
Abstract
Background
Hierarchical clustering is an exploratory data analysis method that reveals the groups (clusters) of similar objects. The result of the hierarchical clustering is a tree structure called dendrogram that shows the arrangement of individual clusters. To investigate the row/column hierarchical cluster structure of a data matrix, a visualization tool called ‘cluster heatmap’ is commonly employed. In the cluster heatmap, the data matrix is displayed as a heatmap, a 2-dimensional array in which the colour of each element corresponds to its value. The rows/columns of the matrix are ordered such that similar rows/columns are near each other. The ordering is given by the dendrogram which is displayed on the side of the heatmap.
Results
We developed InCHlib (Interactive Cluster Heatmap Library), a highly interactive and lightweight JavaScript library for cluster heatmap visualization and exploration. InCHlib enables the user to select individual or clustered heatmap rows, to zoom in and out of clusters or to flexibly modify heatmap appearance. The cluster heatmap can be augmented with additional metadata displayed in a different colour scale. In addition, to further enhance the visualization, the cluster heatmap can be interconnected with external data sources or analysis tools. Data clustering and the preparation of the input file for InCHlib is facilitated by the Python utility script inchlib_clust.
Conclusions
The cluster heatmap is one of the most popular visualizations of large chemical and biomedical data sets originating, e.g., in high-throughput screening, genomics or transcriptomics experiments. The presented JavaScript library InCHlib is a client-side solution for cluster heatmap exploration. InCHlib can be easily deployed into any modern web application and configured to cooperate with external tools and data sources. Though InCHlib is primarily intended for the analysis of chemical or biological data, it is a versatile tool which application domain is not limited to the life sciences only.
Similar content being viewed by others


Background
Clustering is a data exploration technique that identifies groups of objects that are similar to each other but different from objects in other groups [1]. Cluster analysis is widely applied in cheminformatics for the analysis of databases of chemical structures [2],[3]. Its main use is to find representative subsets from high throughput screening (HTS) [4]-[6], to design chemical libraries of diverse structures pertinent to pharmaceutical discovery [7]-[9] and to increase the diversity of these libraries through the selection of additional compounds from other data sets [10],[11]. The most popular approach of cluster analysis is hierarchical clustering [12] in which data are merged together based on a tree structure called dendrogram. The input to a clustering algorithm is a data matrix that contains individual data points in rows and data features in columns. Data can be clustered either by rows or by columns. The data matrix can be visualized as a ‘data heatmap’, a rectangular array that uses colour to represent numerical values of individual matrix cells. The data heatmap augmented with row and/or column dendrograms is known as a ‘cluster heatmap’ [13],[14].
Owing to the wide application of the cluster heatmap in biomedical sciences [15], many software tools for its visualization and exploration are available. Several of them, such as the R programming environment [16] with Bioconductor package [17], CIMminer[18] or Cluster/TreeView[19],[20], generate only static images with fixed appearance and no interactivity. Higher level of interactivity offer standalone programs typically implemented in Java programming language that are, however, usually tailored towards the analysis of specific data [21],[22]. For example, the following packages enable the analysis of gene expression experiments: Java Treeview[23], High-Throughput GoMiner[24], TM4[25], Genesis[26] or PageMan[27]. Similarly, genomics data can be explored by geWorkbench[28], StratomeX[29], GENE-E[30], Qcanvas[31] or Gitools[32]. The main disadvantage of desktop solutions is their limited set of features that cannot be easily enhanced by the user. In addition, desktop applications cannot be readily deployed in modern web-based systems.
In recent years, client-side scripting became very popular for the development of interactive web solutions. The client is the system on which the web browser runs. Client-side scripts are interpreted by the browser and they work in the following steps: (1) the user requests the web page from the server, (2) the server finds the page and sends it to the user, (3) the page is displayed in the browser with any scripts run during or after display. Because all data processing is performed by the client, the speed of the script execution depends on the user’s hardware. Two types of clients exist: thick (fat) and thin clients. The thick clients are written in full-blown programming languages, such as Java or C#. To be executed, thick clients require additional software (e.g., Java Virtual Machine or .NET framework) to be installed on the user machine. On the other hand, the thin client is executed by an engine embedded directly in the web browser. The main scripting language for the thin client programming is JavaScript. JavaScript is a powerful, easy to learn and use language which became an integral part of many existing web technologies. Compared to the thick client, the thin client typically requires less performing user devices equipped with lower amounts of memory.
If the deployment of the cluster heatmap into a web application is required, possibilities are rather limited. Though several web solutions for the analysis of genomics data exist, such as The UCSC Cancer Genomics Browser[33],[34], Expression Profiler[35], Babelomics[36], Next-Generation Clustered Heatmaps[37] or INVEX [38], they work as standalone web applications. It means that they can be used only from their hosting websites and their interface reflects the nature of the data they are designed to work with. The use of such applications for the analysis of, often sensitive, user’s data requires the data to be uploaded to the web server of the application provider. Though a few thick clients exist (e.g., Gitools[32]), the availability of JavaScript solutions for cluster heatmap exploration is rather limited. While jHeatmap[39] and the BioJS HeatmapViewer component [40] can display only the data heatmap without its underlying cluster structure, the Heatmap viewer from the JavaScript library canvasXpress[41] offers only limited functionality. Thus, we developed InCHlib, a free browser independent JavaScript library that facilitates the visualization, exploration and web integration of the cluster heatmap. Though InCHlib is primarily intended for the analysis of chemical or biological data, its application domain is not limited to the life sciences only.
Implementation
InCHlib is a free browser independent JavaScript library which HTML5 canvas-based rendering is handled by the KineticJS[42] framework (version 5.0.0) and HTML elements are processed using the jQuery framework [43] (version 2.0.3). InCHlib enables to interact in real time with other elements on the page or with external data sources. This is achieved by handling the events that occur during the user interaction with the cluster heatmap, such as clicking on a heatmap row or dendrogram node. For each event, a callback function can be defined and invoked if the event is triggered. The interconnection between the visualization and external data sources is realized by the exchange of the IDs of passed objects. The tutorial with commented examples demonstrating all steps of InCHlib deployment is available at http://openscreen.cz/software/inchlib/examples/18.
Input format
InCHlib is a visualization library and is, thus, not responsible for data clustering. Instead, data must be clustered by an external program, such as inchlib_clust (see the ‘inchlib_clust’ paragraph) and then passed into InCHlib either as a JavaScript variable or as a file stored in the InCHlib input format. The InCHlib input conforms the JSON (JavaScript Object Notation) standard [44]. Key elements of the InCHlib input format are demonstrated by the code snippets in this section and the complete example of the input file is given in Additional file 1.
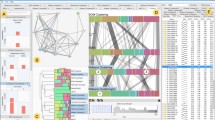
The input format describes three parts cluster heatmap visualization consists of: data, metadata and column dendrogram (Figure 1). The data block contains the data matrix and describes the structure of the row dendrogram. The row dendrogram consists of inner and terminal (usually referred to as leaves) nodes connected by branches. Each leaf is associated with one ‘data item’, i.e., with one heatmap row. Each data item corresponds either to one data point or, if the row reduction is used (see further), to several data points merged into one. Each data item is annotated with the IDs of data points it comprises of. The following code snippet demonstrates how the leaf is described in the InCHlib format.

An example cluster heatmap. The visualization consists of three blocks described in the InCHlib input format. The data block (green) contains feature names and the data for the rendering of the row dendrogram and heatmap. The metadata block (red) contains the additional data that are appended to the original data after clustering. The column dendrogram block (blue) contains the data for the rendering of column dendrogram.
"leaf_1": { //ID of the node (leaf)
"count": 1, //number of objects (heatmap rows) which lie in the dendrogram hierarchy below the given node
"distance": 0, //distance in dendrogram measured from leaves to the root node given by the distance measure used for the clustering
"features": [1.4, 3.5, 5.1], //values of individual features forming a data item
"parent": "node_1", //the ID of a parent node
"objects": ["object_1", “object_2”] // IDs of data points represented by the given row
},
Each node is identified by a unique ID string. While each inner node has two children, no child exists for the leaf. Children of a node are given as the left_child and right_child parameters. ID of the parent’s node is given as the parent parameter. The only node without the parent is the root node of the dendrogram. The following code snippet demonstrates how the node is described in the InCHlib format.
"node_1": { //ID of the node
"count": 3, //number of objects (heatmap rows) which lie in the dendrogram hierarchy below the given node
"distance": 3.32, //distance from the zero base of the dendrogram, given by the distance measure used for clustering
"parent": "node_1", //the ID of a parent node
"left_child": "leaf_1", //ID of a left child
"right_child": "leaf_2" //ID of a right child
},
The metadata block (Figure 1) describes additional information associated with individual data items, such as class membership. The metadata, displayed as additional column(s) in the heatmap, have no influence on the order of data items because they are not subjected to the clustering. The following code snippet shows how the metadata are described in the InCHlib format.
"metadata": { //contains nodes and feature_names section of metadata
"feature_names": ["Numeric", "Categoric"], //names of metadata features
"nodes": { //contains object IDs with metadata features
"leaf_1": [0.03, "positive"], // metadata features
"leaf_2": [0.02, "negative"]
}
},
The column dendrogram block (Figure 1) of the InCHlib input format describes the vertical dendrogram and has the same structure as the row dendrogram. The only difference is that leaves don't have the features and objects parameters inchlib_clust.
To facilitate the preparation of data in the InCHlib format, we developed a utility script inchlib_clust. inchlib_clust is written in Python 2.7 programming language. It performs both data preprocessing, such as data normalization or compression, and hierarchical clustering. Hierarchical clustering in inchlib_clust is accomplished by the fastcluster[45] library that implements several common hierarchical clustering schemes. List of available fastcluster linkages and distances is given in Additional file 2. Clustering results are saved in the InCHlib input file that can be readily passed into InCHlib. inchlib_clust can be easily extended by other hierarchical clustering approaches, such as by the popular Super Paramagnetic Clustering (SPC) [46],[47] which scales more favourably (as O(N)) than the O(N2) implementation of hierarchical clustering in fastcluster.
Data normalization is a preprocessing step used to balance the influence of features measured at different scales. inchlib_clust enables features to be scaled to the range between 0 and 1 using the MinMax scaler. MinMax scaler transforms the original feature x into its normalized version x ' according to the formula
where min(x) and max(x) are minimum and maximum values of the feature x. If the data normalization is used, the order of the heatmap rows (i.e., the row dendrogram) is always given by the clustering of the normalized data. However, the user can choose whether the normalized or original data will be displayed in the heatmap.
Because the speed of rendering decreases as the number of rows increases (see the ‘Performance assessment’ paragraph), inchlib_clust also enables to reduce the size of the data matrix. To increase the speed of visualization, as well as to reveal new data motifs by noise suppression, the number of the data matrix rows can be reduced. In row reduction, similar rows are aggregated into a single vector. Elements of this vector are calculated as the mean or median values of the elements of original rows. The extent of the compression is given as the number of reduced data matrix rows.
Another possibility how to speed up the visualization is to completely hide the data heatmap. In such case, only the dendrogram and metadata are displayed. This option comes in handy when the number of dimensions (columns) is too high, such as in the case of hashed chemical fingerprints.
Results and discussion
In this section, a typical InCHlib use consisting of data preparation and web page deployment is described. In addition, advanced InCHlib capabilities are demonstrated on the clustering of the ligands of estrogen receptor α (ERα). Finally, the speed of both data clustering by inchlib_clust and data visualization by InCHlib is evaluated.
The deployment of InCHlib consists of several steps (Figure 2): data preparation, data clustering, web page integration and cluster heatmap visualization.

The InCHlib deployment pipeline. It consists of three steps: data preparation, clustering and rendering. In the data preparation step, the data matrix consisting of data points and their IDs in rows and their features in columns is stored in the text file. Similarly, metadata are saved in a separate text file. In the clustering step, these files are supplied to the software that performs hierarchical clustering and stores the results in the InCHlib input file. Though any clustering software can be used, a utility script inchlib_clust that uses fastcluster library for clustering and outputs data directly in the InCHlib format was developed. In the third step, the InCHlib input file is read in by InCHlib which renders the cluster heatmap visualization.
Though data can be clustered by inchlib_clust, any clustering software can be used provided that the valid InCHlib input file is generated. Typically, the data matrix is supplied to inchlib_clust in a comma-separated values (csv) file, though other delimiters, such as tab or semicolon, are also possible. The data matrix consists of data points in rows and their features in columns. The first column always contains the IDs of individual rows. Optionally, feature names are given in the first row. The example of the data file is given in Additional file 3. Similarly, metadata are supplemented as a separate file using the same format. More metadata columns can be specified, and the metadata can be both numerical (e.g., EC50) or categorical (e.g., class membership). The metadata are associated with the corresponding data through their respective IDs. The example of the metadata file is given in Additional file 4.
Clustering
The only mandatory input to inchlib_clust is the data matrix stored in the csv file. If default parameters are used, no data scaling or row compression is applied and the data are clustered by rows using Ward’s clustering with the Euclidean distance. For example, to cluster the data stored in the example_data.csv file using the Ward’s clustering with the Euclidean distance, the following command line is used:
python inchlib_clust.py example_data.csv –m example_metadata.csv -dh –mh –a both -o example.json
In this case, the metadata are supplied (option -m) as the example_metadata.csv file, and both data and metadata contain column headers (−dh and -mh options). The data are clustered both by rows and columns (−a both option). The output file example.json (Additional file 1) contains the cluster heatmap in the InCHlib input format. Besides the command line interface, inchlib_clust also offers the application programming interface (API) and can, thus, be invoked from the user code. The use of inchlib_clust API from the Python script is demonstrated in Additional file 5.
Once the InCHlib input file is created, it is read by InCHlib and the cluster heatmap is visualized. Prior calling InCHlib functions, KineticJS and jQuery libraries must be imported. Then, the InCHlib object is instantiated with the settings parameter (given as the JavaScript object), the JSON file is read using the read_data_from_file() method and the cluster heatmap is rendered by calling the draw() method. The only obligatory attribute of the settings parameter is the target attribute that defines the id of the HTML element the cluster heatmap is inserted in. Other optional attributes of the settings parameter influence the appearance of the visualization (e.g., colors or size attributes) or of its individual parts (e.g., row dendrogram, column dendrogram, heatmap or metadata attributes). The example of the HTML/JavaScript code demonstrating InCHlib web page integration is given in Additional file 6. The resulting web page with commented HTML/JavaScript code is shown in Additional file 2.
Use case
In this section, the use of InCHlib for the exploration of the estrogen receptor α (ERα) ligand binding will be demonstrated. ERα belongs to the family of steroid hormone receptors [48], ligand-inducible transcription factors that control essential physiological, developmental, reproductive and metabolic processes [49],[50]. ERs are overexpressed in around 70% of breast cancer cases [51] and have also been implicated in ovarian, colon and prostate cancers. Thus, ERs represent an important target for therapeutic intervention [52].
The analysed data consist of 8 physico-chemical and structural properties of 195 ERα ligands obtained from the ChEMBL database [53]. The ligand properties were calculated by the RDKit cheminformatics toolkit [54] and they include the logarithm of the octanol-water partition coefficient (logP), molar refractivity (SMR), topological polar surface area (TPSA), molecular weight, and number of rotatable bonds, hydrogen-bond donors, hydrogen-bond acceptors and aromatic rings. To each ligand, its metadata represented by the K i value (equilibrium dissociation constant determined in inhibition studies) is also assigned.
The results of the hierarchical Ward’s clustering with the Euclidean distance performed by inchlib_clust are shown in the left panel of Figure 3. In this heatmap, physico-chemical properties and K i show no clear relationship. However, the clustering is biased by the wide range of molecular weight (250 – 600 Da). Because the values of other features are from narrower intervals (e.g., logP has values between 3 and 6), molecular weight prevails and the data are clustered mainly by this descriptor. To remove this artefact, data were normalized to the scale between 0 and 1. After the normalization, the data became more ordered (Figure 3, right panel) and correlated with K i values. Such patterns indicate potential relationships between physico-chemical descriptors and biological activity.

The comparison of ERα ligand clusterings performed with original (left panel) and normalized (right panel) values. The data were clustered by Ward’s clustering with the Euclidean distance. In the case of the clustering of normalized data (right panel), original data values are depicted in the heatmap (parameter --write_original of inchlib_clust).
To facilitate the discovery of the structure-activity relationships, depictions of ligand structures are shown right of the cluster heatmap (Figure 4, left panel). This is achieved by handling the row_onmouseover event. This event is triggered upon hovering the mouse over the row and displays the ligand image. The ligand image, which is pre-generated by the chemoinformatics toolkit RDKit[54], is stored in the file CHEMBLID.png. For example, CHEMBL1276308.png contains the structure of mifepristone, the compound with the CHEMBL1276308 ID. The structure depiction is hyperlinked with the CHEMBL database and, upon clicking the structure image, the corresponding CHEMBL record opens in a new tab.

The cluster heatmap of ERα ligands enhanced by the structure visualization. On the left panel, the row_onmousover event is used for the visualization of molecular structure. On the right panel, the scaffold composition of the selected cluster is shown by handling the dendrogram_node_onclick event. If the scaffold image is clicked, the heatmap rows representing compounds with the selected scaffold are highlighted in red.
Though the depiction of molecular structures is useful, the next step in the discovery of structure-activity relationships is the so-called scaffold analysis. Molecular scaffold is the graph representation of a molecular core structure [55]. Molecular scaffolds were successfully applied, among other, to the diversity analysis [56],[57] of bioactive compounds [58]-[64]. In the ERα use case, molecular scaffolds are revealed when the cluster is selected (Figure 4, right panel). This is achieved by handling the dendrogram_node_onclick event. When the scaffold image is clicked, compounds with the given scaffold are highlighted (Figure 4, right panel). The colour of highlighted rows is set as a highlight_colors settings attribute on InCHlib instantiation; the default colour scheme is Reds. In the presented use case, scaffolds of all 195 ligands are extracted and their images are generated by the RDKit[54] toolkit. A unique ID is assigned to each scaffold and scaffold image is stored in the ID.png file. To display the scaffold images upon node clicking, we implemented the server-side Python function that accepts the list of compound IDs (CHEMBL IDs), extracts the molecular scaffold of each compound and groups the compounds with the identical scaffolds. The function returns an array of scaffold IDs with attached compound IDs. For example, the array [1, ["CHEMBL234638", "CHEMBL278703", "CHEMBL234633"]] contains 3 compounds that share a common scaffold with ID 1.
The ERα use case, as well other examples demonstrating the use of InCHlib for the exploration of protein structures, identification of gene expression patterns or classification of whiskies based on their taste characteristics, are available from http://openscreen.cz/software/inchlib/use_cases/13. In addition, their short description is given in Additional file 2.
Performance assessment
To assess the performance of inchlib_clust and InCHlib, the dependence of the speed of clustering (inchlib_clust) and rendering (InCHlib) on the data size was investigated. The data, consisting of randomly generated integers between 0 and 1 000, were clustered using the Euclidean distance and Ward’s linkage. Experiments were performed using the following computer configuration: Kubuntu 13.10, Chrome 33.0.1750.146, Intel Core i5-2400 CPU 3.10 GHz, 8 GB RAM, 120 GB solid-state drive (SSD).
Clustering time increases quadratically with the number of data points (Figure 5, top left panel) which corresponds to the O(N2) complexity of the implementation of the Ward linkage hierarchical clustering in the fastcluster library [45]. Similarly, memory requirements increase with the number of data points; while clustering of 10,000 data points required 0.5 GB of RAM, memory consumption grew up to 2 GB for clustering of 20,000 data points. Contrary to the quadratic increase in clustering time with the increase of the number of data points (i.e., rows of the data matrix), the dependence of the clustering speed on the number of features (i.e., columns of the data matrix) is linear (Figure 5, top right panel).

The speed of clustering and heatmap rendering. Top panel: the dependence of the speed of clustering by inchlib_clust on the number of data points (i.e., the number of data matrix rows) and on the number of features (i.e., the number of data matrix columns). Bottom panel: the dependence of the speed of rendering by InCHlib on the data size.
In addition to the performance of inchlib_clust, speed of InCHlib rendering was also investigated. InCHlib rendering time depends linearly on the number of data points (Figure 5, bottom panel). While the linear dependence is the feature of the InCHlib implementation, absolute rendering times are greatly influenced by the PC hardware and web browser in which the primary limiting factor is the speed of the JavaScript engine.
Conclusions
InCHlib is a browser independent JavaScript library that facilitates the uncluttered visualization, powerful exploration and easy web integration of the cluster heatmap. InCHlib is an interactive tool that enables the user to select individual or clustered heatmap rows, to zoom in and out of clusters or to flexibly modify heatmap appearance. The InCHlib application programming interface defines a rich set of events through which the visualization can be interconnected with external data sources and analysis tools. The cluster heatmap can be augmented with additional metadata displayed in a different colour scale. To reduce the size of the heatmap and to reveal unique motifs in the data, number of rows can be limited by using several averaging methods. The clustered data are passed into InCHlib in a JSON compliant input data format. To facilitate data clustering and InCHlib input preparation, the Python utility script inchlib_clust can be employed. Though InCHlib is primarily intended for the analysis of chemical or biological data, its application domain is not limited to the life sciences only. InCHlib has already been successfully deployed at the Institute of Molecular Genetics AS CR as the part of an high-throughput screening information management system used at CZ-OPENSCREEN: National Infrastructure for Chemical Biology. InCHlib and inchlib_clust are provided free for download, and InCHlib is also available as the BioJS[65] component.
Availability and requirements
Project name: InCHlib
Project home page:http://openscreen.cz/software/inchlib/home/, https://www.ebi.ac.uk/Tools/biojs/registry/Biojs.InCHlib.html
Operating system(s): platform independent
Programming language: JavaScript
Other requirements: Python 2.7 to run inchlib_clust
License: MIT
Any restrictions to use by non-academics: None
Authors’ contributions
DS and PB instigated the project, participated in the development of the software, collected data sets, proposed the use cases and drafted the manuscript. CŠ is the lead developer of InCHlib. He designed and implemented the application and all use cases, prepared the web pages, performed all tests and calculations and helped to draft the manuscript. All authors read and approved the final manuscript.
Additional files
References
Xu R, Wunsch D: Survey of clustering algorithms. IEEE Trans Neural Netw. 2005, 16 (3): 645-678.
MacCuish JD, MacCuish NE: Chemoinformatics applications of cluster analysis. Wiley Interdiscip Rev Comput Mol Sci. 2013, 4 (1): 34-48.
Downs GM, Barnard JM: Clustering methods and their uses in computational chemistry. Reviews in Computational Chemistry. Edited by: Lipkowitz KB, Boyd DB. 2002, VCH, New York, 1-40.
Gagarin A, Makarenkov V, Zentilli P: Using clustering techniques to improve hit selection in high-throughput screening. J Biomol Screen. 2006, 11 (8): 903-914.
Pu M, Hayashi T, Cottam H, Mulvaney J, Arkin M, Corr M, Carson D, Messer K: Analysis of high-throughput screening assays using cluster enrichment. Stat Med. 2012, 31 (30): 4175-4189.
Stanton DT, Morris TW, Roychoudhury S, Parker CN: Application of nearest-neighbor and cluster analyses in pharmaceutical lead discovery. J Chem Inf Comput Sci. 1999, 39 (1): 21-27.
Bender A, Glen RC: Molecular similarity: a key technique in molecular informatics. Org Biomol Chem. 2004, 2 (22): 3204-3218.
Perez JJ: Managing molecular diversity. Chem Soc Rev. 2005, 34 (2): 143-152.
Petrone PM, Wassermann AM, Lounkine E, Kutchukian P, Simms B, Jenkins J, Selzer P, Glick M: Biodiversity of small molecules–a new perspective in screening set selection. Drug Discov Today. 2013, 18 (13–14): 674-680.
Schuffenhauer A, Popov M, Schopfer U, Acklin P, Stanek J, Jacoby E: Molecular diversity management strategies for building and enhancement of diverse and focused lead discovery compound screening collections. Comb Chem High Throughput Screen. 2004, 7 (8): 771-781.
Olah MM, Bologa CG, Oprea TI: Strategies for compound selection. Curr Drug Discov Technol. 2004, 1 (3): 211-220.
Xu R, Wunsch DC: Clustering algorithms in biomedical research: a review. IEEE Rev Biomed Eng. 2010, 3: 120-154.
Weinstein JN, Myers TG, O'Connor PM, Friend SH, Fornace AJ, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD: An information-intensive approach to the molecular pharmacology of cancer. Science. 1997, 275 (5298): 343-349.
Wilkinson L, Friendly M: The history of the cluster heat map. Am Stat. 2009, 63 (2): 179-184.
Weinstein JN: Biochemistry. A postgenomic visual icon Science. 2008, 319 (5871): 1772-1773.
Team. RDC: R: a language and environment for statistical computing. Vienna, Austria: R Foundation for statistical computing; 2010. ., [http://www.gbif.org/resources/2585]
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5 (10): R80-
CIMminer.., [http://discover.nci.nih.gov/cimminer/home.do]
Eisen MB, Spellman PT, Brown PO, Botstein D: Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998, 95 (25): 14863-14868.
TreeView.., [http://rana.lbl.gov/EisenSoftware.htm]
Schroeder MP, Gonzalez-Perez A, Lopez-Bigas N: Visualizing multidimensional cancer genomics data. Genome Med. 2013, 5 (1): 9-
Dudoit S, Gentleman RC, Quackenbush J: Open source software for the analysis of microarray data.Biotechniques 2003, 34(Supp):45–51. ., [http://www.biotechniques.com/multimedia/archive/00072/Mar03Dudoit_72037a.pdf]
Saldanha AJ: Java Treeview-extensible visualization of microarray data. Bioinformatics. 2004, 20 (17): 3246-3248.
Zeeberg BR, Qin H, Narasimhan S, Sunshine M, Cao H, Kane DW, Reimers M, Stephens RM, Bryant D, Burt SK, Elnekave E, Hari DM, Wynn TA, Cunningham-Rundles C, Stewart DM, Nelson D, Weinstein JN: High-Throughput GoMiner, an 'industrial-strength' integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of Common Variable Immune Deficiency (CVID). BMC Bioinformatics. 2005, 6: 168-
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J: TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003, 34 (2): 374-378.
Sturn A, Quackenbush J, Trajanoski Z: Genesis: cluster analysis of microarray data. Bioinformatics. 2002, 18 (1): 207-208.
Usadel B, Nagel A, Steinhauser D, Gibon Y, Blasing OE, Redestig H, Sreenivasulu N, Krall L, Hannah MA, Poree F, Fernie AR, Stitt M: PageMan: an interactive ontology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinformatics. 2006, 7: 535-
Floratos A, Smith K, Ji Z, Watkinson J, Califano A: geWorkbench: an open source platform for integrative genomics. Bioinformatics. 2010, 26 (14): 1779-1780.
Lex A, Streit M, Schulz HJ, Partl C, Schmalstieg D, Park PJ, Gehlenborg N: StratomeX: visual analysis of large-scale heterogeneous genomics data for cancer subtype characterization. Comput Graph Forum. 2012, 31 (3): 1175-1184.
GENE-E.., [http://www.broadinstitute.org/cancer/software/GENE-E/]
Kim N, Park H, He N, Lee HY, Yoon S: QCanvas: an advanced tool for data clustering and visualization of genomics data. Genomics Inform. 2012, 10 (4): 263-265.
Perez-Llamas C, Lopez-Bigas N: Gitools: analysis and visualisation of genomic data using interactive heat-maps. PLoS One. 2011, 6 (5): e19541-
Zhu J, Sanborn JZ, Benz S, Szeto C, Hsu F, Kuhn RM, Karolchik D, Archie J, Lenburg ME, Esserman LJ, Kent WJ, Haussler D, Wang T: The UCSC cancer genomics browser. Nat Methods. 2009, 6 (4): 239-240.
Goldman M, Craft B, Swatloski T, Ellrott K, Cline M, Diekhans M, Ma S, Wilks C, Stuart J, Haussler D, Zhu J: The UCSC cancer genomics browser: update 2013. Nucleic Acids Res. 2013, 41 (Database issue): D949-D954.
Kapushesky M, Kemmeren P, Culhane AC, Durinck S, Ihmels J, Korner C, Kull M, Torrente A, Sarkans U, Vilo J, Brazma A: Expression Profiler: next generation–an online platform for analysis of microarray data. Nucleic Acids Res. 2004, 32 (Web Server issue): W465-W470.
Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, Conesa A, Tarraga J, Pascual-Montano A, Nogales-Cadenas R, Santoyo J, García F, Marbà M, Montaner D, Dopazo J: Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010, 38 (Web Server issue): W210-W213.
Next-generation clustered heatmaps.., [http://bioinformatics.mdanderson.org/main/NG-CHM:Overview]
Xia J, Lyle NH, Mayer ML, Pena OM, Hancock RE: INVEX–a web-based tool for integrative visualization of expression data. Bioinformatics. 2013, 29 (24): 3232-3234.
Deu-Pons J, Schroeder MP, Lopez-Bigas N: jHeatmap: an interactive heatmap viewer for the web. Bioinformatics. 2014, 30 (12): 2-
Yachdav G, Hecht M, Pasmanik-Chor M, Yeheskel A, Rost B: HeatMapViewer: interactive display of 2D data in biology. F1000Res. 2014, 3: 48-
CanvasXpress.., [http://www.canvasxpress.org/]
KineticJS.., [http://kineticjs.com/]
jQuery.., [http://jquery.com]
JSON (JavaScript Object Notation).., [http://json.org/]
Müllner D: Fastcluster: fast hierarchical, agglomerative clustering routines for r and python. J Stat Softw. 2013, 53 (9): 1-18.
Blatt M, Wiseman S, Domany E: Superparamagnetic clustering of data. Phys Rev Lett. 1996, 76 (18): 3251-3254.
Tetko IV, Facius A, Ruepp A, Mewes HW: Super paramagnetic clustering of protein sequences. BMC Bioinformatics. 2005, 6: 82-
Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM: The nuclear receptor superfamily: the second decade. Cell. 1995, 83 (6): 835-839.
Katzenellenbogen JA, Katzenellenbogen BS: Nuclear hormone receptors: ligand-activated regulators of transcription and diverse cell responses. Chem Biol. 1996, 3 (7): 529-536.
Whitfield GK, Jurutka PW, Haussler CA, Haussler MR: Steroid hormone receptors: evolution, ligands, and molecular basis of biologic function. J Cell Biochem. 1999, 33 (Suppl 32): 110-122.
Ali S, Coombes RC: Estrogen receptor alpha in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia. 2000, 5 (3): 271-281.
Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA: Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007, 87 (3): 905-931.
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP: ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40 (Database issue): D1100-D1107.
RDKit: cheminformatics and machine learning software.., [http://www.rdkit.org/]
Bemis GW, Murcko MA: The properties of known drugs. 1. Molecular frameworks. J Med Chem. 1996, 39 (15): 2887-2893.
Krier M, Bret G, Rognan D: Assessing the scaffold diversity of screening libraries. J Chem Inf Model. 2006, 46 (2): 512-524.
Medina-Franco JL, Martinez-Mayorga K, Bender A, Scior T: Scaffold diversity analysis of compound daft sets using an entropy-based measure. Qsar Comb Sci. 2009, 28 (11–12): 1551-1560.
Hu Y, Bajorath J: Scaffold distributions in bioactive molecules, clinical trials compounds, and drugs. ChemMedChem. 2010, 5 (2): 187-190.
Varin T, Schuffenhauer A, Ertl P, Renner S: Mining for bioactive scaffolds with scaffold networks: improved compound set enrichment from primary screening data. J Chem Inf Model. 2011, 51 (7): 1528-1538.
Grabowski K, Baringhaus KH, Schneider G: Scaffold diversity of natural products: inspiration for combinatorial library design. Nat Prod Rep. 2008, 25 (5): 892-904.
Lee ML, Schneider G: Scaffold architecture and pharmacophoric properties of natural products and trade drugs: application in the design of natural product-based combinatorial libraries. J Comb Chem. 2001, 3 (3): 284-289.
Hu Y, Bajorath J: Structural and potency relationships between scaffolds of compounds active against human targets. ChemMedChem. 2010, 5 (10): 1681-1685.
Hu Y, Bajorath J: Systematic identification of scaffolds representing compounds active against individual targets and single or multiple target families. J Chem Inf Model. 2013, 53 (2): 312-326.
Hu Y, Bajorath J: Many drugs contain unique scaffolds with varying structural relationships to scaffolds of currently available bioactive compounds. Eur J Med Chem. 2014, 76: 427-434.
Gomez J, Garcia LJ, Salazar GA, Villaveces J, Gore S, Garcia A, Martin MJ, Launay G, Alcantara R, Del-Toro N, Dumousseau M, Orchard S, Velankar S, Hermjakob H, Zong C, Ping P, Corpas M, Jiménez RC: BioJS: an open source JavaScript framework for biological data visualization. Bioinformatics. 2013, 29 (8): 1103-1104.
Acknowledgements
The authors would like to thank Dr. M. Kolář and Dr. David Sedlák for their valuable and constructive suggestions during the design and development of the InCHlib library. This work was supported by the research grant LO1220 of the Ministry of Education, Youth and Sports of the Czech Republic.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Škuta, C., Bartůněk, P. & Svozil, D. InCHlib – interactive cluster heatmap for web applications. J Cheminform 6, 44 (2014). https://doi.org/10.1186/s13321-014-0044-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13321-014-0044-4