
Abstract
Owing to the increase in freely available software and data for cheminformatics and structural bioinformatics, research for computer-aided drug design (CADD) is more and more built on modular, reproducible, and easy-to-share pipelines. While documentation for such tools is available, there are only a few freely accessible examples that teach the underlying concepts focused on CADD, especially addressing users new to the field. Here, we present TeachOpenCADD, a teaching platform developed by students for students, using open source compound and protein data as well as basic and CADD-related Python packages. We provide interactive Jupyter notebooks for central CADD topics, integrating theoretical background and practical code. TeachOpenCADD is freely available on GitHub: https://github.com/volkamerlab/TeachOpenCADD.

Similar content being viewed by others

Introduction
Open access resources for cheminformatics and structural bioinformatics as well as public platforms for code deposition such as GitHub are increasingly used in research. This combination facilitates and promotes the generation of modular, reproducible, and easy-to-share pipelines for computer-aided drug design (CADD). Comprehensive lists of open resources are reviewed by Pirhadi et al. [1], or presented in the form of the web-based search tool Click2Drug [2], aiming to cover the full CADD pipeline.
While documentation for open access resources is available, freely accessible teaching platforms for concepts and applications in CADD are rare. Available examples include the following: On the one hand, graphical user interface (GUI) based tutorials teach CADD basics, such as the web-based educational Drug Design Workshop [3, 4]. On the other hand, examples for educational coding tutorials are the Java-based Chemistry Development Kit (CDK) [5,6,7,8,9] and the Teach–Discover–Treat (TDT) initiative [10], which launched challenges to develop tutorials, such as a Python-based virtual screening (VS) workflow to identify malaria drugs [11, 12].
Complementing these resources, we developed the TeachOpenCADD platform to provide students and researchers new to CADD and/or programming with step-by-step tutorials suitable for self-study training as well as classroom lessons, covering both ligand- and structure-based approaches. TeachOpenCADD is a novel teaching platform developed by students for students, using open source data and Python packages to tackle various common tasks in cheminformatics and structural bioinformatics. Interactive Jupyter notebooks [13] are presented for central topics, integrating detailed theoretical background and well-documented practical code. Topics build upon one another in the form of a pipeline, which is illustrated at the example of the epidermal growth factor receptor (EGFR) kinase, but can easily be adapted to other query proteins. TeachOpenCADD is publicly available on GitHub and open to contributions from the community: https://github.com/volkamerlab/TeachOpenCADD (current release: https://doi.org/10.5281/zenodo.2600909).
Methods
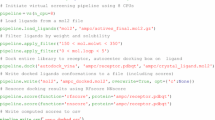
TeachOpenCADD currently consists of ten talktorials covering central topics in CADD, see Fig. 1. Talktorials are offered as interactive Jupyter notebooks that can be used as tutorials but also for oral presentations, e.g. in student CADD seminars (talk + tutorial = talktorial). They start with a topic motivation and learning goals, continue with the main part composed of theoretical background and practical code, and end with a short discussion and quiz, see Fig. 2.
Open data resources employed are the ChEMBL [14] and PDB [15] databases for compound and protein structure data acquisition, respectively. Open source libraries utilized are RDKit [16] (cheminformatics), the ChEMBL webresource client [17] and PyPDB [18] (ChEMBL and PDB application programming interface access), BioPandas [19] (loading and manipulating molecular structures), and PyMOL [20] (structural data visualization). Additionally, basic Python computing libraries employed include numpy [21, 22] and pandas [23, 24] (high-performance data structures and analysis), scikit-learn [25] (machine learning), as well as matplotlib [26] and seaborn [27] (plotting). Furthermore, the user is instructed how to work with conda [28], a widely used package, dependency and environment management tool. A conda yml file is provided to ensure an easy and quick setup of an environment containing all required packages.
The talktorial topics include how to acquire data from ChEMBL (T1), filter compounds for drug-likeness (T2), and identify unwanted substructures (T3). Furthermore, measures for compound similarity are introduced and applied for VS of kinase inhibitor gefitinib (T4) as well as for compound clustering (T5), including the use of maximum common substructures (T6). Machine learning approaches are employed to build models for predicting active compounds (T7). Lastly, protein-ligand complexes are fetched from the PDB (T8), used to generate ligand-based ensemble pharmacophores (T9). Geometry-based binding site comparison of kinase inhibitor imatinib binding proteins is performed to analyse potential off-targets (T10). In summary, the presented talktorials build a pipeline with starting points being (i) a query protein to study associated compound data (T1 and T8) and (ii) a query ligand to investigate associated on- and off-targets (T10), see Fig. 1. These talktorials can be studied independently from each other or as a pipeline.
As an example, the talktorial pipeline is used to identify novel EGFR kinase inhibitors. EGFR kinase is a transmembrane protein, which activates several signaling cascades to convert extracellular signals into cellular responses. Dysfunctional signaling of EGFR is associated with diseases such as cancer, making it a frequent target in drug development projects (the reader is referred to a review by Chen et al. [29] for more information on EGFR). Furthermore, the pipeline can easily be adapted to other examples by simply exchanging the query protein (T1 and T8: protein UniProt ID) and query ligand (T10: ligand names in the PDB).
Results
In the following, the content of each talktorial is briefly discussed and summarized in Fig. 1. If not noted otherwise, tasks are conducted with RDKit or basic Python libraries as stated in the Methods section. Note that reported numbers and results are based on data sets from ChEMBL and PDB queries conducted in November 2018.
T1. Data acquisition from ChEMBL. Compound information on structure, bioactivity and associated targets is organized in databases such as ChEMBL, PubChem [30], or DrugBank [31]. For the query target EGFR (UniProt ID P00533), compound data including molecular structure (SMILES) and bioactivity data is automatically fetched from the ChEMBL database, using the ChEMBL webresource client, and is filtered for e.g. binding assays and IC\(_{50}\) measurements (6,641 compounds). The data set is formatted and further filtered: e.g. duplicates and entries with missing values are dropped and only bioactivity values in molar units are kept and converted to pIC\(_{50}\) values (4,771 compounds retained, referred to as data set T1), see Fig. 1.T1.
T2. Molecular filtering: ADME criteria. Not all compounds are suitable starting points for drug development due to undesirable pharmacokinetic properties, which for instance negatively affect a drug’s absorption, distribution, metabolism, and excretion (ADME). Therefore, such compounds are usually not included in data sets for VS. Data set T1 is filtered by lead-likeness criteria, i.e. Lipinski’s rule of five [32], in order to remove less drug-like molecules from the EGFR data set (4009 compounds retained, referred to as data set T2). This data set is visualized using radar plots demonstrating their ADME properties, see Fig. 1.T2, and serves as starting point for several talktorials discussed in the following.
T3. Molecular filtering: unwanted substructures. Compounds can contain unwanted substructures that may cause mutagenic, reactive, or other unfavorable pharmacokinetic effects [33] or that may lead to non-specific interactions with assays (PAINS) [34]. Such unwanted substructures are detected and highlighted in data set T2. This knowledge can be integrated into cheminformatics pipelines to either perform an additional filtering step before screening (1,951 compounds retained) or – more often – to set alert flags to compounds being potentially problematic. They can be manually evaluated by medicinal chemists if reported as hits after screening, see Fig. 1.T3.
T4. Ligand-based screening: compound similarity. In VS, compounds similar to known ligands of a target under investigation often constitute the starting point for drug development. This approach follows the similar property principle stating that structurally similar compounds are more likely to exhibit similar biological activities [35, 36] (exceptions are so-called activity cliffs [37]). For computational representation and processing, compound properties can be encoded in the form of bit arrays, so-called molecular fingerprints, e.g. MACCS [38] and Morgan fingerprints [39, 40]. Compound similarity can be assessed by comparison measures, such as the Tanimoto and Dice similarity [41]. Using these encoding and comparison methods, VS is conducted based on a similarity search: the EGFR inhibitor gefitinib is used to find its most similar compounds in data set T2. With the data being split into active and inactive compounds based on the chosen pIC\({_{50}}\) cutoff of 6.3, screening results are evaluated with enrichment plots, see Fig. 1.T4. In the top 5% of the compounds ranked by similarity, called the enrichment factor at 5% (EF\(_{5\%}\)), 8.3% of actives can be retrieved, while the random and optimal EF\(_{5\%}\) of this data set are 5.0% and 9.2%, respectively.
T5. Compound clustering. The similar property principle can also be used to identify groups of similar compounds via clustering, in order to pick a set of diverse compounds from these clusters for e.g. non-redundant experimental testing. In this talktorial, Butina clustering [42] based on the RDKFingerprint [43] is applied to cluster data set T2 at a Tanimoto distance cutoff of 0.2, resulting in 988 clusters with the largest cluster consisting of 143 compounds, see Fig. 1.T5. Following the example in the TDT pipeline by Riniker et al. [11], a maximum of 1000 compounds is subsequently picked by selecting the ten most similar compounds per cluster (or 50% for clusters with fewer compounds), starting with the largest cluster. Thereby, compound diversity is ensured (representatives of each cluster), while structure-activity relationship (SAR) information is retained (most similar compounds selected from clusters).
T6. Maximum common substructures. In order to visualize shared scaffolds and thereby emphasize the extent and type of chemical similarities or differences of a compound cluster, the maximum common substructure (MCS) [44] can be calculated and highlighted. The MCS for the largest cluster from T5 is calculated using the FMCS algorithm [45], see Fig. 1.T6. Different parameters can be applied, e.g. a threshold to set the percentage of compounds in the set that need to share the same MCS, or a restriction to match ring bonds only with other ring bonds.
T7. Ligand-based screening: machine learning. With the continuously increasing amount of available data, machine learning (ML) gained momentum in drug discovery and especially in ligand-based VS to predict the activity of novel compounds against a target of interest. The EGFR compound data set is split into active and inactive compounds as described in T4, and used to train ML classifiers based on random forests (RF) [46], support vector machines (SVM) [47], and artificial neural networks (ANN) [48], applying 10-fold cross validation. Models are evaluated using receiver operating characteristic (ROC) curves and mean area under the curve (AUC) values (mean AUC results for RF, SVM, and ANN are 90%, 87%, and 87%, respectively), see Fig. 1.T7. The trained models can be used to perform a classification of an unknown screening data set to predict novel potential EGFR inhibitors.
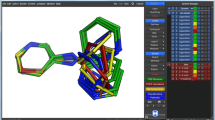
T8. Data acquisition from PDB. The PDB database holds 3D structural data and meta information on experimentally resolved proteins. Using PyPDB, all EGFR structures are automatically fetched from the PDB (by UniProt ID) and filtered by ligand-bound structures resolved with X-ray crystallography, retaining four EGFR-ligand structures with good structural resolution. Using the Python integration of the molecular visualization tool PyMOL, those structures are subsequently aligned to each other in 3D. Ligands are extracted, see Fig. 1.T8, and saved to be used in T9 for the generation of a ligand-based ensemble pharmacophore.
T9. Ligand-based ensemble pharmacophores. Another approach for ligand-based VS – besides a similarity search (T4) or machine learning classifiers (T7) – are ligand-based (ensemble) pharmacophore models. They describe important steric and physicochemical properties of a ligand (or a set of ligands) to bind a target under investigation. Examples for physicochemical properties are so-called donor, acceptor, and hydrophobic pharmacophoric features present in a molecule [49, 50]. For the EGFR ligands selected and aligned in T8, pharmacophoric features are identified for each ligand and subsequently clustered with k-means clustering [51] in order to define an ensemble pharmacophore, see Fig. 1.T9. Such a pharmacophore represents the properties of the set of known EGFR ligands and can be used to search for novel EGFR ligands via VS, as described in an RDKit pharmacophore tutorial by Stiefl et al. [52].
T10. Off-target prediction and binding site comparison. Off-targets are proteins that interact with a drug or (one of) its metabolite(s) without being the designated target, potentially causing unwanted side effects. Off-targets mainly occur because they share similar structural motifs in their binding site with on-targets, and are therefore able to bind similar ligands. Computational off-target prediction using binding site comparison is an established approach in early stages of drug development [53, 54]. In T10, structural similarity is exemplarily accessed using a basic measure, i.e. the geometrical variation between structures by calculating the root mean square deviation (RMSD) between pairs of aligned structures using PyMOL, including either the whole proteins or focusing on their binding sites. Pairwise RMSD comparison of seven protein structures binding imatinib, a small molecule tyrosine kinase inhibitor for cancer treatment, is able to separate tyrosine kinases (on-targets) from quinone reductase (reported off-target [55]), see Fig. 1.T10.

TeachOpenCADD talktorial pipeline. TeachOpenCADD is a teaching platform for open source data and packages, currently offering ten talktorials in the form of Jupyter notebooks on central topics in CADD, ranging from cheminformatics (T1–7) to structural bioinformatics (T8–10). The talktorials are illustrated at the example of EGFR (based on data sets from ChEMBL and PDB queries in November 2018)

Screenshot of TeachOpenCADD talktorial composition. TeachOpenCADD talktorials are Jupyter notebooks that cover one CADD topic each, composed of (i) a topic motivation, (ii) learning goals, (iii) references to literature, (iv) theoretical background, (v) practical code, (vi) a short discussion, and (vii) a quiz—all in one place. Shown here is a screenshot of parts of talktorial T9 to generate pharmacophores
Conclusion
The presented teaching platform TeachOpenCADD aims at introducing interested students and researchers to the ease and benefit of using open access resources for cheminformatics and structural bioinformatics. Jupyter notebooks (talktorials) offer detailed theoretical background and Python code examples, forming an automated pipeline that saves and reloads results from one topic to another. The pipeline is illustrated using the example of EGFR, but can easily be adapted to other examples by exchanging the input protein and ligand. Beyond their teaching purpose for self-study training and classroom lessons, the talktorials can serve as starting point for users’ project-directed modifications and extensions. TeachOpenCADD intends to expand existing and add new topics continuously, and is open for contributions and ideas from the community.
Abbreviations
- CADD:
-
computer-aided drug design
- GUI:
-
graphical user interface
- CDK:
-
Chemistry Development Kit
- TDT:
-
Teach–Discover–Treat
- VS:
-
virtual screening
- EGFR:
-
epidermal growth factor receptor
- ADME:
-
absorption, distribution, metabolism, excretion
- SAR:
-
structure–activity relationship
- MCS:
-
maximum common substructure
- ML:
-
machine learning
- RF:
-
random forest
- SVM:
-
support vector machine
- ANN:
-
artificial neural network
- ROC:
-
receiver operating characteristic
- AUC:
-
area under the curve
- RMSD:
-
root mean square deviation
- EF:
-
enrichment factor
References
Pirhadi S, Sunseri J, Koes DR (2016) Open source molecular modeling. J Mol Graph Modell 69:127–43
Swiss Institute of Bioinformatics (2013) Click2Drug website. http://www.click2drug.org/. Accessed 18 Dec 2018
Daina A, Blatter MC, Baillie Gerritsen V, Palagi PM, Marek D, Xenarios I, Schwede T, Michielin O, Zoete V (2017) Drug design workshop: a web-based educational tool to introduce computer-aided drug design to the general public. J Chem Educ 94:335–344
Swiss Institute of Bioinformatics (2015) Drug Design Workshop website. www.drug-design-workshop.ch. Accessed 18 Dec 2018
Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E (2003) The Chemistry Development Kit (CDK): an open-source java library for chemo- and bioinformatics. J Chem Inf Comput Sci 43:493–500
Steinbeck C, Hoppe C, Kuhn S, Floris M, Guha R, Willighagen EL (2006) Recent developments of the Chemistry Development Kit (CDK)—an open-source java library for chemo- and bioinformatics. Curr Pharm Des 12:2111–20
May JW, Steinbeck C (2014) Efficient ring perception for the Chemistry Development Kit. J Cheminf 6:3
Willighagen EL, Mayfield JW, Alvarsson J, Berg A, Carlsson L, Jeliazkova N, Kuhn S, Pluskal T, Rojas-Chertó M, Torrance G, Evelo CT, Guha R, Steinbeck C (2017) The Chemistry Development Kit (cdk) v2.0: atom typing, depiction, molecular formulas, and substructure searching. J Cheminf 9:33
Chemistry Development Kit (2017) Chemistry Development Kit (CDK) website. https://cdk.github.io/, Accessed 18 Dec 2018
Jansen JM, Cornell W, Tseng YJ, Amaro RE (2012) Teach–Discover–Treat (TDT): collaborative computational drug discovery for neglected diseases. J Mol Graph Modell 38:360–2
Riniker S, Landrum GA, Montanari F, Villalba SD, Maier J, Jansen JM, Walters WP, Shelat AA (2017) Virtual-screening workflow tutorials and prospective results from the Teach–Discover–Treat competition 2014 against malaria. F1000Research 6:1136
Riniker S, Landrum GA, Montanari F, Villalba SD, Maier J, Jansen JM, Walters WP, Shelat AA (2017) Tutorial for the Teach–Discover–Treat (TDT) Competition 2014—Challenge 1: anti-malaria hit finding using classifier-fusion boosted predictive models. https://github.com/sriniker/TDT-tutorial-2014. Accessed 18 Dec 2018
Kluyver T, Ragan-Kelley B, Pérez F, Granger B, Bussonnier M, Frederic J, Kelley K, Hamrick J, Grout J, Corlay S, Ivanov P, Avila D, Abdalla S, Willing C, Team Jupyter Development (2016) Jupyter Notebooks—a publishing format for reproducible computational workflows. Agents and agendas. In: Loizides F, Schmidt B (eds) Positioning and power in academic publishing: players. IOS Press, Amsterdam, pp 87–90
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40:1100–7
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–42
RDKit (2018) RDKit: Open-Source Cheminformatics, Version 2018.09.1. http://www.rdkit.org
Davies M, Nowotka M, Papadatos G, Dedman N, Gaulton A, Atkinson F, Bellis L, Overington JP (2015) ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res 43:W612–W620
Gilpin W (2015) PyPDB: a Python API for the protein data bank. Bioinformatics 32:159–60
Raschka S (2017) BioPandas: working with molecular structures in pandas DataFrames. J Open Source Softw 2:279
Schrödinger L (2015) The PyMOL molecular graphics system. Version 1.8
Oliphant T (2006) A guide to NumPy. Trelgol Publishing
van der Walt S, Colbert SC, Varoquaux G (2011) The NumPy array: a structure for efficient numerical computation. Comput Sci Eng 13(2):22–30
McKinney W (2010) Data structures for statistical computing in Python. In: van der Walt S, Millman J (eds) Proceedings of the 9th Python in science conference, pp 51–56
McKinney W (2011) pandas: a foundational Python library for data analysis and statistics
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E (2011) Scikit-learn: machine learning in Python. J Mach Learn Res 12:2825–2830
Hunter JD (2007) Matplotlib: a 2D graphics environment. Comput Sci Eng 9:90–95
Waskom M (2018) seaborn v0.9.0
Continuum Analytics Inc (dba Anaconda Inc) (2017) conda. https://www.anaconda.com. Accessed 18 Dec 2018
Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang MZ, Harris RC (2016) Expression and function of the epidermal growth factor receptor in physiology and disease. Physiol Rev 96:1025–1069
Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J, Bryant SH (2016) PubChem substance and compound databases. Nucleic Acids Res 44:D1202–D1213
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34:D668–D672
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25
Brenk R, Schipani A, James D, Krasowski A, Gilbert IH, Frearson J, Wyatt PG (2008) Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 3:435–444
Baell JB, Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53:2719–2740
Johnson MA, Maggiora GM (1990) Concepts and applications of molecular similarity, 1st edn. Wiley, New York
Bender A, Glen RC (2004) Molecular similarity: a key technique in molecular informatics. Org Biomol Chem 2:3204
Bajorath J (2017) Representation and identification of activity cliffs. Expert Opin Drug Discov 12:879–883
Accelrys Inc, San Diego, CA, USA (2011) MACCS structural keys
Morgan HL (1965) The generation of a unique machine description for chemical structures—a technique developed at Chemical Abstracts Service. J Chem Doc 5:107–113
Rogers D, Hahn M (2010) Extended-connectivity fingerprints. J Chem Inf Model 50:742–754
Maggiora G, Vogt M, Stumpfe D, Bajorath J (2014) Molecular similarity in medicinal chemistry. J Med Chem 57:3186–3204
Butina D (1999) Unsupervised data base clustering based on Daylight’s fingerprint and Tanimoto similarity: a fast and automated way to cluster small and large data sets. J Chem Inf and Model 39:747–750
RDKit (2018) RDKFingerprint. http://rdkit.org/docs/source/rdkit.Chem.rdmolops.html. Accessed 18 Dec 2018
Raymond JW, Willett P (2002) Maximum common subgraph isomorphism algorithms for the matching of chemical structures. J Comput-Aided Mol Des 16:521–33
Dalke A, Hastings J (2013) FMCS: a novel algorithm for the multiple MCS problem. J Cheminf 5:O6
Ho TK (1995) Random decision forests. In: Proceedings of 3rd international conference on document analysis and recognition, vol 1. IEEE Comput Soc Press, Los Alamitos, California, pp 278–282
Cortes C, Vapnik V (1995) Support-vector networks. Mach Learn 20:273–297
van Gerven M, Bohte S (2017) Editorial: artificial neural networks as models of neural information processing. Front Comput Neurosci 11:114
Wermuth CG, Ganellin CR, Lindberg P, Mitscher LA (1998) Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl Chem 70:1129–1143
Seidel T, Wolber G, Murgueitio MS (2018) Pharmacophore perception and applications. Applied chemoinformatics. Wiley, Weinheim, pp 259–282
Macqueen J (1967) Some methods for classification and analysis of multivariate observations. In: 5th Berkeley symposium on mathematical statistics and probability, pp 281–297
Stiefl N (2016) 3D pharmacophores in the RDKit. https://github.com/rdkit/UGM_2016/blob/master/Notebooks/Stiefl_RDKitPh4FullPublication.ipynb. Accessed 18 Dec 2018
Kellenberger E, Schalon C, Rognan D (2008) How to measure the similarity between protein ligand-binding sites? Curr Comput-Aided Drug Des 4:209–220
Ehrt C, Brinkjost T, Koch O (2016) Impact of binding site comparisons on medicinal chemistry and rational molecular design. J Med Chem 59:4121–4151
Winger JA, Hantschel O, Superti-Furga G, Kuriyan J (2009) The structure of the leukemia drug imatinib bound to human quinone reductase 2 (NQO2). BMC Struct Biol 9:7
Authors' contributions
All authors (DS, AM, MD, and AV) contributed to implementing the platform, finalizing the talktorials, and editing/reviewing the manuscript. DS was responsible for management and major writing, and AV for conceptualization, management, and writing. All authors read and approved the final manuscript.
Acknowledgements
The authors thank the participants of the CADD seminar courses in 2017 and 2018 (joint bioinformatics study program at the Freie Universität Berlin and the Charité) for working on the reported talktorials: Svetlana Leng and Paula Junge (T1), Mathias Wajnberg and Michele Ritschel (T2), Maximilian Driller and Sandra Krüger (T3), Andrea Morger and Franziska Fritz (T4), Gizem Spriewald and Calvinna Caswara (T5), Oliver Nagel (T6), Jacob Gora and Jan Philipp Albrecht (T7), Majid Vafadar and Anja Georgi (T8), Pratik Dhakal and Florian Gusewski (T9), as well as Angelika Szengel and Marvis Sydow (T10). Additionally, the authors acknowledge Greg Landrum and Boran Adas for their feedback on the talktorials. Finally, the authors express their gratitude to the Freie Universität Berlin for supporting the TeachOpenCADD project (SUPPORT für die Lehre: Förderung innovativer Lehrvorhaben).
Competing interests
The authors declare that they have no competing interests.
Availability and requirements
Project name: TeachOpenCADD. Project home page: https://github.com/volkamerlab/TeachOpenCADD. Operating system(s): Platform independent. Programming language: Python. Other requirements: Databases: ChEMBL and PDB. Python packages: RDKit, ChEMBL webresource client, PyPDB, BioPandas, PyMOL, numpy, pandas, scikit-learn, matplotlib, seaborn, and conda. License: http://creativecommons.org/licenses/by/4.0/. Any restrictions to use by non-academics: Not applicable.
Availability of data and materials
TeachOpenCADD talktorial material is available at https://github.com/volkamerlab/TeachOpenCADD. Compound and protein structure data used as EGFR example in the talktorials are fetched from the ChEMBL (query by UniProt ID “P00533”) and PDB (query by UniPort ID “P00533”, “STI”, and “imatinib”) databases.
Funding
The authors receive funding from the Bundesministerium für Bildung und Forschung (AV: Grant Number 031A262C), Deutsche Forschungsgemeinschaft (DFG) (AV and DS: Grant Number 391684253), and the HaVo-Stiftung, Ludwigshafen, Germany (AM). The authors acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité – Universitätsmedizin Berlin.
Publisher's Note
Publisher's Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sydow, D., Morger, A., Driller, M. et al. TeachOpenCADD: a teaching platform for computer-aided drug design using open source packages and data. J Cheminform 11, 29 (2019). https://doi.org/10.1186/s13321-019-0351-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13321-019-0351-x