-up cohort, where IMM-affected cases had a milder degree of myofiber lymphocytic infiltrates and included those with only perivascular lymphocytes. As additional nearby variants segregated with the IMM phenotype at identical frequencies as MYH1 E321G (Table 2), these nine horses were genotyped for these additional variants. All nine horses genotyped homozygous for the reference allele at the two additional variants, identical to the population of unaffected horses. Lymphocytic infiltrates in the skeletal muscle are not specific for immune-mediated myositis and can be found in other inflammatory myopathies [2, 13] with sarcocystosis being the most common equine infectious myopathy characterized by lymphocytic infiltrates [40, 41]. The nine horses lacking the MYH1 E321G variant could very well represent phenocopies due to an inflammatory myopathy. Phenocopies were to be expected because differentiation of immune-mediated versus inflammatory myopathies of infectious origin is difficult in horses based solely on muscle histopathology [2, 13, 42].
In total, 15 variants segregated at identical frequencies with the IMM phenotype (Table 2). While most of these variants were classified as having LOW or MODIFIER effects by SNPEff [23], two were characterized as having MODERATE effects (missense variants in MYH3 and MYH1) when using the Ensembl annotation. We have recently demonstrated that this annotation is not highly accurate in the horse, and we have published a tissue-specific transcriptome for the horse based on RNA-sequencing data and integration with other EquCab2.0 annotations, including NCBI [35]. This custom annotation includes transcriptomes of equine skeletal muscle and embryo. Across all tissues, the MYH3 variant was identified as non-coding. Therefore, we propose that these variants are in linkage disequilibrium with the MYH1 E321G variant.
Mutations in MYH2 have been reported in a small number of human patients. Recessive truncating deletions in MYH2 result in early-onset weakness confined to extraocular, semitendinosus, gracilis, vastus lateralis, and medial gastrocnemius muscles whereas dominant point mutations in MYH2 result in a later onset of mild progressive weakness [43,44,45]. Both recessive and dominant MYH2 mutations result in mild myopathic changes such as variability of fiber size, internalized nuclei, and increased interstitial connective and adipose tissue; however, only recessive deletions produce a total absence of type 2A myofibers [43, 45]. Unlike patients with recessive MYH2 mutations, in horses with IMM, the reduction in type 2X myofibers was dependent upon inflammation and, in the absence of inflammation, homozygous IMM horses had a normal proportion of type 2X fibers. In the small number of samples evaluated from homozygous horses with chronic atrophy, we found a decrease in the proportion of type 2X myofibers with a higher proportion of type 2A and 2AX fibers. Our results suggest that lymphocytic destruction of MyHC 2X fibers appears to be a prerequisite for acute inflammation in IMM horses. Previous studies have not identified immunoglobulin bound to myofibers in IMM horses [3]; however, circulating anti-MYH1 myosin or cytokines have not been studied in equine IMM. The clinical signs and muscle histopathology of IMM appear to be distinct from those previously reported for MYH2 mutations in other species.
In contrast to the low frequency of mutations in MYH1 and MYH2, more than 500 disease-causing point mutations have been described in MYH7, with the majority producing hypertrophic or dilated cardiomyopathy [46, 47]. A minority of MYH7 mutations are reported to cause skeletal myopathies such as myosin storage myopathies or Laing distal myopathy [46, 47]. One family with a p.K1729del in MYH7 had similar inflammatory changes to those seen in equine IMM, although the clinical presentation was that of distal limb weakness not the rapid proximal muscle atrophy seen in equine IMM [48]. Similar to equine IMM, increased skeletal muscle MHC class I expression and perivascular and endomysial lymphocytic infiltrates (CD3+, CD4+, and CD8+) were found in this family with Laing distal myopathy, along with rimmed vacuoles, which are not a feature of equine IMM [6, 48]. Laing distal myopathy has highly variable muscle pathology, however, and inflammation is not a consistent feature of the skeletal muscle in most patients [49].
The best characterized link between myosin, inflammation, and muscle disease would be immune-mediated myocarditis [50]. Fragments of cardiac myosin have been shown to activate Toll-like receptors (TLR2), which strongly drive reactivity to self and subsequently determine the type of adaptive immune response (i.e., Th1, Th2) that occurs [51]. Synergy between the activated innate immune response and the adaptive response of pathogenic T cell epitopes appear to be important in the generation of chronic myocardial inflammation [51]. Similar to human myocarditis, the adaptive immune response could be triggered in IMM horses by shared epitopes between bacteria such as the M protein of group A Streptococcus sp. and myosin (Additional file 3: Figure S2) [52]. The innate immune response could be triggered in IMM by release of the mutant form of MyHC 2X from myofibers following muscle damage (trauma, vaccination). The loss of hydrogen bonds with the MYH1 mutation could possibly lead to conformational changes in myosin that activate TLRs and autoimmunity. Of note, a nonsynonymous mutations in MYH7 (S545A) in DBA/2 mice appears to predispose these mice to immune-mediated myocarditis [53, 54]. When DBA/2 mice with the MYH7 S545A variant and BALB/c mice without the variant are auto-inoculated with cardiac myosin, chronic myocarditis only occurs when serum from the DBA/2 strain is injected into DBA/2 not BALB/c mice [53]. The authors concluded that susceptibility to autoimmune myocarditis was dependent not only on the activation of self-reactive lymphocytes but also on genetically determined target organ sensitivity. Because both the DBA/2 MYH7 mutation and the equine MYH1 variant are located in the globular head of myosin, it is possible that a mutation in this highly conserved region somehow confers target host susceptibility to myositis.
Perhaps the most intriguing link between myosin and autoimmunity comes from canine masticatory muscle myositis (CMM). CMM presents with painful swelling followed by rapid atrophy of masticatory muscles [8]. Similar to equine IMM, biopsies of CMM masseter muscle are characterized by MHC I upregulation, B cells, and a predominance of CD4+ over CD8+ T lymphocytic infiltrates in masticatory muscles [2, 6, 8, 53]. Both within the masseter muscle and in the circulation, autoantibodies for masticatory muscle myosin (2M) or myosin binding protein-C are evident with CMM, suggesting that myosin isoforms unique to masseter muscles have antigenic potential and can serve as target antigens for inflammatory cells [55]. Genetic analysis of CMM dogs has yet to be performed to assess the potential for putative mutations in masticatory muscle myosin encoding genes to enhance susceptible to CMM.
Conclusions
In conclusion, an E321G MYH1 mutation is highly associated with susceptibility to IMM in horses. In the absence of inflammation, type 2X muscle fiber type composition is within normal limits in IMM horses; however, within a particular environment, the MYH1 mutation results in invasion and destruction of type 2X myofibers by lymphocytes and rapid onset of gross muscle atrophy.
Abbreviations
- GWA:
-
Genome-wide association
- HE:
-
Hematoxylin and eosin
- IMM:
-
Immune-mediated myositis
- MHC:
-
Major histocompatibility complex
- MYH:
-
Myosin heavy chain
- QH:
-
Quarter Horse
- SNP:
-
Single nucleotide polymorphism
- WGS:
-
Whole genome sequencing
References
Dalakas MC. Polymyositis, dermatomyositis and inclusion-body myositis. N Engl J Med. 1991;325(21):1487–98.
Evans J, Levesque D, Shelton GD. Canine inflammatory myopathies: a clinicopathologic review of 200 cases. J Vet Intern Med. 2004;18(5):679–91.
Lewis SS, Valberg SJ, Nielsen IL. Suspected immune-mediated myositis in horses. J Vet Intern Med. 2007;21(3):495–503.
Hunyadi L, Sundman EA, Kass PH, Williams DC, Aleman M. Clinical implications and hospital outcome of immune-mediated myositis in horses. J Vet Intern Med. 2017;31(1):170–5.
Shelton GD. From dog to man: the broad spectrum of inflammatory myopathies. Neuromuscul Disord. 2007;17(9–10):663–70.
Durward-Akhurst SA, Finno CJ, Barnes N, Shivers J, Guo LT, Shelton GD, Valberg SJ. Major histocompatibility complex I and II expression and lymphocytic subtypes in muscle of horses with immune-mediated myositis. J Vet Intern Med. 2016;30(4):1313–21.
Schmidt J, Dalakas MC. Inclusion body myositis: from immunopathology and degenerative mechanisms to treatment perspectives. Expert Rev Clin Immunol. 2013;9(11):1125–33.
Shelton GD, Cardinet GH 3rd, Bandman E. Canine masticatory muscle disorders: a study of 29 cases. Muscle Nerve. 1987;10(8):753–66.
Medsger TA Jr, Dawson WN Jr, Masi AT. The epidemiology of polymyositis. Am J Med. 1970;48(6):715–23.
Rothwell S, Cooper RG, Lamb JA, Chinoy H. Entering a new phase of immunogenetics in the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2013;25(6):735–41.
Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001;345(5):340–50.
Massey J, Rothwell S, Rusbridge C, Tauro A, Addicott D, Chinoy H, Cooper RG, Ollier WE, Kennedy LJ. Association of an MHC class II haplotype with increased risk of polymyositis in Hungarian Vizsla dogs. PLoS One. 2013;8(2):e56490.
Durward-Akhurst SA, Valberg SJ. Immune-mediated muscle diseases of the horse. Vet Pathol. 2018;55(1):68-75.
McCue ME, Valberg SJ, Miller MB, Wade C, DiMauro S, Akman HO, Mickelson JR. Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 2008;91(5):458–66.
Finno CJ, Aleman M, Higgins RJ, Madigan JE, Bannasch DL. Risk of false positive genetic associations in complex traits with underlying population structure: a case study. Vet J. 2014;202(3):543–9.
Garbe JR, Da Y. Pedigraph: a software tool for the graphing and analysis of large complex pedigrees. User manual version 2.4. Minneapolis, MN: Department of Animal Science, University of Minnesota; 2008.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11. 10 11-33
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM: A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6(2):80–92.
Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, Lu X. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front Genet. 2012;3:35.
Bordbari MH, Penedo MCT, Aleman M, Valberg SJ, Mickelson J, Finno CJ. Deletion of 2.7 kb near HOXD3 in an Arabian horse with occipitoatlantoaxial malformation. Anim Genet. 2017;48(3):287–94.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115.
Finno CJ, Bordbari MH, Valberg SJ, Lee D, Herron J, Hines K, Monsour T, Scott E, Bannasch DL, Mickelson J, et al. Transcriptome profiling of equine vitamin E deficient neuroaxonal dystrophy identifies upregulation of liver X receptor target genes. Free Radic Biol Med. 2016;101:261–71.
Solomon O, Kunik V, Simon A, Kol N, Barel O, Lev A, Amariglio N, Somech R, Rechavi G, Eyal E. G23D: online tool for mapping and visualization of genomic variants on 3D protein structures. BMC Genomics. 2016;17:681.
Eyal E, Najmanovich R, McConkey BJ, Edelman M, Sobolev V. Importance of solvent accessibility and contact surfaces in modeling side-chain conformations in proteins. J Comput Chem. 2004;25(5):712–24.
McConkey BJ, Sobolev V, Edelman M. Discrimination of native protein structures using atom-atom contact scoring. Proc Natl Acad Sci U S A. 2003;100(6):3215–20.
McConkey BJ, Sobolev V, Edelman M. Quantification of protein surfaces, volumes and atom-atom contacts using a constrained Voronoi procedure. Bioinformatics. 2002;18(10):1365–73.
Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33(Web Server issue):W306–10.
Tulloch LK, Perkins JD, Piercy RJ. Multiple immunofluorescence labelling enables simultaneous identification of all mature fibre types in a single equine skeletal muscle cryosection. Equine Vet J. 2011;43(4):500–3.
Mansour TA, Scott EY, Finno CJ, Bellone RR, Mienaltowski MJ, Penedo MC, Ross PJ, Valberg SJ, Murray JD, Brown CT. Tissue resolved, gene structure refined equine transcriptome. BMC Genomics. 2017;18(1):103.
Oldfors A. Hereditary myosin myopathies. Neuromuscul Disord. 2007;17(5):355–67.
Valberg SJ. Muscle anatomy, physiology, and adaptations to exercise and training. In: Hodgson dR, McGowan C, McKeever K, editors. The athletic horse: principles and practice of equine sports medicine. 2nd ed. London: Elsevier; 2014. p. 174–201.
Weiss A, Schiaffino S, Leinwand LA. Comparative sequence analysis of the complete human sarcomeric myosin heavy chain family: implications for functional diversity. J Mol Biol. 1999;290(1):61–75.
Maccatrozzo L, Caliaro F, Toniolo L, Patruno M, Reggiani C, Mascarello F. The sarcomeric myosin heavy chain gene family in the dog: analysis of isoform diversity and comparison with other mammalian species. Genomics. 2007;89(2):224–36.
Traub-Dargatz JL, Schlipf JW Jr, Granstrom DE, Ingram JT, Shelton GD, Getzy DM, Lappin MR, Baker DC. Multifocal myositis associated with Sarcocystis sp in a horse. J Am Vet Med Assoc. 1994;205(11):1574–6.
Aleman M, Shapiro K, Siso S, Williams DC, Rejmanek D, Aguilar B, Conrad PA. Sarcocystis fayeri in skeletal muscle of horses with neuromuscular disease. Neuromuscul Disord. 2016;26(1):85–93.
Barrott MJ, Brooks HW, McGowan CM. Suspected immune-mediated myositis in a pony. Equine Vet Educ. 2004;16(2):58–61.
Martinsson T, Oldfors A, Darin N, Berg K, Tajsharghi H, Kyllerman M, Wahlstrom J. Autosomal dominant myopathy: missense mutation (Glu-706 --> Lys) in the myosin heavy chain IIa gene. Proc Natl Acad Sci U S A. 2000;97(26):14614–9.
Tajsharghi H, Darin N, Rekabdar E, Kyllerman M, Wahlstrom J, Martinsson T, Oldfors A. Mutations and sequence variation in the human myosin heavy chain IIa gene (MYH2). Eur J Hum Genet. 2005;13(5):617–22.
Tajsharghi H, Hilton-Jones D, Raheem O, Saukkonen AM, Oldfors A, Udd B. Human disease caused by loss of fast IIa myosin heavy chain due to recessive MYH2 mutations. Brain. 2010;133(Pt 5):1451–9.
Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013;125(1):3–18.
Colegrave M, Peckham M. Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat Rec (Hoboken). 2014;297(9):1670–80.
Roda RH, Schindler AB, Blackstone C, Mammen AL, Corse AM, Lloyd TE. Laing distal myopathy pathologically resembling inclusion body myositis. Ann Clin Transl Neurol. 2014;1(12):1053–8.
Lamont PJ, Udd B, Mastaglia FL, de Visser M, Hedera P, Voit T, Bridges LR, Fabian V, Rozemuller A, Laing NG. Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psychiatry. 2006;77(2):208–15.
Root-Bernstein R, Fairweather D. Unresolved issues in theories of autoimmune disease using myocarditis as a framework. J Theor Biol. 2015;375:101–23.
Zhang P, Cox CJ, Alvarez KM, Cunningham MW. Cutting edge: cardiac myosin activates innate immune responses through TLRs. J Immunol. 2009;183(1):27–31.
Pahlman LI, Morgelin M, Eckert J, Johansson L, Russell W, Riesbeck K, Soehnlein O, Lindbom L, Norrby-Teglund A, Schumann RR, et al. Streptococcal M protein: a multipotent and powerful inducer of inflammation. J Immunol. 2006;177(2):1221–8.
Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med. 1995;181(3):1123–31.
Quinn-Laquer BK, Kennedy JE, Wei SJ, Beisel KW. Characterization of the allelic differences in the mouse cardiac alpha-myosin heavy chain coding sequence. Genomics. 1992;13(1):176–88.
Wu X, Li ZF, Brooks R, Komives EA, Torpey JW, Engvall E, Gonias SL, Shelton GD. Autoantibodies in canine masticatory muscle myositis recognize a novel myosin binding protein-C family member. J Immunol. 2007;179(7):4939–44.
Acknowledgements
We would like to acknowledge the technical assistance of Marisa Henry and the assistance of Katherine Williamson, DVM.
Funding
Funding for this study was provided by the American Quarter Horse Association. Funding for one of the researchers (CJF) is supported by NIH K01OD015134 and L40 TR001136. Graduate funding for EB is supported by USDA NIFA National Need Fellowship Award #20143842021796.
Availability of data and materials
All sequencing reads for this study have been submitted to NCBI Sequence Read Archive, SUB3102481. All SNP genotyping data is available by request as there is currently no publically available repository for equine SNP genotype results.
Author information
Authors and Affiliations
Contributions
All authors contributed to the experimental design oversight. CJF and SJV developed the research hypothesis, performed all the included experiments, analyzed the results, wrote the manuscript, and provided oversight to all students involved. GG performed the DNA extractions and genotyping. SP performed the in silico predictions of the altered amino acid. ZW, MB, EB, and KLG performed the DNA extractions. KLG performed the muscle fluorescent immunohistochemistry. SP and GG performed the SRA database search. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All blood samples were collected with approval from the Animal Care and Use Committee at the University of Minnesota and University of California, Davis.
Consent for publication
All data is publically available, and consent for use of all data is available.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Across breed genotyping. (XLSX 9 kb)
Additional file 2:
Figure S1. Within the cohort of genome-wide association horses affected with IMM, 23/36 had available pedigree information. All affected horses (red) could be traced back to a common sire within eight generations. Pedigree analysis supported either an autosomal dominant or autosomal recessive mode of inheritance. Circles = females, squares = males. (TIFF 7954 kb)
Additional file 3:
Figure S2. Protein alignment of MYH1 gene and M protein of Streptococcus equi (S. equi) (AHI46575.1) showing similarities in regions of the S. equi alignment and the MYH1 gene. Sequences were aligned using CLUSTALX (version 2). (TIFF 15425 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Finno, C.J., Gianino, G., Perumbakkam, S. et al. A missense mutation in MYH1 is associated with susceptibility to immune-mediated myositis in Quarter Horses. Skeletal Muscle 8, 7 (2018). https://doi.org/10.1186/s13395-018-0155-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13395-018-0155-0