
Abstract
Mass spectrometry based non-target analysis is increasingly adopted in environmental sciences to screen and identify numerous chemicals simultaneously in highly complex samples. However, current data processing software either lack functionality for environmental sciences, solve only part of the workflow, are not openly available and/or are restricted in input data formats. In this paper we present patRoon, a new R based open-source software platform, which provides comprehensive, fully tailored and straightforward non-target analysis workflows. This platform makes the use, evaluation and mixing of well-tested algorithms seamless by harmonizing various common (primarily open) software tools under a consistent interface. In addition, patRoon offers various functionality and strategies to simplify and perform automated processing of complex (environmental) data effectively. patRoon implements several effective optimization strategies to significantly reduce computational times. The ability of patRoon to perform time-efficient and automated non-target data annotation of environmental samples is demonstrated with a simple and reproducible workflow using open-access data of spiked samples from a drinking water treatment plant study. In addition, the ability to easily use, combine and evaluate different algorithms was demonstrated for three commonly used feature finding algorithms. This article, combined with already published works, demonstrate that patRoon helps make comprehensive (environmental) non-target analysis readily accessible to a wider community of researchers.
Similar content being viewed by others

Introduction
Chemical analysis is widely applied in environmental sciences such as earth sciences, biology, ecology and environmental chemistry, to study, e.g. geomorphic processes (chemical) interaction between species or the occurrence, fate and effect of chemicals of emerging concern in the environment. The environmental compartments investigated include air, water, soil, sediment and biota, and exhibit a highly diverse chemical composition and complexity. The number and quantities of chemicals encountered within samples may span several orders of magnitude relative to each other. Therefore, chemical analysis must discern compounds at ultra-trace levels, a requirement that can be largely met with modern analytical instrumentation such as liquid or gas chromatography coupled with mass spectrometry (LC-MS and GC–MS). The high sensitivity and selectivity of these techniques enable accurate identification and quantification of chemicals in complex sample materials.
Traditionally, a ‘target analysis’ approach is performed, where identification and quantitation occur by comparing experimental data with reference standards. The need to pre-select compounds of interest constrains the chemical scope of target analysis, and hampers the analysis of chemicals with (partially) unknown identities such as transformation products and contaminants of emerging concern (CECs). In addition, the need to acquire or synthesize a large number of analytical standards may not be feasible for compounds with a merely suspected presence. Recent technological advancements in chromatography and high resolution MS (HRMS) allows detection and tentative identification of compounds without the prior need of standards [1]. This ‘non-target’ analysis (NTA) approach is increasingly adopted to perform simultaneous screening of up to thousands of chemicals in the environment, such as finding new CECs [1,2,3,4,5,6], identifying chemical transformation (by)products [7,8,9,10,11,12] and identification of toxicants in the environment [13,14,15,16].
Studies employing environmental NTA typically allow the detection of hundreds to thousands of different chemicals [17, 18]. Effectively processing such data requires workflows to automatically extract and prioritize NTA data, perform chemical identification and assist in interpreting the complex resulting datasets. Currently available tools often originate from other research domains such as life sciences and may lack functionality or require extensive optimization before being suitable for environmental analysis. Examples include handling chemicals with low sample-to-sample abundance, recognition of halogenated compounds, usage of data sources with environmentally relevant substances, or temporal and spatial trends [1, 2, 5, 6, 9, 19].
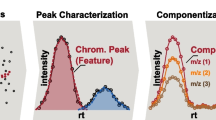
An NTA workflow can be generalized as a four step process (Fig. 1) [1]. Firstly, data from LC or GC-HRMS is either acquired or retrieved retrospectively, and pre-treated for subsequent analysis (Fig. 1a). This pre-treatment may involve conversion to open data formats (e.g. mzML [20] or mzXML [21]) to increase operability with open-source software, re-calibration of mass spectra to improve accuracy and centroiding [22] or other raw data reduction steps to conserve space such as trimming chromatographs or filtering mass scans (e.g. with the functionality from the ProteoWizard suite [23]). Secondly (Fig. 1b), features with unique chromatographic and mass spectral properties (e.g. retention time, accurate mass, signal intensity) are automatically extracted and features considered equivalent across sample analyses are grouped to allow qualitative and (semi-) quantitative comparison further down the workflow. Thirdly (Fig. 1c), the feature dataset quality is refined, for instance, via rule-based filters (e.g. minimum intensity and absence in sample blanks) and grouping of features based on a defined relationship such as adducts or homologous series (e.g. “componentization”). Further prioritization during this step of the workflow is often required for efficient data analysis, for instance, based on chemical properties (e.g. mass defect and isotopic pattern), suspected presence (i.e. “suspect screening”) or intensity trends in time and/or space (e.g. reviewed in [1]). Finally (Fig. 1d), prioritized features are annotated, for instance by assigning chemical formulae or compounds from a chemical database (e.g. PubChem [24] or CompTox [25]) based on the exact mass of the feature. The resulting candidates are ranked by conformity with MS data, such as match with theoretical isotopic pattern and in silico or library MS fragmentation spectra, and study-specific metadata, such as number of scientific references and toxicity data [1, 19].

Generic workflow for environmental non-target analysis
Various open and closed software tools are already available to implement (parts of) the NTA workflow. Commercial software tools such as MetaboScape [26], UNIFI [27], Compound Discoverer [28] and ProGenesis QI [29] provide a familiar and easy to use graphical user interface, may contain instrument specific functionality and optimizations and typically come with support for their installation and usage. However, they are generally not open-source or open-access and are often restricted to proprietary and specific vendor data formats. This leads to difficulties in data sharing, as exact algorithm implementations and parameter choices are hidden, while maintenance, auditing or code extension by other parties is often not possible. Many open-source or open-access tools are available to process mass spectrometry data, such as CFM-ID [30, 31], enviMass [32], enviPick [33], nontarget [34], GenForm [35], MetFrag [36], FOR-IDENT [37], MS-DIAL [38], MS-FINDER [39], MZmine [40], OpenMS [41], ProteoWizard [23], RAMClustR [42], SIRIUS and CSI:FingerID [43,44,45,46,47], XCMS [48], CAMERA [49] and XCMS online [50] (Table 1, further reviewed in [51, 52]). Various open tools are easily interfaced with the R statistical environment [53] (Table 1). Leveraging this open scripting environment inherently allows defining highly flexible and reproducible workflows and increases the accessibility of such workflows to a wider audience as a result of the widespread usage of R in data sciences. While many tools were originally developed to process metabolomics and proteomics data, approaches such as XCMS and MZmine have also been applied to environmental NTA studies [6, 54]. However, as stated above, these tools can lack the specific functionality and optimizations required for effective environmental NTA data processing. While a complete environmental NTA workflow requires several steps from data pre-processing through to automated annotation (see Fig. 1), existing software approaches designed for processing environmental data (e.g. enviMass and nontarget) and most others only implement part of the required functionality, as indicated in Table 1. Furthermore, only few workflow solutions support automated compound annotation. Moreover, available tools often overlap in functionality (Table 1), and are implemented with differing algorithms or employing different data sources. Consequently, tools may generate different results, as has been shown when generating feature data [55,56,57,58,59] or performing structural annotations [19, 60]. Hence, the need to learn, combine, optimize and sometimes develop or adapt various specialized software tools, and perform tedious transformation of datasets currently hinders further adoption of NTA, especially in more routine settings lacking appropriate in-house computational expertise. Thus, before NTA is fully “ready to go” [1], a new platform is necessary that (a) is independent of closed MS vendor input data, (b) incorporates optimizations and functionality necessary for a complete environmental NTA workflow and (c) allows researchers to seamlessly combine and evaluate existing and well-tested algorithms in order to tailor an optimal NTA workflow to the particular study types and methodological characteristics.
Here, we present an R based open-source software platform called patRoon (‘pattern’ in Dutch) providing comprehensive NTA data processing from HRMS data pre-treatment, detection and grouping of features, through to molecular formula and compound annotation. This is achieved by harmonizing various commonly used (and primarily open) tools in a consistent and easy to use interface, which provides access to well-established algorithms without aforementioned limitations when used alone. Complementary and novel functionality is implemented, such as automated chemical annotation, visualization and reporting of results, comparing and combining results from different algorithms, and data reduction and prioritization strategies, which further improve and simplify effective NTA data processing. The architecture of patRoon is designed to be extendable in order to accommodate for rapid developments in the NTA research field.
Implementation
The implementation section starts with an overview of the patRoon workflows. Subsequent sections provide details on novel functionality implemented by patRoon, which relate to data processing, annotation, visualization and reporting. Finally, a detailed description is given of the software architecture. patRoon is then demonstrated in the Results and discussion section. The software tools and databases used for the implementation of patRoon are summarized in Additional file 1.
Workflow in patRoon
patRoon encompasses a comprehensive workflow for HRMS based NTA (Fig. 2). All steps within the workflow are optional and the order of execution is largely customizable. Some steps depend on data from previous steps (blue arrows) or may alter or amend data from each other (red arrows). The workflow commonly starts with pre-treatment of raw HRMS data. Next, feature data is generated, which consists of finding features in each sample, an optional retention time alignment step, and then grouping into “feature groups”. Finding and grouping of features may be preceded by automatic parameter optimization, or followed by suspect screening. The feature data may then finally be used for componentization and/or annotation steps, which involves generation of MS peak lists, as well as formula and compound annotations. At any moment during the workflow, the generated data may be inspected, visualized and treated by, e.g. rule based filtering. These operations are discussed in the next section.

Overview of the NTA patRoon workflow. All steps are optional. Steps that are connected by blue and straight arrows represent a one-way data dependency, whereas steps connected with red curved and dashed arrows represent steps with two-way data interaction
Several commonly used open software tools, such as ProteoWizard [23], OpenMS [41], XCMS [48], MetFrag [36] and SIRIUS [43,44,45,46,47], and closed software tools, such as Bruker DataAnalysis [61] (chosen due to institutional needs), are interfaced to provide a choice between multiple algorithms for each workflow step (Additional file 3: Table S1). Customization of the NTA workflow may be achieved by freely selecting and mixing algorithms from different software tools. For instance, a workflow that uses XCMS to group features allows that these features originate from other algorithms such as OpenMS, a situation that would require tedious data transformation when XCMS is used alone. Furthermore, the interface with tools such as ProteoWizard and DataAnalysis provides support to handle raw input data from all major MS instrument vendors.
To ease parameter selection over the various feature finding and grouping algorithms, an automated feature optimization approach was adopted from the isotopologue parameter optimization (IPO) R package [62], which employs design of experiments to optimize LC–MS data processing parameters [63]. IPO was integrated in patRoon, and its code base was extended to (a) support additional feature finding and grouping algorithms from OpenMS, enviPick and usage of the new XCMS 3 interface, (b) support isotope detection with OpenMS, (c) perform optimization of qualitative parameters and (d) provide a consistent output format for easy inspection and visualization of optimization results.
In patRoon, componentization refers to consolidating different (grouped) features with a prescribed relationship, which is currently either based on (a) highly similar elution profiles (i.e. retention time and peak shape), which are hypothesized to originate from the same chemical compound (based on [42, 49]), (b) participation in the same homologous series (based on [64]) or (c) the intensity profiles across samples (based on [4, 5, 65]). Components obtained by approach (a) typically comprise adducts, isotopologues and in-source fragments, and these are recognized and annotated with algorithms from CAMERA [49] or RAMClustR [42]. Approach (b) uses the nontarget R package [34] to calculate series from aggregated feature data from replicates. The interpretation of homologous series between replicates is assisted by merging series with overlapping features in cases where this will not yield ambiguities to other series. If merging would cause ambiguities, instead links are created that can then be explored interactively and visualized by a network graph generated using the igraph [66] and visNetwork [67] R packages (see Additional file 2: Figure S1).
During the annotation step, molecular formulae and/or chemical compounds are automatically assigned and ranked for all features or feature groups. The required MS peak list input data are extracted from all MS analysis data files and subsequently pre-processed, for instance, by averaging multiple spectra within the elution profile of the feature and by removing mass peaks below user-defined thresholds. All compound databases and ranking mechanisms supported by the underlying algorithms are supported by patRoon and can be fully configured. Afterwards, formula and structural annotation data may be combined to improve candidate ranking and manual interpretation of annotated spectra. More details are outlined in the section “MS peak list retrieval, annotation and candidate ranking”.
Data reduction, comparison and conversion
Various rule-based filters are available for data-cleanup or study specific prioritization of all data obtained through the workflow (see Table 2), and can be inverted to inspect the data that would be removed (i.e. negation). To process feature data, multiple filters are often applied, however, the order may influence the final result. For instance, when features were first removed from blanks by an intensity filter, a subsequent blank filter will not properly remove these features in actual samples. Similarly, a filter may need a re-run after another to ensure complete data clean-up. To reduce the influence of order upon results, filters for feature data are executed by default as follows:
-
1.
An intensity pre-filter, to ensure good quality feature data for subsequent filters;
-
2.
Filters not affected by other filters, such as retention time and m/z range;
-
3.
Minimum replicate abundance, blank presence and ‘regular’ minimum intensity;
-
4.
Repetition of the replicate abundance filter (only if previous filters affected results);
-
5.
Other filters that are possibly influenced by prior steps, such as minimum abundance in feature groups or sample analyses.
Note that the above scheme only applies to those filters requested by the user, and the user can apply another order if desired.
Further data subsetting allows the user to freely select data of interest, for instance, following a (statistical) prioritization approach performed by other tools. Similarly, features that are unique or overlapping in different sample analyses may be isolated, which is a straightforward but common prioritization technique for NTA studies that involve the comparison of different types of samples.
Data from feature groups, components or annotations that are generated with different algorithms (or parameters thereof) can be compared to generate a consensus by only retaining data with (a) minimum overlap, (b) uniqueness or (c) by combining all results (only (c) is supported for data from components). Consensus data are useful to remove outliers, for inspection of algorithmic differences or for obtaining the maximum amount of data generated during the workflow. The consensus for formula and compound annotation data are generated by comparison of Hill-sorted formulae and the skeleton layer (first block) of the InChIKey chemical identifiers [72], respectively. For feature groups, where different algorithms may output deviating retention and/or mass properties, such a direct comparison is impossible. Instead, the dimensionality of feature groups is first reduced by averaging all feature data (i.e. retention times, m/z values and intensities) for each group. The collapsed groups have a similar data format as ‘regular’ features, where the compared objects represent the ‘sample analyses’. Subjection of this data to a feature grouping algorithm supported by patRoon (i.e. from XCMS or OpenMS) then allows straightforward and reliable comparison of feature data from different algorithms, which is finally used to generate the consensus.
Hierarchical clustering is utilized for componentization of features with similar intensity profiles or to group chemically similar candidate structures of an annotated feature. The latter “compound clustering” assists the interpretation of features with large numbers of candidate structures (e.g. hundreds to thousands). This method utilizes chemical fingerprinting and chemical similarity methods from the rcdk package [73] to cluster similar structures, and subsequent visual inspection of the maximum common substructure then allows assessment of common structural properties among candidates (methodology based on [74]). Cluster assignment for both componentization and compound annotation approaches is performed automatically using the dynamicTreeCut R package [75]. However, clusters may be re-assigned manually by the desired amount or tree height.
Several data conversion methods were implemented to allow interoperability with other software tools. All workflow data types are easily converted to commonly used R data types (e.g. data.frame or list), which allows further processing with other R packages. Furthermore, feature data may be converted to and from native XCMS objects (i.e. xcmsSet and XCMSnExp) or exported to comma-separated values (CSV) formats compatible with Bruker ProfileAnalysis or TASQ, or MZmine.
MS peak list retrieval, annotation and candidate ranking
Data for MS and MS/MS peak lists for a feature are collected from spectra recorded within the chromatographic peak and averaged to improve mass accuracies and signal to noise ratios. Next, peak lists for each feature group are assigned by averaging the mass and intensity values from peak lists of the features in the group. Mass spectral averaging can be customized via several data clean-up filters and a choice between different mass clustering approaches, which allow a trade-off between computational speed and clustering accuracy. By default, peak lists for MS/MS data are obtained from spectra that originate from precursor masses within a certain tolerance of the feature mass. This tolerance in mass search range is configurable to accommodate the precursor isolation window applied during data acquisition. In addition, the precursor mass filter can be completely disabled to accommodate data processing from data-independent MS/MS experiments, where all precursor ions are fragmented simultaneously.
The formula annotation process is configurable to allow a tradeoff between accuracy and calculation speeds. Candidates are assigned to each feature group, either directly by using group averaged MS peak list data, or by a consensus from formula assignments to each individual feature in the group. While the latter inherently consumes more time, it allows removal of outlier candidates (e.g. false positives due to features with poor spectra). Candidate ranking is improved by inclusion of MS/MS data in formula calculation (optional for GenForm [35] and DataAnalysis).
Formula calculation with GenForm ranks formula candidates on isotopic match (amongst others), where any other mass peaks will penalize scores. Since MS data of “real-world” samples typically includes many other mass peaks (e.g. adducts, co-eluting features, background ions), patRoon improves the scoring accuracy by automatic isolation of the feature isotopic clusters prior to GenForm execution. A generic isolation algorithm was developed, which makes no assumptions on elemental formula compositions and ion charges, by applying various rules to isolate mass peaks that are likely part of the feature isotopic cluster (see Additional file 2: Figure S2). These rules are configured to accommodate various data and study types by default. Optimization is possible, for instance, to (a) improve studies of natural or anthropogenic compounds by lowering or increasing mass defect tolerances, respectively, (b) constrain cluster size and intensity ranges for low molecular weight compounds or (c) adjust to expected instrumental performance such as mass accuracy. Note that precursor isolation can be performed independently of formula calculation, which may be useful for manual inspection of MS data.
Compound annotation is usually the most time and resource intensive process during the non-target workflow. As such, instead of annotating individual features, compound assignment occurs for the complete feature group. All compound databases supported by the underlying algorithms, such as PubChem [24], ChemSpider [76] or CompTox [25] and other local CSV files, as well as the scoring terms present in these databases, such as in silico and spectral library MS/MS match, references in literature and presence in suspect lists, can be utilized with patRoon. Default scorings supported by the selected algorithm/database or sets thereof are easily selectable to simplify effective compound ranking. Furthermore, formula annotation data may be incorporated in compound ranking, where a ‘formula score’ is calculated for each candidate formula, which is proportional to its ranking in the formula annotation data. Execution of unattended sessions is assisted by automatic restarts after occurrence of timeouts or errors (e.g. due to network connectivity) and automatic logging facilities.
Visualization, reporting and graphical interface
In patRoon, visualization functionality is provided for feature and annotation data (e.g. extracted ion chromatograms (EICs) and annotated spectra), to compare workflow data (i.e. by means of Venn, chord and UpSet [77] diagrams, using the VennDiagram [78], circlize [79] and UpSetR [80] R packages, respectively) and others such as plotting results from automatic feature optimization experiments and hierarchical clustering data. Reports can be generated in a common CSV text format or in a graphical format via export to a portable document file (PDF) or hypertext markup language (HTML) format. The latter are generated with the R Markdown [81, 82] and flexdashboard [83] R packages, and provide an easy to use interface for interactive sorting, searching and browsing reported data. As plotting and reporting functionalities can be performed at any stage during the workflow, the data that is included in the reports is fully configurable.
While patRoon is primarily interfaced through R, several graphical user interface tools are provided to assist the (novice) user. Most importantly, patRoon provides a Shiny [84] based graphical user interface tool that automatically generates a commented template R script from visual user parameter input selection, such as MS data input files, workflow algorithms and other common workflow parameters (Fig. 3a). Secondly, chromatographic data of features may be inspected either by automatic addition of EICs in a Bruker DataAnalysis session or with a Shiny graphical based interface (Fig. 3b).

Graphical user interface tools in patRoon. Tools are provided a to create a new patRoon data analysis project and b to inspect feature chromatography data
Software architecture
patRoon is distributed as an R package. Its source code is primarily written in the R language, with some support code written in C++ and JavaScript. Both Microsoft Windows (hereafter referred to as Windows) and Linux platforms are supported (support for macOS is envisaged in the future). Several external dependencies are required; notable examples are in Additional file 3: Table S1. GenForm is automatically compiled during package installation. For Windows platforms, an installation script is provided to install and configure patRoon and all of its dependencies automatically. Documentation includes a handbook, tutorial and full reference manual [85,86,87,88], which are produced with the bookdown [89, 90], R Markdown and roxygen2 [91] R packages, respectively. Example data is contained in the patRoonData R package [92, 93].
An important design goal was to provide a consistent, generic and easy to use interface that does not require the user to know the implementation and interfacing details of the supported algorithms. Each workflow step is executed by a generator function that takes the desired algorithm and its parameters as input and returns objects from a common set of data formats (see Fig. 4). Names for commonly used parameters supported by multiple algorithms are standardized for consistency and defaults are set where reasonable. Furthermore, the format of input data such as retention time units as well as formula and adduct specifications are harmonized and automatically converted to the format expected by the algorithm. Nearly all parameters from the underlying algorithm can be set by the user, hence, full configurability of the workflow is retained wherever possible. Generic naming schemes are applied to output data, which assist the user in comparing results originating from different algorithms. All exported functions from patRoon verify user input with the checkmate [94] package, which efficiently performs tests such as correctness of value range and type, and prints descriptive messages if input is incorrect.

Interface for the patRoon workflow. The workflow steps are performed by a set of functions that execute the selected algorithm and return the data in a harmonized format by utilizing the ‘S4’ object oriented programming approach of R. These objects all derive from a common base class and may be further sub-classed in algorithm specific classes (as is exemplified for features). Generic functions are defined for all workflow classes to implement further data processing functionality in a predictable and algorithm independent manner (see also Table 3). Further information is provided in the reference manual [85, 86]
A set of generic methods are defined for workflow classes that perform general data inspection, selection, conversion and visualization, irrespective of the algorithm that was used to generate the object (see Table 3). Consequently, the implementation of common function names for multiple output classes allows a predictable and consistent user interface.
Several optimization strategies are employed in patRoon to reduce computational requirements and times. Firstly, external command line (CLI) tools are executed in parallel to reduce overall execution times for repetitive (e.g. per sample analysis or per feature) calculations. Commands are queued (first in, first out) and their execution is handled with the processx package [95]. Secondly, functions employing time intensive algorithms automatically cache their (partial) results in a local SQLite database file, which is accessed via the DBI [96] and RSQLite [97] R packages. Thirdly, performance critical code dealing with OpenMS data files and loading chromatographic data was written in C++ (interfaced with Rcpp [98,99,100]) to significantly reduce times needed to read or write data. Fourthly, the output files from OpenMS tools are loaded in chunks using the pugixml software library [101] to ensure a low memory footprint. Finally, reading, writing and processing (large) internal tabular data is performed with the data.table R package, which is a generally faster and more memory efficient drop-in replacement to the native tabular data format of R (data.frame), especially for large datasets [102].
Interfacing with ProteoWizard [23], OpenMS, GenForm, SIRIUS and MetFrag occurs by wrapper code that automatically executes the CLI tools and perform the data conversions necessary for input and output files. An alternative interface to MetFrag is also provided by employing the metfRag R package [103], however, in our experience this option is currently significantly slower than the CLI and therefore not used by default. For tools that are not readily controllable from R (i.e. ProfileAnalysis, TASQ and MZmine), interfacing occurs via importing or exporting CSV files (only export is supported for MZmine). Finally, the RDCOMClient R package [104] is used to interface with Bruker DataAnalysis via the distributed component object model, which allows automation of DataAnalysis functionality from R that otherwise would only be available via its integrated visual basic scripting environment.
A continuous integration pipeline performs automated tests during development and delivers files to simplify installation of patRoon and all its dependencies (Additional file 2: Figure S3). More than 900 unit tests are performed (> 80% code coverage) with the testthat and vdiffr R packages [105, 106]. After successful test completion, the final step involves building (a) Windows binary R packages of patRoon and its dependencies and (b) Linux Docker images with a complete working environment of patRoon and the RStudio integrated development environment [107] (based on [108]), which both facilitate installation of patRoon with tested and compatible dependencies.
Results and discussion
This section starts with benchmarks of important optimization strategies implemented in patRoon, and concludes with demonstrations on how patRoon can implement a common NTA workflow and the algorithm consensus functionality. Since the implementation of individual workflow steps, such as obtaining feature data and annotations, heavily rely on well-established algorithms that have been evaluated elsewhere, further evaluations have not been performed here. Furthermore, an objective comparison of patRoon with other NTA workflows is currently being performed as part of a collaborative trial organized by the NORMAN Network [109]. Recent applications of complete environmental NTA studies performed with patRoon are already described in several publications [7, 12, 14, 71, 110].
Benchmark and demonstration data
The data used to benchmark and demonstrate patRoon were obtained with an LC-HRMS analysis of influent and effluent samples from two drinking water treatment pilot installations and a procedural blank. The pilot installations were fed by surface water (Meuse and IJsselmeer, the Netherlands) that were subjected to various pre-treatment steps (e.g. rapid and slow sand filtration, drum sieves and dune filtration). Effluent samples investigated in this study were produced after advanced oxidation utilizing O3 and H2O2 or ultrafiltration and reverse osmosis. Sample blanks were obtained from tap water. All samples were filtered in triplicate by 0.2 µm regenerated-cellulose filters. Influent samples were spiked with a set of 18 common environmental contaminants (see Table 5). The analyses were performed using an LC-HRMS Orbitrap Fusion system (ThermoFisher Scientific, Bremen, Germany) operating with positive electrospray ionization. Further details of the pilot installations and analytical conditions are described in [11]. The raw data files can be obtained from [111].
Parallelization benchmarks
Several benchmarks were performed to test the multiprocessing functionality of patRoon. Tests were performed on a personal computer equipped with an Intel® Core™ i7-8700 K CPU (6 cores, 12 threads), 32 gigabyte RAM, SATA SSD storage and the Windows 10 Enterprise operating system. Benchmarks were performed in triplicate using the microbenchmark R package [112]. Standard deviations were below ten percent (see Fig. 5a). Benchmarking was performed on msConvert, FeatureFinderMetabo, GenForm, SIRIUS and MetFrag. The multiprocessing functionality was compared to native multithreading for the tools that supported this (FeatureFinderMetabo, SIRIUS and MetFrag). In addition, the performance of batch calculations with multiprocessing was compared with native batch calculation modes of tools where possible (msConvert and SIRIUS). Parallelization methods were tested with 1-12 parallel processes or threads (i.e. up to full utilization of both CPU threads of each core). Input conditions were chosen to simulate “simple” and “complex” workflows, where the latter resulted in more demanding calculations with ~ 2–10 × longer mean execution times (Table 4). The caching functionality of patRoon was disabled, where appropriate, to obtain representative and reproducible test results. Prior to benchmarking, candidate chemical compounds from PubChem for MetFrag tests were cached in a local database to exclude influences from network connectivity. Similarly, general spectral data required to post-process FeatureFinderMetabo results were cached, as this is usually loaded once during a workflow, even with varying input parameters. The input features for GenForm tests that resulted in very long individual run times (i.e. > 30 s) were removed to avoid excessive benchmark runtimes. Generating feature and MS peaklist input data for annotation related tests was performed with patRoon using algorithms from OpenMS and mzR [113], respectively. Pre-treatment of feature data consisted of removal of features with low intensity and lacking MS/MS data. The number of features for SIRIUS (except tests with native batch mode) and MetFrag benchmarks were further reduced by application of blank, replicate and intensity filters to avoid long total runtimes due to their relatively high individual run times. Finally, the feature dataset was split in low (0-500) and high (500-1000) m/z portions, which were purposed for execution of “simple” and “complex” experiments, respectively. For more details of the workflow and input parameters see the R script code in Additional file 4. The software tools used for benchmarking are summarized in Additional file 1.

Parallelization benchmark results. a Benchmark results for commonly used CLI tools applied in patRoon workflows under varying parallelization conditions. The tested tools were msConvert, FeatureFinderMetabo (FFM), GenForm, SIRIUS and MetFrag. Tests were performed with “simple” (left) and “complex” (right) input conditions (Table 4) to simulate varying workflow complexity. Parallelization was performed with the multiprocessing functionality of patRoon (top) or by using native multithreading (bottom, for tools that supported this). Graphs represent number of processes or threads versus relative execution time (normalized to sequential results). The dotted grey lines represent the theoretical trend if maximum parallelization performance is achieved. The dashed blue line represents the number of physical cores that became the default selection in patRoon based on these results. b Comparison of execution times (normalized to the execution times of the unoptimized results) when tools are executed without optimizations (green), executed with native multithreading (FeatureFinderMetabo, SIRIUS and MetFrag) or batch mode (GenForm) (orange), executed with multiprocessing (purple) or a combination of the latter two (pink), using simple (left) and complex (right) input conditions. c Overview of execution times for a complete patRoon workflow executed under optimized versus unoptimized conditions. All results for msConvert and SIRIUS were obtained without enabling their native batch mode
When multiprocessing was used all tests (except GenFormsimple, discussed below) showed a clear downward trend in execution times (down to ~ 200–500%), and optimum conditions were generally reached when the number of parallel processes equaled the number of physical cores (six, see Fig. 5a). When algorithms are fully parallelized, execution times are expected to follow an inverse relationship with the number of parallel process (i.e. 1/n) and this was observed most closely with msConvert, whereas execution times for other tools show a less steep reduction. Furthermore, utilizing multiple threads per core (i.e. hyperthreading) did not reduce execution times further and even slowed down in some cases (e.g. MetFragcomplex). These deviations in scalability were not investigated in detail. Since they were more noticeable under complex conditions, it is expected that this may be caused by (a) more involved post-processing results after each execution, which is currently not parallelized, and (b) increased memory usage, which may raise the overhead of context switches performed by the operating system. Nevertheless, the experiments performed here clearly show that the multiprocessing functionality of patRoon can significantly reduce execution times of various steps in an NTA workflow.
An exception, however, was the test performed with GenFormsimple, which exhibited no significant change in execution times with multiprocessing (Fig. 5a). Due to the particularly small mean run times (0.2 s) of this test, it was hypothesized that the overhead of instantiating a new process from R (inherently not parallelized) dominated the overall run times. To mitigate this, a ‘batch mode’ was implemented, where such process initiation occurs from a command shell sub-process instead. Here, multiple commands are executed by the sub-process in series, and the desired degree of parallelization is then achieved by launching several of these sub-processes and evenly dividing commands amongst them. The maximum size of each series (or “batch size”) is configurable, and represents a balance between reduction of process initiation overhead and potential loss of effectively load balancing of, for instance, commands with highly deviating execution times. Next, various batch sizes were tested for GenForm, both with and without multiprocessing parallelization (Additional file 2: Figure S4). For GenFormsimple, execution times clearly decreased with increasing batch sizes, however, no further reduction was observed with parallelism. In contrast, serial execution of GenFormcomplex was not affected by varying batch size, whereas added parallelism reduced execution times for small batch sizes (≤ 8), but significantly increased such times for larger sizes. The results demonstrate that the typical short lived GenForm executions clearly benefit from batch mode. In addition, it is expected that by further increasing the batch size for GenFormsimple, overall lifetimes of batch sub-processes may increase sufficiently to allow better utilization of parallelization. However, since GenFormcomplex results for larger batch sizes clearly show possible performance degradation for more complex calculations (e.g. due to suboptimal load balancing), eight was considered as a ‘safe’ default which improves overall performance for both simple and complex calculation scenarios (Fig. 5b).
Utilizing native multithreading for FeatureFinderMetabo, SIRIUS (without native batch mode) and MetFrag yields only relatively small reductions in their execution times (Fig. 5b). Under optimum conditions (6-8 threads), the most significant drop was observed for SIRIUScomplex (~ 40%), followed by FeatureFinderMetabosimple, FeatureFinderMetabocomplex and MetFragcomplex-C (~ 20%). These results suggest that native multithreading only yields partial parallelization, which primarily occurs with complex input conditions. Note that SIRIUS supports different linear programming solvers (Gurobi [114], CPLEX [115] and the default GLPK [116]), which may influence overall performance and parallelization [117]. Nevertheless, a comparison between these solvers did not reveal significant changes with our experimental conditions (Additional file 2: Figure S5). Combining the multiprocessing functionality with native multithreading under optimum conditions (i.e. 6 parallel processes/threads) only reduces execution times for SIRIUScomplex (Fig. 5b). As such, both performance improvements and scalability of the multiprocessing implementation of patRoon appear highly effective at this stage.
The native batch modes of msConvert and SIRIUS allow calculations from multiple inputs within a single execution. This reduces the total number of tool executions, which may (1) lower the accumulated overhead associated with starting and finishing tool executions and (2) hamper effective parallelization from multiprocessing, especially if executions are less than the available CPU cores. The combination of multiprocessing (optimum conditions) and native batch mode was benchmarked with increasing number of inputs per tool execution (i.e. the native batch size; Additional file 2: Figure S6). For msConvert, execution times were largely unaffected by the input batch size if multiprocessing was disabled, which indicates a low execution overhead. Lowest execution times were observed when multiprocessing was enabled with small batch sizes (≤ 25% of the total inputs), which indicates a lack of native parallelization support. In contrast, SIRIUS showed significantly lower overall execution times with increasing batch sizes (up to ~ 7000% and ~ 320% for SIRIUSsimple and SIRIUScomplex, respectively), while enabling multiprocessing did not reduce execution times for batch sizes > 1. These results show that (1) SIRIUS has a relative large execution overhead, which impairs multiprocessing performance gains, and (2) supports effective native parallelized batch execution. Thus, SIRIUS performs most optimal if all calculations are performed within a single execution. Similar to previous SIRIUS benchmarks, no significant differences were found across different linear solvers (Additional file 2: Figure S7). The results demonstrate that multiprocessing may improve efficiency for batch calculations with tools with low execution overhead and/or lack of native parallelization. Nonetheless, the dramatic improvement in SIRIUS calculation times when using the native batch mode indicates that software authors should generally consider implementing native threaded batch mode functionality if large batch calculations are an expected use case.
Finally, the implemented optimization strategies were tested for a complete patRoon NTA workflow consisting of typical data processing steps and using all previously tested tools. The chosen input conditions roughly fell in between the aforementioned “simple” and “complex” conditions (see code in Additional file 4). Note that optimization strategies were unavailable for some steps (e.g. grouping of features and collection of MS peak lists), and native batch mode was not used in order to demonstrate the usefulness of multiprocessing for tools that do not support this (e.g. other tools than msConvert and SIRIUS and those potentially available in future versions of patRoon). Regardless, the benchmarks revealed a reduction in total run times of ~ 50% (from ~ 200 to ~ 100 min; Fig. 5c). Since execution times of each step may vary significantly, the inclusion of different combinations of steps may significantly influence overall execution times.
The use of multiprocessing for all tools (except SIRIUS), the implemented batch mode strategies for GenForm and the use of the native batch mode supported by SIRIUS were set as default in patRoon with the determined optimal parameters from the benchmarks results. However, the user can still freely configure all these options to potentially apply further optimizations or otherwise (partially) disable parallelization to conserve system resources acquired by patRoon.
As a final note, it is important to realize that a comparison of these benchmarks with standalone execution of investigated tools is difficult, since reported execution times here are also influenced by (a) preparing input and processing output and (b) other overhead such as process creation from R. However, (b) is probably of small importance, as was revealed by the highly scalable results of msConvert where the need to perform (a) is effectively absent. Furthermore, the overhead from (a) is largely unavoidable, and it is expected that handling of input and output data is still commonly performed from a data analysis environment such as R. Nonetheless, the various optimization strategies employed by patRoon minimize such overhead, and it was shown that the parallelization functionality often provide a clear advantage in efficiency when using typical CLI tools in an R based NTA workflow, especially considering the now widespread availability of computing systems with increasing numbers of cores.
Demonstration: suspect screening
The previous section investigated several parallelization strategies implemented in patRoon for efficient data processing. A common method in environmental NTA studies to increase data processing efficiency and reducing the data complexity is by merely screening for chemicals of interest. This section demonstrates such a suspect screening workflow with patRoon, consisting of (a) raw data pre-treatment, (b) extracting, grouping and suspect screening of feature data, and finally (c) annotating features to confirm their identity. During the workflow several rule-based filters are applied to improve data quality. The ‘suspects’ in this demonstration are, in fact, a set of compounds spiked to influent samples (Table 5), therefore, this brief NTA primarily serves for demonstration purposes. After completion of the suspect screening workflow, several methods are demonstrated to inspect the resulting data.
Suspect screening: workflow
The code described here can easily be generated with the newProject() function, which automatically generates a ready-to-use R script based on user input (section “Visualization, reporting and graphical interface”).
First, the patRoon R package is loaded and a data.frame is generated with the file information of the sample analyses and their replicate and blank assignments. Next, this information is used to centroid and convert the raw analyses files to the open mzML file format, a necessary step for further processing.

The next step involves finding features and grouping them across samples. This example uses the OpenMS algorithms and sets several algorithm specific parameters that were manually optimized for the employed analytical instrumentation to optimize the workflow output. Other algorithms (e.g. enviPick, XCMS) are easily selected by changing the algorithm function parameter.

Several rule-based filters are then applied for general data clean-up, followed by the removal of sample blanks from the feature dataset.

Next, features are screened with a given suspect list, which is a CSV file read into a data.frame containing the name, SMILES and (optionally) retention time for each suspect (see Additional file 5). While the list in this demonstration is rather small (18 compounds, see Table 5), larger lists containing several thousands of compounds such as those available on the NORMAN network Suspect List Exchange [118] can also be used. The screening results are returned in a data.frame, where each row is a hit (a suspect may occur multiple times) containing the linked feature group identifier and other information such as detected m/z and retention time (deviations). Finally, this table is used to transform the original feature groups object (fGroups) by removing any unassigned features and tagging remainders by their suspect name.

In the final step of this workflow annotation is performed, which consists of (a) generation of MS peak list data, (b) general clean-up to only retain significant MS/MS mass peaks, automatic annotation of (c) formulae and (d) chemical compounds, and (e) combining both annotation data to improve ranking of candidate compounds. As with previous workflow steps, the desired algorithms (mzR, GenForm and MetFrag in this example) are set using the algorithm function parameter. Similarly, the compound database used by MetFrag (here CompTox via a local CSV file obtained from [119]) can easily be changed to other databases such as PubChem, ChemSpider or another local file.

Suspect screening: data inspection
All data generated during the workflow (e.g. features, peak lists, annotations) can be inspected by overloads of common R methods.

Furthermore, all workflow data can easily be subset with, e.g. the R subset operator (“[“), for instance, to perform a (hypothetical) prioritization of features that are most intense in the effluent samples.

Visualization of data generated during the workflow, such as an overview of features, chromatograms, annotated MS spectra and uniqueness and overlap of features, can be performed by various plotting functions (see Fig. 6).

Common visualization functionality of patRoon applied to the demonstrated workflow. From left to right: an m/z vs retention time plot of all feature groups uniquely present in the samples, an EIC for the tramadol suspect, a compound annotated spectrum for the benzotriazole suspect and comparison of feature presence between sample groups using UpSet [77], Venn (influent/effluent A) and chord diagrams

The final step in a patRoon NTA workflow involves automatic generation of comprehensive reports of various formats which allow (interactive) exploration of all data (see Additional file 2: Figure S8).

Suspect screening: results
A summary of data generated during the NTA workflow demonstrated here is shown in Tables 5 and 6. The complete workflow finished in approximately 8 min (employing a laptop with an Intel® Core™ I7-8550U CPU, 16 gigabyte RAM, NVME SSD and the Windows 10 Pro operating system). While nearly 60,000 features were grouped into nearly 20,000 feature groups, the majority (97%, 678 remaining) were filtered out during the various pre-treatment filter steps. Regardless, most suspects were found (17/18 attributed to 19/20 individual chromatographic peaks, Table 5), and the missing suspect (aniline) could be detected when lowering the intensity threshold of the filter() function used to post-filter feature groups in the workflow. The majority of suspects (17) were annotated with the correct chemical compound as first candidate (Table 6), the two n-methylbenzotriazole isomer suspects were ranked as second or fourth. Results for formulae assignments were similar, with the exception of dimethomorph, where the formula was ranked in only the top 25 (the candidate chemical compound was ranked first, however).
While this demonstration conveys a relative simple NTA with ‘known suspects’, the results show that patRoon is (a) time-efficient on conventional computer hardware, (b) allows a straightforward approach to perform a complete and tailored NTA workflow, (c) provides powerful general data clean-up functionality to prioritize data and (d) performs effective automated annotation of detected features.
Demonstration: algorithm consensus
This section briefly demonstrates how the consensus functionality of patRoon can be used to compare and combine output from the supported algorithms from OpenMS, XCMS and enviPick. The MS data from the suspect screening demonstration above was also used here. The full processing script can be found as Additional file 6.
To obtain the feature data the findFeatures(), groupFeatures() and filter() functions were used as was demonstrated previously (see Additional file 6). The first step is to create a comparison from this data, which is then used to create a consensus (discussed in section “Data reduction, comparison and conversion”). The consensus can be formed from combining all data or from overlapping or unique data, which can then be inspected with the aforementioned data inspection functionality.

A summary of the results is shown in Table 7 and Additional file 2: Figure S9. While the number of features prior to grouping and filtering varied significantly between algorithms (~ 10 000 to ~ 60 000), they were roughly equal after pre-treatment: 678 (OpenMS), 801 (XCMS) and 836 (enviPick). Combining these resulted in 1243 grouped features, of which 541 (44%) were unique to one algorithm, 332 (27%) were shared amongst two algorithms and 370 (30%) fully overlapped. Application of the suspect screening workflow from the previous section revealed that the same 17 out of 18 suspects were present in all the algorithm specific, combined and overlapping feature datasets. Still, the results from this demonstration indicates that each algorithm generates unique results. Dedicated efforts such as ENTACT [120,121,122] will help to unravel the importance of unique and overlapping algorithm results, however, such studies are out of the scope of this article. Regardless, this demonstration showed how patRoon provides researchers the tools needed to easily use and combine workflow data from different algorithms to perform such an evaluation for their use cases.
Conclusions
This paper presents patRoon, a fully open source platform that provides a comprehensive MS based NTA data processing workflow developed in the R environment. Major workflow functionality is implemented through the usage of existing and well-tested software tools, connecting primarily open and a few closed approaches. The workflows are easily setup for common use cases, while full customization and mixing of algorithms allows for execution of completely tailored workflows. In addition, extensive functionality related to data processing, annotation, visualization, reporting and others was implemented in patRoon to provide an important toolbox for effectively handling complex NTA studies. The easy and predictable interface of patRoon lowers the computational expertise required of users, making it available for a broad audience. It was shown that the optimization strategies implemented reduced the computational times. Furthermore, it was demonstrated how patRoon can be used to perform a straightforward and effective suspect screening workflow and how it can easily generate, compare and combine results from different NTA workflow algorithms.
patRoon has been under development for several years and has already been applied in a variety of studies, such as the characterization of organic matter [71], elucidation of transformation products of biocides [7, 12], assessment of removal of polar organics by reversed-osmosis drinking water treatment [14] and the investigation of endocrine disrupting chemicals in human breast milk [110]. patRoon will be maintained to stay compatible with its various dependencies and further development is planned. This includes extension of integrated workflow algorithms for new and less commonly used ones and the implementation of additional componentization strategies to help prioritizing data. Addition of new workflow functionality is foreseen, such as usage of ion-mobility spectrometry data to assist annotation, automated screening of transformation products (e.g. utilizing tools such as BioTransformer [123]), prediction of feature quantities for prioritization purposes (recently reviewed in [124]) and automated chemical classification (e.g. through ClassyFire [125]). Finally, interfacing with other R based mass spectrometry software such as those provided by the “R for Mass Spectrometry” initiative [126] is planned to further improve the interoperability of patRoon. The use in real-world studies, feedback from users and developments within the non-target analysis community, are all critical in determining future directions and improvements of patRoon. We envisage that the open availability, straightforward usage, vendor independence and comprehensive functionality will be useful to the community and result in a broad adoption of patRoon.
Availability and requirements
Project name: patRoon.
Project home page: https://github.com/rickhelmus/patRoon.
Operating system(s): Platform independent (tested on Microsoft Windows and Linux).
Programming language(s): R, C ++, JavaScript.
Other requirements: Depending on utilized algorithms (see installation instructions in [85, 88]).
License: GNU GPL version 3.
Any restrictions to use by non-academics: none.
Definitions
Features: data points assigned with unique chromatographic and mass spectral information (e.g. retention time, peak area and accurate m/z), which potentially described a compound in a sample analysis.
Feature group: A group of features considered equivalent across sample analyses.
MS peak list: tabular data (m/z and intensity) for MS or MS/MS peaks attributed to a feature and used as input data for annotation purposes.
Formula/Compound: a chemical formula or compound candidate revealed during feature annotation.
Component: A collection of feature groups that are somehow linked, such as MS adducts, homologous series or highly similar intensity trends.
Availability of data and materials
The source code of patRoon and online versions of its manuals are available for download from https://github.com/rickhelmus/patRoon and archived in [85, 127]. The raw data used for benchmarking and demonstration purposes in this manuscript is archived in [111]. The scripts used to perform benchmarking and the input suspect list for demonstration purposes are provided as Additional file 4 and 5, respectively.
Abbreviations
- CECs:
-
Contaminants of emerging concern
- CLI:
-
Command-line interface
- CSV:
-
Comma-separated value
- DBI:
-
The database interface
- EIC:
-
Extracted ion chromatogram
- GC:
-
Gas chromatography
- GC-MS:
-
GC coupled to mass spectrometry
- HTML:
-
Hypertext markup language
- HRMS:
-
High resolution mass spectrometry
- IPO:
-
Isotopologue parameter optimization
- LC:
-
Liquid chromatography
- LC–MS:
-
LC coupled to mass spectrometry
- MS/MS:
-
Tandem mass spectrometry
- NTA:
-
Non-target analysis
- PDF:
-
Portable document format
- XCMS:
-
Various forms (X) of chromatography mass spectrometry (R package MS data processing)
References
Hollender J, Schymanski EL, Singer HP, Ferguson PL (2017) Nontarget screening with high resolution mass spectrometry in the environment: ready to go? Environ Sci Technol 51:11505–11512. https://doi.org/10.1021/acs.est.7b02184
Chiaia-Hernandez AC, Schymanski EL, Kumar P, Singer HP, Hollender J (2014) Suspect and nontarget screening approaches to identify organic contaminant records in lake sediments. Anal Bioanal Chem 406:7323–7335. https://doi.org/10.1007/s00216-014-8166-0
Sjerps RMA, Vughs D, van Leerdam JA, ter Laak TL, van Wezel AP (2016) Data-driven prioritization of chemicals for various water types using suspect screening LC-HRMS. Water Res 93:254–264. https://doi.org/10.1016/j.watres.2016.02.034
Chiaia-Hernández AC, Günthardt BF, Frey MP, Hollender J (2017) Unravelling contaminants in the anthropocene using statistical analysis of liquid chromatography–high-resolution mass spectrometry nontarget screening data recorded in lake sediments. Environ Sci Technol 51:12547–12556. https://doi.org/10.1021/acs.est.7b03357
Albergamo V, Schollée JE, Schymanski EL, Helmus R, Timmer H, Hollender J, de Voogt P (2019) Nontarget screening reveals time trends of polar micropollutants in a riverbank filtration system. Environ Sci Technol 53:7584–7594. https://doi.org/10.1021/acs.est.9b01750
Hernández F, Bakker J, Bijlsma L, de Boer J, Botero-Coy AM, Bruinen de Bruin Y, Fischer S, Hollender J, Kasprzyk-Hordern B, Lamoree M, López FJ, ter Laak TL, van Leerdam JA, Sancho JV, Schymanski EL, de Voogt P, Hogendoorn EA (2019) The role of analytical chemistry in exposure science: focus on the aquatic environment. Chemosphere 222:564–583. https://doi.org/10.1016/j.chemosphere.2019.01.118
Wagner TV, Helmus R, Quiton Tapia S, Rijnaarts HHM, de Voogt P, Langenhoff AAM, Parsons JR (2020) Non-target screening reveals the mechanisms responsible for the antagonistic inhibiting effect of the biocides DBNPA and glutaraldehyde on benzoic acid biodegradation. J Hazard Mater 386:121661. https://doi.org/10.1016/j.jhazmat.2019.121661
Kolkman A, Martijn BJ, Vughs D, Baken KA, van Wezel AP (2015) Tracing nitrogenous disinfection byproducts after medium pressure UV water treatment by stable isotope labeling and high resolution mass spectrometry. Environ Sci Technol 49:4458–4465. https://doi.org/10.1021/es506063h
Schollée JE, Schymanski EL, Avak SE, Loos M, Hollender J (2015) Prioritizing unknown transformation products from biologically-treated wastewater using high-resolution mass spectrometry, multivariate statistics, and metabolic logic. Anal Chem 87:12121–12129. https://doi.org/10.1021/acs.analchem.5b02905
Brunner AM, Vughs D, Siegers W, Bertelkamp C, Hofman-Caris R, Kolkman A, ter Laak T (2019) Monitoring transformation product formation in the drinking water treatments rapid sand filtration and ozonation. Chemosphere 214:801–811. https://doi.org/10.1016/j.chemosphere.2018.09.140
Brunner AM, Bertelkamp C, Dingemans MML, Kolkman A, Wols B, Harmsen D, Siegers W, Martijn BJ, Oorthuizen WA, ter Laak TL (2020) Integration of target analyses, non-target screening and effect-based monitoring to assess OMP related water quality changes in drinking water treatment. Sci Total Environ 705:135779. https://doi.org/10.1016/j.scitotenv.2019.135779
Wagner TV, Helmus R, Becker E, Rijnaarts HHM, de Voogt P, Langenhoff AAM, Parsons JR (2020) Impact of transformation, photodegradation and interaction with glutaraldehyde on the acute toxicity of the biocide DBNPA in cooling tower water. Environ Sci 6:1058–1068. https://doi.org/10.1039/C9EW01018A
Jonker W, Lamoree MH, Houtman CJ, Hamers T, Somsen GW, Kool J (2015) Rapid activity-directed screening of estrogens by parallel coupling of liquid chromatography with a functional gene reporter assay and mass spectrometry. J Chromatogr A 1406:165–174. https://doi.org/10.1016/j.chroma.2015.06.012
Albergamo V, Escher BI, Schymanski EL, Helmus R, Dingemans MML, Cornelissen ER, Kraak MHS, Hollender J, de Voogt P (2019) Evaluation of reverse osmosis drinking water treatment of riverbank filtrate using bioanalytical tools and non-target screening. Environ Sci 6:103–116. https://doi.org/10.1039/C9EW00741E
Brunner AM, Dingemans MML, Baken KA, van Wezel AP (2019) Prioritizing anthropogenic chemicals in drinking water and sources through combined use of mass spectrometry and ToxCast toxicity data. J Hazard Mater 364:332–338. https://doi.org/10.1016/j.jhazmat.2018.10.044
Zwart N, Jonker W, ten Broek R, de Boer J, Somsen G, Kool J, Hamers T, Houtman CJ, Lamoree MH (2020) Identification of mutagenic and endocrine disrupting compounds in surface water and wastewater treatment plant effluents using high-resolution effect-directed analysis. Water Res 168:115204. https://doi.org/10.1016/j.watres.2019.115204
Schymanski EL, Singer HP, Slobodnik J, Ipolyi IM, Oswald P, Krauss M, Schulze T, Haglund P, Letzel T, Grosse S, Thomaidis NS, Bletsou A, Zwiener C, Ibáñez M, Portolés T, de Boer R, Reid MJ, Onghena M, Kunkel U, Schulz W, Guillon A, Noyon N, Leroy G, Bados P, Bogialli S, Stipaničev D, Rostkowski P, Hollender J (2015) Non-target screening with high-resolution mass spectrometry: critical review using a collaborative trial on water analysis. Anal Bioanal Chem 407:6237–6255. https://doi.org/10.1007/s00216-015-8681-7
Peisl BYL, Schymanski EL, Wilmes P (2018) Dark matter in host-microbiome metabolomics: tackling the unknowns—a review. Anal Chim Acta 1037:13–27. https://doi.org/10.1016/j.aca.2017.12.034
Blaženović I, Kind T, Torbašinović H, Obrenović S, Mehta SS, Tsugawa H, Wermuth T, Schauer N, Jahn M, Biedendieck R, Jahn D, Fiehn O (2017) Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: database boosting is needed to achieve 93% accuracy. J Cheminf 9:32. https://doi.org/10.1186/s13321-017-0219-x
Martens L, Chambers M, Sturm M, Kessner D, Levander F, Shofstahl J, Tang WH, Römpp A, Neumann S, Pizarro AD, Montecchi-Palazzi L, Tasman N, Coleman M, Reisinger F, Souda P, Hermjakob H, Binz P-A, Deutsch EW (2011) mzML—a community standard for mass spectrometry data. Mol Cell Proteomics. https://doi.org/10.1074/mcp.R110.000133
Pedrioli PGA, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, Aebersold R (2004) A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol 22:1459–1466. https://doi.org/10.1038/nbt1031
Urban J, Afseth NK, Štys D (2014) Fundamental definitions and confusions in mass spectrometry about mass assignment, centroiding and resolution. TrAC 53:126–136. https://doi.org/10.1016/j.trac.2013.07.010
Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak M-Y, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30:918–920. https://doi.org/10.1038/nbt.2377
PubChem National Center for Biotechnology Information PubChem Database. https://pubchem.ncbi.nlm.nih.gov/. Accessed 6 Feb 2020
Williams AJ, Grulke CM, Edwards J, McEachran AD, Mansouri K, Baker NC, Patlewicz G, Shah I, Wambaugh JF, Judson RS, Richard AM (2017) The CompTox chemistry dashboard: a community data resource for environmental chemistry. Journal of Cheminformatics 9:61. https://doi.org/10.1186/s13321-017-0247-6
Bruker MetaboScape. https://www.bruker.com/products/mass-spectrometry-and-separations/ms-software/metaboscape.html. Accessed 6 Feb 2020
Waters UNIFI Scientific Information System. https://www.waters.com/waters/en_US/UNIFI-Scientific-Information-System/nav.htm?cid=134801359&locale=en_US. Accessed 6 Feb 2020
Thermo Scientific Compound Discoverer Software. https://www.thermofisher.com/uk/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/compound-discoverer-software.html. Accessed 6 Feb 2020
Progenesis QI. http://www.nonlinear.com/progenesis/qi/. Accessed 6 Feb 2020
Allen F, Pon A, Wilson M, Greiner R, Wishart D (2014) CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res 42:W94–W99. https://doi.org/10.1093/nar/gku436
Allen F, Greiner R, Wishart D (2015) Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 11:98–110. https://doi.org/10.1007/s11306-014-0676-4
Loos M (2018) enviMass version 3.5 LC-HRMS trend detection workflow—R package. https://doi.org/10.5281/zenodo.1213098
Loos M (2016) enviPick: Peak Picking for High Resolution Mass Spectrometry Data. https://CRAN.R-project.org/package=enviPick. Accessed 2 Oct 2018
Loos M (2016) nontarget: Detecting Isotope, Adduct and Homologue Relations in LC–MS Data. https://CRAN.R-project.org/package=nontarget
Meringer M, Reinker S, Zhang J, Muller A MS/MS data improves automated determination of molecular formulas by mass spectrometry. MATCH Commun Math Comput Chem 259–290
Ruttkies C, Schymanski EL, Wolf S, Hollender J, Neumann S (2016) MetFrag relaunched: incorporating strategies beyond in silico fragmentation. J Cheminf 8:3. https://doi.org/10.1186/s13321-016-0115-9
FOR-IDENT LC. https://water.for-ident.org/#!home. Accessed 7 Feb 2020
Tsugawa H, Cajka T, Kind T, Ma Y, Higgins B, Ikeda K, Kanazawa M, VanderGheynst J, Fiehn O, Arita M (2015) MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat Methods 12:523–526. https://doi.org/10.1038/nmeth.3393
Tsugawa H, Kind T, Nakabayashi R, Yukihira D, Tanaka W, Cajka T, Saito K, Fiehn O, Arita M (2016) Hydrogen rearrangement rules: computational ms/ms fragmentation and structure elucidation using MS-FINDER Software. Anal Chem 88:7946–7958. https://doi.org/10.1021/acs.analchem.6b00770
Pluskal T, Castillo S, Villar-Briones A, Orešič M (2010) MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11:395. https://doi.org/10.1186/1471-2105-11-395
Röst HL, Sachsenberg T, Aiche S, Bielow C, Weisser H, Aicheler F, Andreotti S, Ehrlich H-C, Gutenbrunner P, Kenar E, Liang X, Nahnsen S, Nilse L, Pfeuffer J, Rosenberger G, Rurik M, Schmitt U, Veit J, Walzer M, Wojnar D, Wolski WE, Schilling O, Choudhary JS, Malmström L, Aebersold R, Reinert K, Kohlbacher O (2016) OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat Methods 13:741–748. https://doi.org/10.1038/nmeth.3959
Broeckling CD, Afsar FA, Neumann S, Ben-Hur A, Prenni JE (2014) RAMClust: a novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal Chem 86:6812–6817. https://doi.org/10.1021/ac501530d
Böcker S, Letzel MC, Lipták Z, Pervukhin A (2009) SIRIUS: decomposing isotope patterns for metabolite identification. Bioinformatics 25:218–224. https://doi.org/10.1093/bioinformatics/btn603
Dührkop K, Shen H, Meusel M, Rousu J, Böcker S (2015) Searching molecular structure databases with tandem mass spectra using CSI:fingerID. PNAS 112:12580–12585. https://doi.org/10.1073/pnas.1509788112
Dührkop K, Böcker S (2015) Fragmentation Trees Reloaded. In: Przytycka TM (ed). Research in computational molecular biology. Springer International Publishing, pp 65–79
Böcker S, Dührkop K (2016) Fragmentation trees reloaded. Journal of Cheminformatics 8:5. https://doi.org/10.1186/s13321-016-0116-8
Dührkop K, Fleischauer M, Ludwig M, Aksenov AA, Melnik AV, Meusel M, Dorrestein PC, Rousu J, Böcker S (2019) SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat Methods 16:299–302. https://doi.org/10.1038/s41592-019-0344-8
Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G (2006) XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem 78:779–787. https://doi.org/10.1021/ac051437y
Kuhl C, Tautenhahn R, Böttcher C, Larson TR, Neumann S (2012) CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal Chem 84:283–289. https://doi.org/10.1021/ac202450g
Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G (2012) XCMS online: a web-based platform to process untargeted metabolomic data. Anal Chem 84:5035–5039. https://doi.org/10.1021/ac300698c
Misra BB, Mohapatra S (2019) Tools and resources for metabolomics research community: a 2017–2018 update. Electrophoresis 40:227–246. https://doi.org/10.1002/elps.201800428
Stanstrup J, Broeckling CD, Helmus R, Hoffmann N, Mathé E, Naake T, Nicolotti L, Peters K, Rainer J, Salek RM, Schulze T, Schymanski EL, Stravs MA, Thévenot EA, Treutler H, Weber RJM, Willighagen E, Witting M, Neumann S (2019) The metaRbolomics toolbox in bioconductor and beyond. Metabolites 9:200. https://doi.org/10.3390/metabo9100200
R Core Team (2019) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Hohrenk LL, Itzel F, Baetz N, Tuerk J, Vosough M, Schmidt TC (2020) Comparison of software tools for liquid chromatography–high-resolution mass spectrometry data processing in nontarget screening of environmental samples. Anal Chem 92:1898–1907. https://doi.org/10.1021/acs.analchem.9b04095
Lange E, Tautenhahn R, Neumann S, Gröpl C (2008) Critical assessment of alignment procedures for LC–MS proteomics and metabolomics measurements. BMC Bioinf 9:375. https://doi.org/10.1186/1471-2105-9-375
Niu W, Knight E, Xia Q, McGarvey BD (2014) Comparative evaluation of eight software programs for alignment of gas chromatography–mass spectrometry chromatograms in metabolomics experiments. J Chromatogr A 1374:199–206. https://doi.org/10.1016/j.chroma.2014.11.005
Myers OD, Sumner SJ, Li S, Barnes S, Du X (2017) Detailed investigation and comparison of the XCMS and MZmine 2 chromatogram construction and chromatographic peak detection methods for preprocessing mass spectrometry metabolomics data. Anal Chem 89:8689–8695. https://doi.org/10.1021/acs.analchem.7b01069
Hao L, Wang J, Page D, Asthana S, Zetterberg H, Carlsson C, Okonkwo OC, Li L (2018) Comparative evaluation of MS-based metabolomics software and its application to preclinical Alzheimer’s disease. Sci Rep 8:9291. https://doi.org/10.1038/s41598-018-27031-x
Myers OD, Sumner SJ, Li S, Barnes S, Du X (2017) One step forward for reducing false positive and false negative compound identifications from mass spectrometry metabolomics data: new algorithms for constructing extracted ion chromatograms and detecting chromatographic peaks. Anal Chem 89:8696–8703. https://doi.org/10.1021/acs.analchem.7b00947
Schymanski EL, Neumann S (2013) CASMI: and the winner is…. Metabolites 3:412–439. https://doi.org/10.3390/metabo3020412
Bruker DataAnalysis. https://www.bruker.com/. Accessed 20 Mar 2020
Libiseller G, Dvorzak M, Kleb U, Gander E, Eisenberg T, Madeo F, Neumann S, Trausinger G, Sinner F, Pieber T, Magnes C (2015) IPO: a tool for automated optimization of XCMS parameters. BMC Bioinformatics 16:118. https://doi.org/10.1186/s12859-015-0562-8
Eliasson M, Rännar S, Madsen R, Donten MA, Marsden-Edwards E, Moritz T, Shockcor JP, Johansson E, Trygg J (2012) Strategy for optimizing LC–MS data processing in metabolomics: a design of experiments approach. Anal Chem 84:6869–6876. https://doi.org/10.1021/ac301482k
Loos M, Singer H (2017) Nontargeted homologue series extraction from hyphenated high resolution mass spectrometry data. J Cheminform 9:12. https://doi.org/10.1186/s13321-017-0197-z
Schollée JE, Bourgin M, von Gunten U, McArdell CS, Hollender J (2018) Non-target screening to trace ozonation transformation products in a wastewater treatment train including different post-treatments. Water Res 142:267–278. https://doi.org/10.1016/j.watres.2018.05.045
Csardi G, Nepusz T (2006) The igraph software package for complex network research. InterJournal Complex Systems. 1695
Almende BV, Thieurmel B, Robert T (2019) visNetwork: Network Visualization using “vis.js” Library. https://CRAN.R-project.org/package=visNetwork
Kujawinski EB, Behn MD (2006) Automated analysis of electrospray ionization Fourier transform ion cyclotron resonance mass spectra of natural organic matter. Anal Chem 78:4363–4373. https://doi.org/10.1021/ac0600306
Koch BP, Dittmar T (2006) From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun Mass Spectrom 20:926–932. https://doi.org/10.1002/rcm.2386
Koch BP, Dittmar T (2016) From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun Mass Spectrom 30:250–250. https://doi.org/10.1002/rcm.7433
Brock O, Helmus R, Kalbitz K, Jansen B Non-target screening of leaf litter-derived dissolved organic matter using liquid chromatography coupled to high-resolution mass spectrometry (LC-QTOF-MS). Eur J Soil Sci. https://doi.org/10.1111/ejss.12894
Heller SR, McNaught A, Pletnev I, Stein S, Tchekhovskoi D (2015) InChI, the IUPAC international chemical identifier. J Cheminf 7:23. https://doi.org/10.1186/s13321-015-0068-4
Guha R (2007) Chemical Informatics Functionality in R. J Stat Softw 18:1–16
Schymanski EL, Gerlich M, Ruttkies C, Neumann S (2014) Solving CASMI 2013 with MetFrag, MetFusion and MOLGEN-MS/MS. Mass Spectrom 3:S0036–S0036. https://doi.org/10.5702/massspectrometry.S0036
Langfelder P, Zhang B (2016) dynamicTreeCut: Methods for Detection of Clusters in Hierarchical Clustering Dendrograms. https://CRAN.R-project.org/package=dynamicTreeCut
Royal Society of Chemistry ChemSpider. http://www.chemspider.com. Accessed 6 Feb 2020
Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H (2014) UpSet: visualization of intersecting sets. IEEE Trans Visual Comput Graphics 20:1983–1992. https://doi.org/10.1109/TVCG.2014.2346248
Chen H, Boutros PC (2011) VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinf 12:35. https://doi.org/10.1186/1471-2105-12-35
Gu Z, Gu L, Eils R, Schlesner M, Brors B (2014) circlize implements and enhances circular visualization in R. Bioinformatics 30:2811–2812
Gehlenborg N (2019) UpSetR: A More Scalable Alternative to Venn and Euler Diagrams for Visualizing Intersecting Sets. https://CRAN.R-project.org/package=UpSetR
Xie Y, Allaire JJ, Grolemund G (2018) R markdown: the definitive guide. Chapman and Hall/CRC, Boca Raton
Allaire JJ, Xie Y, McPherson J, Luraschi J, Ushey K, Atkins A, Wickham H, Cheng J, Chang W, Iannone R (2019) rmarkdown: Dynamic Documents for R
Iannone R, Allaire JJ, Borges B (2018) flexdashboard: R markdown format for flexible dashboards. https://CRAN.R-project.org/package=flexdashboard
Chang W, Cheng J, Allaire JJ, Xie Y, McPherson J (2019) shiny: web application framework for R. https://CRAN.R-project.org/package=shiny
Helmus R (2020) patRoon manuals. Zenodo. https://doi.org/10.5281/zenodo.3889936
patRoon reference. https://rickhelmus.github.io/patRoon/reference/index.html. Accessed 11 Jun 2020
patRoon tutorial. https://rickhelmus.github.io/patRoon/articles/tutorial.html. Accessed 11 Jun 2020
Helmus R patRoon handbook. https://rickhelmus.github.io/patRoon/handbook_bd/index.html. Accessed 11 Jun 2020
Xie Y (2016) Bookdown: authoring books and technical documents with R markdown. Chapman and Hall/CRC, Boca Raton
Xie Y (2019) Bookdown: authoring books and technical documents with R markdown
Wickham H, Danenberg P, Csárdi G, Eugster M (2019) roxygen2: in-line documentation for R. https://CRAN.R-project.org/package=roxygen2
Helmus R (2020) patRoonData. https://github.com/rickhelmus/patRoonData. Accessed 18 Mar 2020
Helmus R, Albergamo V (2020) patRoonData: 1.0.0. Zenodo. https://doi.org/10.5281/zenodo.3743266
Lang M (2017) checkmate: fast argument checks for Defensive R programming. R J 9:437–445
Csárdi G, Chang W (2019) processx: execute and control system processes. https://CRAN.R-project.org/package=processx
R Special Interest Group on Databases (R-SIG-DB), Wickham H, Müller K (2019) DBI: R database interface. https://CRAN.R-project.org/package=DBI
Müller K, Wickham H, James DA, Falcon S (2019) RSQLite: “SQLite” Interface for R. https://CRAN.R-project.org/package=RSQLite
Eddelbuettel D, François R (2011) Rcpp: seamless R and C++ integration. journal of statistical software 40:1–18. https://doi.org/10.18637/jss.v040.i08
Eddelbuettel D (2013) Seamless R and C++ integration with rcpp. Springer, New York
Eddelbuettel D, Balamuta JJ (2017) extending R with C++: a brief introduction to Rcpp. PeerJ Preprints 5:e3188v1. https://doi.org/10.7287/peerj.preprints.3188v1
Kapoulkine A pugixml. https://pugixml.org/. Accessed 6 Feb 2020
Dowle M, Srinivasan A (2019) data.table: Extension of ‘data.frame’. https://CRAN.R-project.org/package=data.table
MetFragR. http://ipb-halle.github.io/MetFrag/projects/metfragr/. Accessed 6 Feb 2020
Lang DT (2019) RDCOMClient: R-DCOM client
Wickham H (2011) testthat: get started with testing. R J 3:5–10
Henry L, Sutherland C, Hong D, Luciani TJ, Decorde M, Lise V (2019) vdiffr: visual regression testing and graphical diffing. https://CRAN.R-project.org/package=vdiffr
RStudio| Open source & professional software for data science teams. https://rstudio.com/. Accessed 19 Oct 2020
Boettiger C, Eddelbuettel D (2017) An introduction to rocker: docker containers for R. arXiv:171003675 [cs]
NORMAN network. https://www.norman-network.net/. Accessed 6 Oct 2018
Collet B, van Vugt-Lussenburg BMA, Swart K, Helmus R, Naderman M, de Rijke E, Eggesbø M, Brouwer A, van der Burg B (2020) Antagonistic activity towards the androgen receptor independent from natural sex hormones in human milk samples from the Norwegian HUMIS cohort. Environ Int 143:105948. https://doi.org/10.1016/j.envint.2020.105948
Helmus R (2020) patRoon benchmarking & demonstration data. Zenodo. https://doi.org/10.5281/zenodo.3885448
Mersmann O (2019) microbenchmark: Accurate Timing Functions. https://CRAN.R-project.org/package=microbenchmark
Fischer B, Neumann S, Gatto L, Kou Q, Rainer J (2020) mzR: parser for netCDF, mzXML, mzData and mzML and mzIdentML files (mass spectrometry data). https://bioconductor.org/packages/mzR/. Accessed 6 Apr 2020
Gurobi. https://www.gurobi.com/. Accessed 6 Feb 2020
CPLEX Optimizer. https://www.ibm.com/analytics/cplex-optimizer. Accessed 6 Feb 2020
GNU Project—free software foundation (FSF) GLPK (GNU Linear Programming Kit). https://www.gnu.org/software/glpk/. Accessed 6 Feb 2020
Böcker S, Dührkop K, Fleischauer M, Ludwig M (2019) SIRIUS Documentation Release 4.0.1
NORMAN Suspect List Exchange—NORMAN SLE. https://www.norman-network.com/nds/SLE/. Accessed 13 Mar 2020
CompTox March 2019 CSV file. ftp://newftp.epa.gov/COMPTOX/Sustainable_Chemistry_Data/Chemistry_Dashboard/MetFrag_metadata_files/CompTox_17March2019_SelectMetaData.csv
Ulrich EM, Sobus JR, Grulke CM, Richard AM, Newton SR, Strynar MJ, Mansouri K, Williams AJ (2019) EPA’s non-targeted analysis collaborative trial (ENTACT): genesis, design, and initial findings. Anal Bioanal Chem 411:853–866. https://doi.org/10.1007/s00216-018-1435-6
Newton SR, Sobus JR, Ulrich EM, Singh RR, Chao A, McCord J, Laughlin-Toth S, Strynar M (2020) Examining NTA performance and potential using fortified and reference house dust as part of EPA’s non-targeted analysis collaborative trial (ENTACT). Anal Bioanal Chem 412:4221–4233. https://doi.org/10.1007/s00216-020-02658-w
Singh RR, Chao A, Phillips KA, Xia XR, Shea D, Sobus JR, Schymanski EL, Ulrich EM (2020) Expanded coverage of non-targeted LC-HRMS using atmospheric pressure chemical ionization: a case study with ENTACT mixtures. Anal Bioanal Chem 412:4931–4939. https://doi.org/10.1007/s00216-020-02716-3
Djoumbou-Feunang Y, Fiamoncini J, Gil-de-la-Fuente A, Greiner R, Manach C, Wishart DS (2019) BioTransformer: a comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J Cheminf 11:2. https://doi.org/10.1186/s13321-018-0324-5
Kruve A (2019) Semi-quantitative non-target analysis of water with liquid chromatography/high-resolution mass spectrometry: how far are we? Rapid Commun Mass Spectrom 33:54–63. https://doi.org/10.1002/rcm.8208
Djoumbou Feunang Y, Eisner R, Knox C, Chepelev L, Hastings J, Owen G, Fahy E, Steinbeck C, Subramanian S, Bolton E, Greiner R, Wishart DS (2016) ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J Cheminform 8:61. https://doi.org/10.1186/s13321-016-0174-y
R for Mass Spectrometry. www.rformassspectrometry.org. Accessed 13 Mar 2020
Rick Helmus (2020) patRoon. Zenodo. https://doi.org/10.5281/zenodo.3889855
Acknowledgements
The many authors involved in the open mass spectrometry software development community are highly acknowledged as their contributions are the foundation for the development of patRoon. In addition, Vittorio Albergamo, Andrea Brunner, Thomas Wagner, Olaf Brock and other users of patRoon are thanked for testing and providing feedback for future developments. We thank the Dutch drinking water companies Dunea and PWN for sharing the raw HRMS data that was used for benchmarking and demonstration purposes. Markus Fleischauer is acknowledged for his feedback on execution of batch execution of SIRIUS. Finally, Olaf Brock is acknowledged for the design of some of the visualizations of benchmarking data.
Funding
This work was internally funded by the Institute of Biodiversity and Ecosystem Dynamics (University of Amsterdam). ELS is supported by the Luxembourg National Research Fund (FNR) for project A18/BM/12341006.
Author information
Authors and Affiliations
Contributions
RH wrote the manuscript, source code, designed the experiments and interpreted the results. ELS provided valuable feedback to improve the software. ELS and other authors supervised this work and contributed to writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Overview of software and databases that are used in the implementation in patRoon. This table summarizes all the software and databases that are described in the implementation section of the main text.
Additional file 2.
Additional figures that illustrate implementation details of patRoon and miscellaneous benchmarking and demonstration results.
Additional file 3.
Additional tables with more details on the implementation.
Additional file 4.
Source code for benchmarks. Archive with several R scripts that were used to perform the parallelization benchmarks.
Additional file 6.
Algorithm consensus demonstration. Script that was used to generate the results for the feature algorithm consensus demonstration.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Helmus, R., ter Laak, T.L., van Wezel, A.P. et al. patRoon: open source software platform for environmental mass spectrometry based non-target screening. J Cheminform 13, 1 (2021). https://doi.org/10.1186/s13321-020-00477-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13321-020-00477-w