
Abstract
Background
Chronic lung disease of prematurity, called bronchopulmonary dysplasia (BPD), lacks effective therapies, stressing the need for preclinical testing systems that reflect human pathology for identifying causal pathways and testing novel compounds. Alveolar organoids derived from human pluripotent stem cells (hPSC) are promising test platforms for studying distal airway diseases like BPD, but current protocols do not accurately replicate the distal niche environment of the native lung. Herein, we investigated the contributions of cellular constituents of the alveolus and fetal respiratory movements on hPSC-derived alveolar organoid formation.
Methods
Human PSCs were differentiated in 2D culture into lung progenitor cells (LPC) which were then further differentiated into alveolar organoids before and after removal of co-developing mesodermal cells. LPCs were also differentiated in Transwell® co-cultures with and without human fetal lung fibroblast. Forming organoids were subjected to phasic mechanical strain using a Flexcell® system. Differentiation within organoids and Transwell® cultures was assessed by flow cytometry, immunofluorescence, and qPCR for lung epithelial and alveolar markers of differentiation including GATA binding protein 6 (GATA 6), E-cadherin (CDH1), NK2 Homeobox 1 (NKX2-1), HT2-280, surfactant proteins B (SFTPB) and C (SFTPC).
Results
We observed that co-developing mesenchymal progenitors promote alveolar epithelial type 2 cell (AEC2) differentiation within hPSC-derived lung organoids. This mesenchymal effect on AEC2 differentiation was corroborated by co-culturing hPSC-NKX2-1+ lung progenitors with human embryonic lung fibroblasts. The stimulatory effect did not require direct contact between fibroblasts and NKX2-1+ lung progenitors. Additionally, we demonstrate that episodic mechanical deformation of hPSC-derived lung organoids, mimicking in situ fetal respiratory movements, increased AEC2 differentiation without affecting proximal epithelial differentiation.
Conclusion
Our data suggest that biophysical and mesenchymal components promote AEC2 differentiation within hPSC-derived distal organoids in vitro.
Similar content being viewed by others


Introduction
One of the leading morbidities in preterm infants is bronchopulmonary dysplasia (BPD). This disease has many causes, and the incidence has not decreased in over 4 decades. To lessen the burden of disease we need robust preclinical platforms to mimic lung pathology and to test novel compounds. Human alveolar organoids created by directed differentiation of human pluripotent stem cells (hPSC) are promising platforms to investigate well-known BPD risk factors like hyperoxia and overstretch on human alveolar growth and differentiation. Ample studies have demonstrated differentiation of hPSC (ESC and iPSC) towards alveolar progeny in 2D and 3D culture systems [1,2,3,4]. In general, published protocols recapitulate chemical cues present during development in a stepwise fashion. First, hPSCs are differentiated into NKX2-1+ lung progenitors [1, 2, 5,6,7,8]. Alveolar epithelial cells (AEC) are then derived from these lung progenitors [1, 7]. The most effective method involves sorting the progenitor cells into a homogenous population and culturing them in Matrigel [1, 4, 9]. In this hydrogel substratum, lung progenitors self-assemble into 3-dimensional (3D) structures called organoids [10]. These organoids, with appropriate chemical cues, promote the differentiation of alveolar epithelial type II cells (AEC2). However, the efficiency varies widely, ranging from 3 to 85%, depending on the specific protocol, the hPSC line used, as well as the seeding density [1, 2, 4, 9, 11, 12].
Ideally, alveolar organoids should mimic the alveolus in situ [13]. To generate such alveolar organoids a better understanding and replication of the in situ distal niche during development is needed. Most published protocols for generating alveolar organoids have ignored mesenchymal and biophysical cues. In vivo, epithelial-mesenchymal crosstalk is pivotal for proper lung development and AEC2 differentiation [14,15,16,17]. Several studies have shown that lung organoids derived from unsorted hPSC-endodermal cells undergoing directed differentiation to AEC2 contain mesenchymal cells [11, 12, 18,19,20]. Whether this emerging mesenchymal population has a positive effect on epithelial differentiation within the organoids is unknown. During development, the epithelium secretes fluid, which is circulated by the contraction of embryonic airway smooth muscles, creating a phasic hydrostatic distension of the developing airways [21]. Additionally, episodic fetal breathing movements (FBM) begin in the first trimester of pregnancy and change the distal lung surface area [22, 23]. Drainage of lung fluid volume [24] or abolition of FBM [25] in experimental animal models leads to lung hypoplasia, underscoring the importance of physical forces in lung development. It is unknown whether cyclic mechanical deformation represents a crucial biophysical cue for optimal AEC2 differentiation within hPSC-derived organoids. In the present study, we hypothesized that (1) co-developing mesenchymal cells within hPSC-derived lung organoids and (2) cyclic mechanical deformation, mimicking respiratory movements of the fetal lung, would enhance the differentiation of human lung progenitors into AEC2. Experimentally, we found evidence that both biophysical and mesenchymal cues stimulated AEC2 differentiation. These findings may aid in the development of alveolar organoid testing platforms for distal lung diseases including BPD.
Methods
Maintenance of human pluripotent cells
The human pluripotent cell lines were utilized in accordance with the guidelines provided by the Stem Cell Oversight Committee of The Canadian Institute of Health Research. Human CA1 ES and NCRM1 iPS cells were generous gifts of Dr. Andras Nagy, Lunenfeld Research Institute, Sinai Health, Toronto, and the Center for Commercialization of Regenerative Medicine, Toronto, respectively. The cells were cultured in feeder-free conditions on Matrigel-coated (Matrigel®GFR; Corning, #354230) plates in mTeSR™ Plus media (StemCell Technologies, #05825) with 1% (v/v) penicillin/streptomycin (Gibco, #15070-063). Cells were passaged weekly using ReLeSR™ (StemCell Technologies, #05825) and Gentle Cell Dissociation Reagent (GCDR, Stemcell, #07174), respectively. Cultures were maintained at 5% CO2 in ambient air (21% O2) at 37 °C. Lentiviral infection of ES and iPS cells with hSFTPCGFP reporter is detailed in the online supplement.
Differentiation into NKX2-1+ lung progenitor cells
The differentiation protocol (Fig. 1A) was adapted with minor modifications from Yamamoto et al. [4, 26] and is described in detail in the online supplement. Briefly, human ES and/or iPS cells were seeded at a density of 1.5 × 106 cells/well onto 6-well ultra-low attachment plates and differentiated into definitive endoderm in RPMI1640 (Gibco, #118755-119) supplemented with 2% B27 without vitamin A (Gibco, #12587-010), 100 ng/mL Activin A (Stem Cell Technologies, #78132), 1 µM CHIR99021 (Tocris, #4423) and 1% (v/v) penicillin/streptomycin (Gibco, #15140-122). On Day 6 of differentiation, cells were harvested with TrypLE™ Express (Gibco, #12605028) and analysed by flow cytometry for definitive endoderm (DE) markers c-KIT and CRCX4 (Fig. S1B). Efficiency of DE induction (% c-KIT+/CXCR4+ cells) was consistently greater than 85%. DE cells were further differentiated to ventral anterior foregut endoderm (VAFE) by seeding unsorted cells onto 6-well Matrigel-coated plates (1.0 × 106 cells/well) in serum-free differentiation media (SFDM) containing 0.5 µM dorsomorphin homologue 1 (DMH1) (StemCell Technologies, #73634) and 10 µM SB431542 (Tocris, #1614) for anterior foregut (AFE) patterning. SFDM consisted of 1:1 (v/v) DMEM/F12 with 2% B27, 1% N2 (Gibco, #17502-048), 0.05 mg/mL l-ascorbic acid (Sigma, #A92902), 1% (v/v) Glutamax (Gibco, #35050-061), 0.4 mM 1-thioglycerol (Sigma, #M6145) 1% (v/v) penicillin/streptomycin. On Day 11 of differentiation, cells were analysed by flow cytometry for the AFE surface markers CD56 and CD271 (Fig. S1B). Efficiency of AFE induction (% CD56+/CD271+ cells) was consistently greater than 65%. At day 11, medium was changed to SFDM supplemented with BMP4 (R&D system, #314-BP), 0.5 μM retinoic acid (Sigma, #R2625) and 3.5 μM CHIR99021 for ventral anterior foregut endoderm (VAFE) induction. On Day 15, cells were harvested with TrypLE™ Express, analysed for NKX2-1 expression by flow cytometry, re-seeded on Matrigel-coated plates (1.0 × 106 cells/well) and incubated for 7 days in SFDM without N2, supplemented with 10 ng/mL FGF7 (StemCell Technologies, #78046.1), 10 ng/mL FGF10 (StemCell Technologies, #78037.1) and 3 μM CHIR99021 for lung progenitor cell (LPC) formation. Medium was changed every other day. For Notch-inhibited cultures, 20 µM DAPT (Stem Cell Technologies) was added. All differentiations were performed at 37 °C in a 5% CO2 and 5% O2 environment. On Day 22 of differentiation, cells were dissociated with TrypLE™ Express in single cells, passed through a 40-μm pore size cell strainer, and analysed by flow cytometry for CD26/CD47, CPM and NKX2-1 expression—markers of LPC (Fig. S1B). For source and dilution of antibodies see Tables S1A,B in the online supplement.

Organoid size depends on lung progenitor cell seeding density. A Directed differentiation schematic for derivation of NKX2-1+ lung progenitor (LPC) cells. B Efficiency of NKX2-1 specification at day 22 (n = 25 biological replicates; a.k.a. separate differentiations) analyzed via flow cytometry and immunofluorescence. A threshold of ≥ 50% NKX2-1+ cells was used for experiments involving further differentiation to alveolar epithelial type II (AEC2) cells. C Schematic depicting the directed differentiation of D22 LPC to HT2-280+/SFTPC+ AEC2 cells in organoid culture. D Representative brightfield images of organoid cultures throughout days 28 to 42 of differentiation from different seeding densities. E Representative H&E-stained organoid sections derived from different seeding densities at day 42 showing hollow structure. F Average organoid diameter in day 29 and day 36 organoid cultures from 2.5 × 104 or 10 × 104 LPCs. Calculations are based on the measurement of ≥ 180 organoids per condition for three biological replicates. G Percentage of Ki-67+ cells, as measured by flow cytometry, in day 29 and day 36 organoids from different seeding densities (mean ± SEM, n = 3 biological replicates. *p < 0.05 by one-way ANOVA)
Culture and irradiation of human embryonic lung fibroblasts
Human embryonic lung fibroblasts (hELF, 17.5 weeks of gestation, DV Biologics) were cultured in DMEM (Gibco, #11965118) containing 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin. Cells were passaged maximally 5 times. At 80–90% confluency, hELF were irradiated (Gammacell 40 SN 264) with 31,000 rad and then frozen in 40% DMEM, 50% FBS and 10% DMSO. Four days before experimentation, irradiated fibroblasts (ihELF) were thawed, seeded onto 12-well plates (3 × 105 cells/well) in DMEM supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin. Based on VivaFix 410/450 (Bio-Rad, #1351112) viability measurements > 80% of the ihELF cells remained viable for at least 3 weeks in culture.
Differentiation of NKX2-1+ lung progenitor cells into AEC2 cells
Only differentiations that yielded > 50% NKX2-1+ progenitors were used. The cells (2.5 × 104 or 10 × 104) were resuspended in a 400 μL 1:1 mixture of Matrigel® GFR and distalizing medium consisting of Ham’s F12-based SFDM containing 100 μM 8-Br-cAMP (Sigma-Aldrich, #B7880), 100 μM 3-Isobutyl-1-methylxanthine (IBMX) (Sigma-Aldrich, #I5879), 50 nM dexamethasone (Sigma, #D4902), 50–100 ng/mL FGF7, 10 µM SB431542, 3 μM CHIR99021, 0.25% (v/v) bovine albumin (Gibco, #15260-037), 15 mM HEPES (Sigma, #H-0891), 0.8 mM CaCl2 (Sigma-Aldrich, #449709), 0.1% (v/v) ITS Premix (Sigma, #13146) and 1% (v/v) penicillin/streptomycin. The cell suspension was loaded onto a Transwell® insert, placed in a well of a 12-well plate and 0.5 mL of AEC2 induction medium was added to the well and changed every other day (Fig. 1C).
For mechanical stretching, lung progenitor (> 50% NKX2-1+) cells (2.5 × 105/mL) were resuspended in a mixture of AEC2 induction medium and 80% Matrigel® GFR. One mL of cell suspension was then seeded into each well of a 6-well tissue train® circular foam culture plate (FlexCell, TTCF-5001U) and incubated at 37 °C for 2 h to facilitate gelation. Three mL of induction medium was added which was changed every 2–3 days. Mechanical stretching was implemented 3 days post-seeding, the tissue train circular foam culture plate was connected to the FX-4000 Tension System (FlexCell). Settings were set to mimic fetal breathing movements, i,e, 5% elongation, frequencies of 1 Hz, square waveform, continuous cycle of 15 min ON and 45 min OFF [27]. Organoids were subjected to this phasic strain regimen for 21 days. All cultures were maintained at 37 °C in a 5% CO2 and 5% O2 environment.
Sorting CPM+ lung progenitor cells
On Day 22 of differentiation, cells were harvested with TrypLE™ Express and passed through a 40-μm pore size cell strainer. Cells were stained with unconjugated mouse anti-human CPM monoclonal antibody for 30 min at 4 °C and subsequently incubated with Alexa Fluor 647 donkey anti-mouse IgG and VivaFix 410/450 viability dye for 30 min at 4 °C. A MoFlo XDP U/VBR (Beckman Coulter) sorter was used for isolating viable CPM+ cells. For source and dilution of antibodies see Tables S1A/B in the online supplement.
Flow cytometry and immunofluorescence analysis
Flow cytometry and immunofluorescence was conducted as previously described [28, 29]. The procedure of both methods is detailed in the online supplement. Antibodies and their concentrations are described in Tables S1A/B in the online supplement.
Electron microscopy
Samples processing and microscopy was performed as previously reported [30]. Details are described in the online Supplement.
RNA isolation, cDNA preparation and real-time PCR
Total RNA was isolated from cells and organoids using the PureLink RNA Micro kit (Thermo Fisher Scientific, #12183016) and reverse transcribed with the SuperScript™ IV SuperScript IV First-Strand Synthesis System (Invitrogen, #18091050). PCR amplification was conducted using the SYBR Select Master Mix (Applied Biosystems, #34472908) and probes targeting various genes of interest are listed in Table S2 in the online supplement. Gene expression was normalized to beta-actin (ACTB) and expressed relative to selected appropriate positive or negative controls.
Measuring organoid diameter
Organoid diameter was measured using the built-in scale bar function on the EVOS M5000 imaging system (ThermoFisher Scientific). Most organoids had a circular appearance (Fig. 1D) with an aspect ratio (width over length) ranging between 0.85 and 1.15. To obtain the mean of length diameter of each biological replicate, more than 150 organoids from each condition were imaged and measured.
Matrigel stiffness
The mechanical properties of 50 versus 80% Matrigel prior to cell seeding were assessed by atomic force microscopy. The 50 and 80% Matrigel samples were produced by mixing pure Matrigel with ice-cold base media used for cell differentiation. The Matrigel mixtures were cold-pipetted into 3D-printed wells mounted on AFM specimen discs. Specimens were made approximately 2 mm thick to avoid substrate effects during indentation. The samples were placed in Petri dishes sealed with paraffin film to avoid evaporation and were allowed to polymerize at 37 °C overnight. A JPK atomic force microscope (Nano-wizard 4, Bruker Nano, Berlin, Germany) coupled to an inverted Zeiss Axiovert 200 M microscope (Carl Zeiss Microscopy, Göttingen, Germany) was used for the indentation tests. Soft tip-less non-conductive silicon nitride probes (NP-O10-D nominal spring constant of 0.06 N/m, and a nominal resonance frequency of 18 kHz) (Bruker, Mannheim, Germany) were functionalized using 10 µm radius spherical polystyrene beads (Phosphorex Inc. Hopkinton, MA). Prior to each test, the deflection sensitivity of the cantilever was calibrated by engaging the cantilever on the surface of a clean glass slide. The precise spring constant of the cantilever was calibrated with the thermal noise fluctuations in air by fitting the first free resonance peak of the AFM cantilever to that of a simple harmonic oscillator using the JPK software. Using the contact mode force spectroscopy of the JPK AFM, 4 by 4 points on the gel were indented in each of at least 6 different locations (each location an area of 60 micron by 60 micron). All AFM measurements were performed at room temperature with the samples being submerged in PBS. Elastic moduli were estimated from the force–deflection extend curves for each sample, analyzed based on the Hertz/Sneddon model using JPK Data Processing software (version 6.3.11).
Statistical analysis
All data are presented as mean ± standard error of mean (SEM). Statistical analysis was performed using GraphPad Prism Version 7.0.1 software. For multiple groups, one-way ANOVA followed by Tukey’s Multiple Comparisons Test was used. For less than three groups, a paired or unpaired Student t-test was used if both distributions were normal while a Mann–Whitney test was used if normality was not confirmed. Single asterisks (*) indicate statistical significance of p < 0.05.
Results
Higher density seeding of lung progenitor cells improves AEC2 differentiation in organoids
Using an adapted protocol [4, 26], we differentiated human CA1 ES cells to NKX2-1+ lung progenitor (LPC) cells (Figs. 1A and S1). In contrast to the published protocol, we did not observe any benefit from adding DAPT, a γ-secretase inhibitor of the NOTCH pathway, to the media for differentiating ventral anterior foregut endoderm (VAFE) to LPCs (Fig. S2). We introduced DAPT at different time points and durations during the VAFE to LPC differentiation period (Fig. S2A) and validated its inhibition of NOTCH (Fig. S2B) but did not find any effect on NKX2-1+ LPC induction (Fig. S2C). However, extending the original duration of differentiation of AFE to LPC from D11-15 [4, 26] to D11-22 doubled the induction efficiency of NKX2.1+ cells from 21.79 ± 4.21% at D15 (n = 14 biological replicates) to 44.35 ± 3.32% at D22 (n = 25 biological replicates; a.k.a. 25 separate differentiations) (Fig. 1B). Similar NKX2-1 induction efficiencies (45–55%) at D22 of differentiation were observed with CA1 ES and NCRM1 iPS cells that both were stably transfected with a hSFTPCGFP reporter (Fig. S3B). The flow cytometry data was corroborated by nuclear immunostaining of the cells for NKX2-1 (Figs. 1B and S3B). Flow cytometry and real-time PCR analysis also indicated a greater commitment of LPCs to SOX9+ than SOX2+ progenitor lineages (Fig. S3C). When more than 50% of the LPC population at D22 was positive for NKX2-1 [19], cells were embedded in a 1:1 diluted (50%) Matrigel for organoid formation and then incubated in a distalizing medium promoting AEC2 induction (Fig. 1C).
Organoid size and number, but not morphology, were dependent on the seeding density of CA1 LPCs (Fig. 1D, E). Cultures started with 2.5 × 104 LPCs yielded less organoids than a fourfold higher concentration of LPCs. Moreover, organoids in cultures started with 2.5 × 104 LPCs were on average double the size of those found in the higher density seeded cultures (215.95 ± 11.45 μm vs. 129.12 ± 5.66 μm diameter at D36) (Fig. 1F). Independent of seeding density, cells within the organoids were highly proliferative based on Ki-67 expression (Fig. 1G). In both cultures, we observed a slight but non-significant decrease in Ki-67+ cells at D36 versus D29, suggesting a small reduction in proliferation as differentiation progressed. Thus, seeding density of LPCs affects the size of the organoids, especially on D36 of differentiation.
We next investigated whether seeding density (i.e., size of the organoid) impacted AEC2 differentiation. Flow cytometry for HT2-280, a human AEC2 surface marker [31], revealed an increase in HT2-280 expression over time under both seeding conditions (Fig. 2A, B). The percentage of HT2-280+ cells at D43 of differentiation was significantly greater in high-density versus low-density seeded (i.e., smaller vs. larger) organoid cultures (20.15 ± 6.05% vs. 7.20 ± 1.20%, p < 0.05). However, the percentage of CDH1 (E-cadherin) positive epithelial cells was significantly lower in high-density compared to low-density seeded organoid cultures. The efficiency in AEC2 differentiation was validated with CA1 and NCRM1 cells transduced with a hSFTPCGFP reporter (Fig. S4). The hSFTPC promoter has previously been validated to label murine AEC2 in situ [32, 33]. Immunostaining of D43 CA1-SFTPCGFP organoid cells (Fig. S4A) revealed that GFP+ cells, representing SFTPC expression, were positive for CDH1 (see inlet), and that 16.81 ± 1.22% (n = 3 biological replicates) of the organoid cells stained positive for HT2-280 (Fig. S4C). Flow cytometry for GFP (Fig. S4A-right panel) of D43 organoids formed from 105 CA1-hSFTPCGFP or NCRM1-hSFTPCGFP LPCs demonstrated that 20.42 ± 2.76% and 21.06 ± 4.40%, respectively, of the organoid cells expressed GFP (Fig. S4B), matching the differentiation efficiency of LPCs into AEC2 when using HT2-280 as readout (Fig. 2B) and was in line with the immunostaining findings.

Higher density seeding of lung progenitor cells improves AEC2 differentiation in organoids. A Percentage of HT2-280+ and CDH1+ cells from different seeding densities in D29, D36 and D43 organoids assessed by flow cytometry (mean ± SEM, n ≥ 3 biological replicates, *p < 0.05 by one-way ANOVA). Inviable cells were identified and removed from flow cytometric analysis using the VivaFix 410/450 viability stain. B Representative flow cytometry density plots of HT2-280+ cells in D43 organoid cultures from 2.5 × 104 and 10 × 104 LPCs. Left plot: control density plots—PE-conjugated IgG1 isotype only. (C) Gene expression measured by real-time PCR of distal (AEC2: SFTPB, SFTPC) and proximal (FOXJ1: ciliated cells; MUC5AC: goblet cells; SCGB1A1: club cells) lung cell markers in day 43 organoids from different seeding densities relative to day 22 LPCs (mean ± SEM, n ≥ 3 biological replicates, *p 0.05 or **p < 0.01 by unpaired Student t-test)
We then assessed whether time in culture increased AEC2 differentiation. We dissociated the organoids and passaged them into fresh Matrigel at D14 after the initial start of organoid culture from LPCs, extending the duration of organoid differentiation by another week (D50 instead of D43). This did not result in a further increase hSFTPC-driven GFP expression (Fig. S4D). Real-time PCR analysis of GATA6, a transcription factor essential for distal lung epithelial differentiation [34, 35], and AEC2 markers SFTPB and SFTPC substantiated the HT2-280 and GFP cytometry findings. In both low- and high-density seeded organoid cultures, GATA6, SFTPB, and SFTPC mRNA expression increased compared to either ESC/iPSC (Fig. S5) or D22 LPCs (Fig. 2C), with high-density seeded organoids exhibiting the highest expression.
To visualise the diversity of cell types within D43 organoid cultures, we conducted immunofluorescence confocal microscopy for distal and proximal epithelial lung markers. Consistent with the gene expression findings, we identified pro-SFTPB+ and HT-280+ cells (Fig. S6A-bottom panels) within the CDH1+/NKX2-1+ (Fig. S6A-top panels) organoids at D43. Positive immunostaining for mature SFTPB (mSFTB, see inlets of pro-SFTB images) indicated the presence of cells capable of processing pro-SFTPB into its mature form. HTII-280 staining was apical within the lumen of the organoids, in line with an apical-in orientation. Immunostaining for SCGB1A1 (club cell marker) and TUBB4A (ciliated cell marker) was negative, aligning with the reduction in proximal gene expression Fig. S6A-middle panels). Ultrastructural electron microscopy (EM) analysis of the D43 organoids revealed cell junctions between epithelial cells with microvilli at the apical surface (Fig. S6B-left panels). Immuno-gold EM visualized mature SFTPB within cytoplasmic pre-lamellar multivesicular structures (Fig. S6B-left panels), consistent with previous reports for fetal AEC2 in situ [36].
Removal of mesenchymal progenitor cells limits AEC2 differentiation within organoids
We next enriched the CA1 NKX2-1+ expressing population before organoid formation using carboxypeptidase M (CPM), an enzyme present on the surface of lung progenitor cells [1]. Sorting for CPM+ cells at D22 (43.43 ± 3.48% of total cells were CPM+ at D22, n = 8 biological replicates; Fig. 3A) resulted in an enrichment of the NKX2-1 expressing LPCs from 44.35 ± 3.32% to 80.54 ± 4.05% (Fig. 3B). NKX2-1 immunostaining further confirmed the enrichment in the number of NKX2-1+ LPCs (Fig. 3B). As depicted in Fig. 3C, 105 CPM+-sorted LPCs formed organoids within a few days of seeding [1, 37] that were similar in shape and size to organoids formed from unsorted (> 50% NKX-2-1+) LPCs (Figs. 1D, 2, and 3C). Surprisingly, the percentage of HT2-280+ cells in D43 organoids established with CPM+-sorted LPCs was significantly lower compared to organoids formed with unsorted LPCs (Fig. 3D), while the percentage of CDH1+ epithelial cells remained unchanged. We hypothesized that the reduction of HT2-280+ cells in the organoids formed from CPM+-sorted LPCs occurred due to the loss of mesodermal cells, which are known to be essential for distal epithelial differentiation [16, 38,39,40]. In our differentiation protocol (Fig. 1A), we did not sort for a pure definitive endoderm population as ≥ 85% of the cells at D6 of differentiation were double positive for c-KIT and CXCR4 (Figs. S1B and S3A). Therefore, we started our differentiation towards AEC2 with a definitive endoderm population that was contaminated by other lineages, including mesoderm [41, 42]. By sorting the D22 cell population for CPM+ cells, we likely removed most of the mesodermal progenitor cells that had emerged during the differentiation protocol [11, 12, 18, 20]. To verify, we compared the expression of mesenchymal markers vimentin (VIM) and actin alpha 2 (ACTA2) in our CPM+-sorted versus unsorted organoid cultures [40, 43, 44]. Immunofluorescence confocal microscopy revealed the presence of VIM+ cells in the unsorted organoid cultures, while VIM+ cells were sparse in the CPM+-sorted organoid cultures (Fig. 4A). Reduced VIM and ACTA2 expression in the CPM+-sorted compared to unsorted organoid cultures corroborated the immunofluorescence findings (Fig. 4B). Together, the findings suggest that organoids formed with CPM+-sorted LPCs contain fewer mesenchymal progenitor cells. The loss in VIM+ cells could also be due to less epithelial-mesenchymal transition in the sorted LPC organoid cultures, but the similar percentages of CDH1+ epithelial cells in the unsorted and sorted LPC organoid cultures (Fig. 3D) argue against epithelial dedifferentiation. Mesenchymal support for LPC differentiation into AEC2 was corroborated by incubating LPCs in the absence and presence of human embryonic lung fibroblasts (hELF). Human ELFs were mitotically inactivated by irradiation (ihELF) to prevent overgrowth during co-culturing without influencing their secretome [45, 46]. D22 NKX2-1+ LPCs were grown on Transwell® inserts pre-coated with either Matrigel, Laminin, or Collagen type IV (the latter two matrices are the main components of Matrigel). The inserts were cultured in distalizing medium with and without ihELFs seeded in the bottom wells of the Transwell® plates. The percentage of CDH1+ cells did not differ between LPCs cultured for 22 days with or without ihELFs (Fig. 5A). However, independent of the coated matrix, co-culture with ihELFs increased the percentage of HT2-280+ cells (Fig. 5A), confirming the importance of mesenchymal-epithelial crosstalk for fetal AEC2 differentiation [15, 16, 38]. Direct contact between ihELF and LPCs within organoids did not further improve HT-280 induction above indirect co-culturing (Fig. 5B).

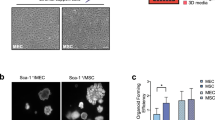
Enriching the NKX2-1+ lung progenitor population does not increase HT-280+ AEC2 differentiation in organoids. A Gating strategy to sort for CPM+ lung progenitor cells on day 22 (D22) of differentiation. B Left panel: Percentage of NKX2-1 positive cells of CPM+-sorted and unsorted D22 LPCs (n = 4 biological replicates, *p < 0.05 by unpaired Student t-test). Right panel: Representative immunofluorescence staining for NKX2-1 of CPM+-sorted and unsorted D22 LPCs. C Representative brightfield images of organoid formation by 105 CPM+ sorted LPCs at days 28–39. D Percentage of HT2-280+ and CDH1+ cells in D43 organoids formed with 105 CPM+ sorted versus unsorted LPCs. Unviable cells were identified and removed from flow cytometric analysis using the VivaFix 410/450 viability stain (n = 4 biological replicates, *p < 0.05 by unpaired Student t-test)

Mesenchymal cells are reduced in organoids formed with CPM+ (NKX2-1 enriched) sorted lung progenitor cells. A Representative immunofluorescence images of epithelial (CDH1) and mesenchymal (VIM) cell markers in day 43 organoids formed with CPM+-sorted versus unsorted LPCs. B Gene expression measured by real-time PCR of VIM and ACTA2 in day 43 organoids from CPM+-sorted versus unsorted LPCs relative to day 22 lung progenitors (mean ± SEM, n = 4 biological replicates. *p < 0.05 by paired Student t-test)

Embryonic lung fibroblasts stimulate the differentiation of lung progenitors into HT-280+ AEC2 cells. A Top panel: Schematic of the indirect Transwell® co-culture system to determine the effect of irradiated human embryonic lung fibroblasts (ihELF) on the differentiation of D22 NKX2-1+ LPCs into AEC2. Bottom panel: Flow cytometry analysis for CDH1 and HT2-280 of organoids grown in different matrices. Data are expressed as mean ± SEM, n = 4 biological replicates. Dashed lines indicate mean % of HT2-280 positive cells, respectively, after combining the matrix data in both groups. *p < 0.05 by Mann–Whitney U test, no ihELF versus ihELF. B Left panel: Schematic of experimental design to determine the effect of direct versus indirect contact of LPCs with ihELF on the differentiation of D22 NKX2-1+ LPCs to AEC2. Right panel: Flow cytometry analysis for HT2-280 of organoids grown directly versus indirectly with ihELF after 20 days of culture. Data are expressed as mean ± SEM, n ≥ 10 biological replicates
Biophysical forces stimulate AEC2 differentiation within organoids
To examine whether mechanical strain affects the differentiation of LPC into AEC2 within organoids, we recapitulated fetal breathing movements in vitro using a 3D stretching device (Fig. 6A). Within 3 days of culture of D22 unsorted (> 50% NKX2-1+) LPCs in 80% Matrigel (concentration required for applying strain to organoids using the FlexCell® system), organoid structures began to form. No apparent differences were observed in the morphology of organoids in static compared to stretched conditions (Fig. 6B). The size of the organoids did not change with periodic stretching compared to static (non-stretched) controls (128.10 ± 7.54 vs. 134.02 ± 22.12 mm, mean ± range, n = 3 biological repeats, static vs. stretch at D42). Flow cytometry for Ki-67 and CDH1 in episodically stretched organoids and paired static (non-stretched) controls demonstrated no significant differences in proliferation and epithelial lineage expression, respectively (Figs. 6C and S7). However, flow cytometry for HT2-280 revealed that episodically stretched organoids had significantly more HT2-280+ cells than organoids from static (non-stretched) cultures (Figs. 6D and S7). Increased gene expression of SFTPC in the stretched organoids corroborated these HT2-280 findings (Fig. 6D). Gene expression of FOXJ1 and SCGB1A1 was similar between static and stretched organoids (Fig. 6D), suggesting that episodic stretching does not contribute to the commitment of LPCs to proximal epithelial lung lineages. To exclude that LPC to AEC2 differentiation varied with the stiffness of the original Matrigel, we determined the elastic moduli of Matrigel prior to cell seeding by atomic force microscopy (AFM) micro-indentation. Two different batches of Matrigel gave similar results. The 80% Matrigel used in the episodic stretch experiments had a similar elastic modulus at room temperature as 50% Matrigel used in all other experiments (150 ± 13 Pa vs. 125 ± 12 Pa, n ≥ 17 measurements). Also, organoid size (128.10 ± 7.54 vs. 129.12 ± 5.66 μm diameter) and SFTPC induction (Fig. 6D vs. Fig. 2C) under static (non-stretched) conditions did not differ between 80 and 50% Matrigel.

Phasic mechanical stretch improves AEC2 differentiation in organoids. A Schematic outlining the parameters for episodic mechanical stretch from day 22 to 43. B Representative brightfield images of organoids formed under stretched and static conditions from day 25 to 42. C Flow cytometry analysis of Ki-67 and CDH1 in D43 stretched versus static organoid cultures. D Top panels: Flow cytometry and gene expression of AEC2 markers HT2-280 and SFTPC, respectively, in D43 stretched and static organoid cultures. Flow data for Ki-67 and HT2-280 data are presented as a ratio compared to paired static controls. Data are mean ± SEM, n = 4 biological replicates, *p < 0.05 by paired Student t-test. Bottom panels: Gene expression of proximal ciliated (FOXJ1) and club (SCGB1A1) cell markers in D43 stretched and static organoid cultures (mean ± SEM, n = 4 biological replicates)
Discussion
Replicating the alveolar microenvironment for lung regenerative purposes has been challenging. Cellular and chemical cues of the distal niche have been used to create complex human alveolar organoids using either fetal lung bud tip progenitors [47, 48] or adult AEC2 cells [10, 49]. The impact of cellular and biophysical cues on AEC2 differentiation within hPSC-derived organoids is not well understood. Here, we demonstrate that seeding density, mesenchymal presence and mechanical strain promote AEC2 differentiation within hPSC-derived organoids.
In the present study, our adapted directed differentiation protocol yielded 44–55% NKX2-1+ lung progenitor cells without sorting. This was significantly lower than the 85% reported by Yamamoto et al. [4], but it matched or exceeded what has been reported for other human cell lines [1, 2]. The variation in induction efficiency could be attributed to factors such as the cell line used, the composition of the media and growth factors, the type of extracellular matrix coating, or the methodology for evaluating NKX2-1+ expression. Immunostaining for NKX2-1+ consistently yielded higher percentages [1, 4] compared to NKX2-1GFP reporters [2]. We observed significant oscillation in the percentage of NKX2-1+ progenitors between experiments, consistent with previous reports [2]. This inter-experiment variability was independent of media composition, hPSC passage number, efficiency of differentiation into definitive endoderm, or cell seeding density. The induction efficiencies of NKX2-1+ LPCs at D22 were validated using CD26low/CD47high and CPM+ flow cytometry.
Herein, we demonstrated that the concentration of Matrigel did not affect organoid formation from LPCs. While Hawkins et al. [2] used pure Matrigel, we utilized 50% and 80% Matrigel to enrobe the cells. Despite this difference, we did not observe any significant variations in organoid formation between 50 and 80% Matrigel. Previous studies have shown that lineage differentiation of human lung progenitors can be influenced by matrix stiffness [50]. To investigate this further, we used AFM micro-indentation to measure the mechanical properties of 50% and 80% Matrigel prior to cell seeding at room temperature. Interestingly, we found that the elastic moduli of both concentrations were very similar, measuring around 120–150 Pa. These values closely align with those reported for pure Matrigel (~ 120 Pa) taken with AFM [51]. Thus, it is unlikely that variations in LPC differentiation towards AEC2 between the various published reports are due to differences in original Matrigel stiffness, although stiffness might change with culturing. It’s worth noting that the measured values are significantly lower than the elastic moduli (~ 4.5–10 kPa) reported for an alveolar wall [52] and decellularized alveolar ECM [53, 54]. This suggests that stiffer matrices than Matrigel may be required to accurately replicate lineage differentiation in the in situ distal niche.
In this study, we evaluated the efficiency of AEC2 differentiation using HT2-280 protein expression. HT2-280 is present on the surface of both fetal and adult human AEC2 [31, 55, 56], and its expression aligns with that of SFTPC [40]. Several studies have used HT2-280 to sort for human AEC2s for the generation of alveolar organoid cultures [10, 40, 57]. While a recent study reported challenges in sorting AEC2 with HT2-280 from hiPSC-derived alveolar organoids [55], it’s important to note that this difficulty may be due to certain PSC lines, as other studies have successfully identified AEC2s using HT2-280 in hPSC-derived alveolar organoids [18]. To validate our findings, we compared the HT2-280 flow cytometry results with those obtained from the GFP expression in organoids established with hPSCs that were stably transduced with a hSFTPCGFP reporter. Both flow cytometric readouts, HT2-280 and hSFTPC-GFP, consistently identified comparable viable AEC2 populations within the organoids, which were further confirmed through immunostaining for HT2-280. Our induction efficiency of 20–30% HT2-280+ cells falls within reported values ranging from 4 to 50% using SFTPC reporters [1, 4, 58]. Our starting population consisted of 50–60% NKX2-1+ LPCs. Surprisingly, enriching the NKX2-1+ LPC population to more than 80% with CPM+ sorting resulted in a decrease in HT2-280+ AEC2 induction within the organoids. The absence of any enrichment in SFTPB and SFTPC gene expression corroborated that CPM sorting did not lead to improved AEC2 differentiation (not shown). By contrast, previous studies have reported that enriching the NKX2-1 population resulted in a higher yield of SFTPC+ cells within the organoids [1, 4, 9]. However, these yield gains were always in comparison to the NKX2-1 negative populations [1, 2, 4, 9], while we compared our findings to the unsorted > 50% NKX2-1+ LPC population. To complicate straight comparisons even further, some studies included direct co-cultures with fetal lung fibroblast cells [1, 4].
We also observed that our CA1 organoids comprised heterogenous cell types. Approximately one-third of the CDH1+ epithelial population in our organotypic cultures were positive for HT2-280, which raises questions about the identity of the remaining 65% CDH1+ cells. Some studies have reported positive staining for AEC1 markers in their hPSC-derived organoids [4, 12], while others showed negative staining [9]. Recently, Kanagaki and colleagues reported that presence of mesenchymal cells was crucial for AEC1 differentiation in organoids [59]. Our CA1 organoid cultures contained hPSC-derived mesenchymal VIM+ cells but stained negative for AEC1 markers PDPN and AQP5 (not shown). Also, unlike in other studies, no TUBB4+ ciliated and SCGB1A1+ secretory club cells were detected, [4, 12]. We speculate that a portion of the CDH+ cells in the organoids represent non-lung endodermal lineages [60]. Ultrastructural and immuno-gold EM analysis revealed cell junctions between epithelial cells within the organoids and presence of SFTPB-positive pre-lamellar multivesicular bodies, but no lamellar bodies, suggesting an immature AEC2 phenotype in HT2-280+ cells at D43 within the CA1 organoids. It has been reported that 14 days post-NKX2-1 enrichment, the NKX2-1+ population within lung organoids fell to 60–70% [9], a level comparable to our starting population, which raises questions about the advantage of enriching the NKX2-1+ population for distal differentiation within organoids. The heterogenicity in organoid cell composition among various studies is likely due to variability in differentiation protocols, cellular plasticity, contaminating cell populations, and hPSC lines used, highlighting the need for standardized protocols to facilitate better comparisons.
One major difference between protocols is whether the epithelial LPCs are co-cultured with mesenchyme. Similar to other studies [1, 4], our organoids and Transwell cultures showed significantly more HT2-280+ cells when mesenchymal cells were present. We highlighted the importance of mesenchymal cells for AEC2 differentiation within organoids by removing co-developed VIM+ mesodermal cells from our LPC population using CPM sorting. This resulted in less HT2-280+ cells within the organoids during distal differentiation. Co-culturing LPCs with irradiated embryonic lung fibroblasts (iELF) in Transwell plates had the opposite effect and resulted in more HT2-280+ cells. The stimulatory effect on HT2-280 induction was independent of the substratum used and did not require direct contact between LPCs and iELF. The ELF-secretome is unaffected by irradiation [45, 46] and contains various factors, including FGFs and WNTs, that are key to alveolar organoid formation [17, 39, 61, 62]. The cocktail of ELF factors stimulating optimal AEC differentiation remains yet unresolved.
Finally, we demonstrated the importance of mechanical strain in alveolar development. In utero, the lung is exposed to mechanical forces generated by fetal breathing movements (FBM) that stimulate epithelial lung growth and differentiation [27, 63, 64]. In a recent study using a postnatal lung organoid model of CD326+ epithelial cells and fibroblasts, it was shown that static stretch increased cell proliferation, while cycle stretch promoted mesenchymal lineage gene expression [65]. In our study, we did not observe an increase in progenitor cell proliferation in organoids subjected to a stretch regimen mimicking FBMs. This is not surprising, as more than 70% of the progenitors in static organoid cultures are proliferating. Despite no change in proliferation, we observed a marked increase in the number of HT2-280+ cells and SFTPC expression within the organoids. This aligns with previous studies demonstrating that a similar stretch regimen increased the differentiation of primary fetal epithelial cells into AEC2 based on surfactant phospholipid [66, 67] and SFTPC [68] production. The mechanotransduction pathways stimulating AEC2 differentiation are unknown. Recently, it has been reported that ROCK-Yap/Taz signaling is essential to regulate AEC1 differentiation in response to mechanical loading (stretching) of the fetal lung [69]. The role of this mechanotransduction pathway in episodic stretch stimulated AEC2 differentiation remains to be elucidated.
Our study had several strengths, including the utilization of both iPSC and ESC lines, as well as the incorporation of SFTPC-GFP reporter lines. The exploration of mechanical strain in a 3D culture system, rather than a 2D system, is a novel approach with clinical relevance. Future applications could involve investigations in the impact of injurious strain combined with hyperoxia, which are well-known risk factors for BPD, on human AEC2 proliferation and differentiation in 3D organoid culture. However, there were also several limitations of our findings. One limitation is our reliance on HT2-280+ AEC2 marker expression. Single-cell transcriptomics might have provided a more comprehensive understanding of the distal epithelial population within our organoids and identified non-lung cell populations within them [60]. Additionally, a longer duration of culture might be necessary to improve alveolar differentiation efficiency. Also, we applied a periodic mechanical strain constantly during organoid formation. The employed strain settings were based upon previous studies with fetal lung cells in 2D culture [31]. In situ, fetal breathing movements start when the pulmonary bronchial tree is already partially established and their frequency and amplitude increase at the time AT2 differentiation occurs [27]. Further studies are needed to investigate whether the episodic strain should be applied when organoids already have been formed and if the frequency and amplitude of the strain should be altered over time to mimic better the in situ strain environment of the developing distal niche.
Conclusions
Our study highlights the critical factors influencing efficient and validated AEC2 differentiation from hPSCs, including lung progenitor concentration, mesenchymal population, and mechanical strain. While these factors increase the yield of AECs, further investigation is needed to understand the mechanisms driving AEC1 generation in hPSC-derived organoids to achieve a more accurate modeling of the human alveolus in situ. Organoids that replicate closely the alveolar niche will aid in understanding human alveolar pathology in lung diseases like BPD.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- ACTA2:
-
Actin alpha 2
- AEC:
-
Alveolar epithelial cell
- AEC2:
-
Alveolar epithelial type 2 cell
- AEC1:
-
Alveolar epithelial type 1 cell
- AFE:
-
Anterior foregut endoderm
- AFM:
-
Atomic force microscopy
- AQP5:
-
Aquaporin 5
- BPD:
-
Bronchopulmonary dysplasia
- BMP4:
-
Bone morphogenic protein 4
- CDH1:
-
E-cadherin
- CPM:
-
Carboxypeptidase M
- c-Kit:
-
KIT proto-oncogene, receptor tyrosine kinase
- CXCR4:
-
C-X-C chemokine receptor type 4
- DMH1:
-
Dorsomorphin homologue 1
- ECM:
-
Extracellular matrix
- EM:
-
Electron microscopy
- ESC:
-
Embryonic stem cell
- FBM:
-
Fetal breathing movements
- FOXJ1:
-
Forkhead box J1
- GATA6:
-
Gata binding protein 6
- GFP:
-
Green fluorescent protein
- hELF:
-
Human embryonic lung fibroblasts
- hPSC:
-
Human pluripotent stem cell
- ihELF:
-
Irradiated human embryonic lung fibroblast
- LPC:
-
Lung progenitor cells
- NKX2-1:
-
NK2 homeobox 1
- PDPN:
-
Podoplanin
- PSC:
-
Pluripotent stem cells
- iPSC:
-
Induced pluripotent stem cell
- PCR:
-
Polymerase chain reaction
- qPCR:
-
Quantitative PCR
- SCGB1A1:
-
Secretoglobin family 1A member 1
- SFDM:
-
Serum free differentiation medium
- SOX2:
-
SRY-box transcription factor 2
- SOX9:
-
SRY-box transcription factor 9
- STFPB:
-
Surfactant protein B
- SFTPC:
-
Surfactant protein C
- TUBB4A:
-
Tubulin beta 4A class IVa
- VAFE:
-
Ventral anterior foregut endoderm
- VIM:
-
Vimentin
References
Gotoh S, Ito I, Nagasaki T, Yamamoto Y, Konishi S, Korogi Y, et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Rep. 2014;3(3):394–403.
Hawkins F, Kramer P, Jacob A, Driver I, Thomas DC, McCauley KB, et al. Prospective isolation of NKX2-1-expressing human lung progenitors derived from pluripotent stem cells. J Clin Investig. 2017;127(6):2277–94.
Huang SX, Islam MN, O’Neill J, Hu Z, Yang YG, Chen YW, et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol. 2014;32(1):84–91.
Yamamoto Y, Gotoh S, Korogi Y, Seki M, Konishi S, Ikeo S, et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat Methods. 2017;14(11):1097–106.
Green MD, Chen A, Nostro MC, d’Souza SL, Schaniel C, Lemischka IR, et al. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat Biotechnol. 2011;29(3):267–72.
Huang SX, Green MD, de Carvalho AT, Mumau M, Chen YW, D’Souza SL, et al. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat Protoc. 2015;10(3):413–25.
Longmire TA, Ikonomou L, Hawkins F, Christodoulou C, Cao Y, Jean JC, et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell. 2012;10(4):398–411.
Mou H, Zhao R, Sherwood R, Ahfeldt T, Lapey A, Wain J, et al. Generation of multipotent lung and airway progenitors from mouse ESCs and patient-specific cystic fibrosis iPSCs. Cell Stem Cell. 2012;10(4):385–97.
Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, et al. Differentiation of human pluripotent stem cells into functional lung alveolar epithelial cells. Cell Stem Cell. 2017;21(4):472–88.
Barkauskas CE, Chung MI, Fioret B, Gao X, Katsura H, Hogan BL. Lung organoids: current uses and future promise. Development. 2017;144(6):986–97.
Dye BR, Hill DR, Ferguson MA, Tsai YH, Nagy MS, Dyal R, et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife. 2015;4: e05098.
Leibel SL, Winquist A, Tseu I, Wang J, Luo D, Shojaie S, et al. Reversal of surfactant protein b deficiency in patient specific human induced pluripotent stem cell derived lung organoids by gene therapy. Sci Rep. 2019;9(1):13450.
Crapo JD, Young SL, Fram EK, Pinkerton KE, Barry BE, Crapo RO. Morphometric characteristics of cells in the alveolar region of mammalian lungs. Am Rev Respir Dis. 1983;128(2 Pt 2):S42–6.
Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18(1):8–23.
Caniggia I, Tseu I, Han RN, Smith BT, Tanswell K, Post M. Spatial and temporal differences in fibroblast behavior in fetal rat lung. Am J Physiol. 1991;261(6 Pt 1):L424–33.
Deimling J, Thompson K, Tseu I, Wang J, Keijzer R, Tanswell AK, et al. Mesenchymal maintenance of distal epithelial cell phenotype during late fetal lung development. Am J Physiol Lung Cell Mol Physiol. 2007;292(3):L725–41.
Fox E, Shojaie S, Wang J, Tseu I, Ackerley C, Bilodeau M, et al. Three-dimensional culture and FGF signaling drive differentiation of murine pluripotent cells to distal lung epithelial cells. Stem Cells Dev. 2015;24(1):21–35.
Chen YW, Huang SX, de Carvalho A, Ho SH, Islam MN, Volpi S, et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat Cell Biol. 2017;19(5):542–9.
Leibel SL, McVicar RN, Winquist AM, Niles WD, Snyder EY. Generation of complete multi-cell type lung organoids from human embryonic and patient-specific induced pluripotent stem cells for infectious disease modeling and therapeutics validation. Curr Protoc Stem Cell Biol. 2020;54(1): e118.
Leibel SL, McVicar RN, Winquist AM, Snyder EY. Generation of 3D whole lung organoids from induced pluripotent stem cells for modeling lung developmental biology and disease. J Vis Exp. 2021;170: e62456.
Jesudason EC. Airway smooth muscle: An architect of the lung? Thorax. 2009;64(6):541–5.
Harding R. Fetal pulmonary development: the role of respiratory movements. Equine Vet J Suppl. 1997;24:32–9.
Kitterman JA. The effects of mechanical forces on fetal lung growth. Clin Perinatol. 1996;23(4):727–40.
Moessinger AC, Harding R, Adamson TM, Singh M, Kiu GT. Role of lung fluid volume in growth and maturation of the fetal sheep lung. J Clin Investig. 1990;86(4):1270–7.
Wigglesworth JS, Desai R. Effect on lung growth of cervical cord section in the rabbit fetus. Early Hum Dev. 1979;3(1):51–65.
Yamamoto Y, Korogi Y, Hirai T, Gotoh S. A method of generating alveolar organoids using human pluripotent stem cells. Methods Cell Biol. 2020;159:115–41.
Liu M, Skinner SJ, Xu J, Han RN, Tanswell AK, Post M. Stimulation of fetal rat lung cell proliferation in vitro by mechanical stretch. Am J Physiol. 1992;263(3 Pt 1):L376–83.
Shojaie S, Ermini L, Ackerley C, Wang J, Chin S, Yeganeh B, et al. Acellular lung scaffolds direct differentiation of endoderm to functional airway epithelial cells: requirement of matrix-bound HS proteoglycans. Stem Cell Rep. 2015;4(3):419–30.
Litvack ML, Wigle TJ, Lee J, Wang J, Ackerley C, Grunebaum E, et al. Alveolar-like stem cell-derived Myb(−) macrophages promote recovery and survival in airway disease. Am J Respir Crit Care Med. 2016;193(11):1219–29.
Bilodeau C, Shojaie S, Goltsis O, Wang J, Luo D, Ackerley C, et al. TP63 basal cells are indispensable during endoderm differentiation into proximal airway cells on acellular lung scaffolds. NPJ Regen Med. 2021;6(1):12.
Gonzalez RF, Allen L, Gonzales L, Ballard PL, Dobbs LG. HTII-280, a biomarker specific to the apical plasma membrane of human lung alveolar type II cells. J Histochem Cytochem. 2010;58(10):891–901.
van Tuyl M, Groenman F, Kuliszewski M, Ridsdale R, Wang J, Tibboel D, et al. Overexpression of lunatic fringe does not affect epithelial cell differentiation in the developing mouse lung. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L672–82.
Tibboel J, Groenman FA, Selvaratnam J, Wang J, Tseu I, Huang Z, et al. Hypoxia-inducible factor-1 stimulates postnatal lung development but does not prevent O2-induced alveolar injury. Am J Respir Cell Mol Biol. 2015;52(4):448–58.
Keijzer R, van Tuyl M, Meijers C, Post M, Tibboel D, Grosveld F, et al. The transcription factor GATA6 is essential for branching morphogenesis and epithelial cell differentiation during fetal pulmonary development. Development. 2001;128(4):503–11.
Yang H, Lu MM, Zhang L, Whitsett JA, Morrisey EE. GATA6 regulates differentiation of distal lung epithelium. Development. 2002;129(9):2233–46.
Ridsdale R, Post M. Surfactant lipid synthesis and lamellar body formation in glycogen-laden type II cells. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):L743–51.
McCauley KB, Hawkins F, Serra M, Thomas DC, Jacob A, Kotton DN. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt signaling. Cell Stem Cell. 2017;20(6):844–57.
Shannon JM. Induction of alveolar type II cell differentiation in fetal tracheal epithelium by grafted distal lung mesenchyme. Dev Biol. 1994;166(2):600–14.
Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, et al. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell. 2017;170(6):1134–48.
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Investig. 2013;123(7):3025–36.
Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661–80.
Cheng X, Ying L, Lu L, Galvao AM, Mills JA, Lin HC, et al. Self-renewing endodermal progenitor lines generated from human pluripotent stem cells. Cell Stem Cell. 2012;10(4):371–84.
Farahani RM, Xaymardan M. Platelet-derived growth factor receptor alpha as a marker of mesenchymal stem cells in development and stem cell biology. Stem Cells Int. 2015;2015: 362753.
Broers JL, de Leij L, Rot MK, ter Haar A, Lane EB, Leigh IM, et al. Expression of intermediate filament proteins in fetal and adult human lung tissues. Differentiation. 1989;40(2):119–28.
Eiselleova L, Peterkova I, Neradil J, Slaninova I, Hampl A, Dvorak P. Comparative study of mouse and human feeder cells for human embryonic stem cells. Int J Dev Biol. 2008;52(4):353–63.
Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 2017;12(2):439–51.
Miller AJ, Hill DR, Nagy MS, Aoki Y, Dye BR, Chin AM, et al. In vitro induction and in vivo engraftment of lung bud tip progenitor cells derived from human pluripotent stem cells. Stem Cell Rep. 2018;10(1):101–19.
Lim K, Donovan APA, Tang W, Sun D, He P, Pett JP, et al. Organoid modeling of human fetal lung alveolar development reveals mechanisms of cell fate patterning and neonatal respiratory disease. Cell Stem Cell. 2023;30(1):20–37.
Liao D, Li H. Dissecting the niche for alveolar type II cells with alveolar organoids. Front Cell Dev Biol. 2020;8:419.
Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–89.
Alcaraz J, Xu R, Mori H, Nelson CM, Mroue R, Spencer VA, et al. Laminin and biomimetic extracellular elasticity enhance functional differentiation in mammary epithelia. EMBO J. 2008;27(21):2829–38.
Bou Jawde S, Takahashi A, Bates JHT, Suki B. An analytical model for estimating alveolar wall elastic moduli from lung tissue uniaxial stress–strain curves. Front Physiol. 2020;11:121.
Luque T, Melo E, Garreta E, Cortiella J, Nichols J, Farre R, et al. Local micromechanical properties of decellularized lung scaffolds measured with atomic force microscopy. Acta Biomater. 2013;9(6):6852–9.
Jorba I, Beltran G, Falcones B, Suki B, Farre R, Garcia-Aznar JM, et al. Nonlinear elasticity of the lung extracellular microenvironment is regulated by macroscale tissue strain. Acta Biomater. 2019;92:265–76.
Korogi Y, Gotoh S, Ikeo S, Yamamoto Y, Sone N, Tamai K, et al. In vitro disease modeling of Hermansky–Pudlak syndrome type 2 using human induced pluripotent stem cell-derived alveolar organoids. Stem Cell Rep. 2019;12(3):431–40.
Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555(7695):251–5.
Konda B, Mulay A, Yao C, Beil S, Israely E, Stripp BR. Isolation and enrichment of human lung epithelial progenitor cells for organoid culture. J Vis Exp. 2020;161: e61541.
Ghaedi M, Calle EA, Mendez JJ, Gard AL, Balestrini J, Booth A, et al. Human iPS cell-derived alveolar epithelium repopulates lung extracellular matrix. J Clin Investig. 2013;123(11):4950–62.
Kanagaki S, Ikeo S, Suezawa T, Yamamoto Y, Seki M, Hirai T, et al. Directed induction of alveolar type I cells derived from pluripotent stem cells via Wnt signaling inhibition. Stem Cells. 2021;39(2):156–69.
Hurley K, Ding J, Villacorta-Martin C, Herriges MJ, Jacob A, Vedaie M, et al. Reconstructed single-cell fate trajectories define lineage plasticity windows during differentiation of human PSC-derived distal lung progenitors. Cell Stem Cell. 2020;26(4):593–608.
Shiraishi K, Shichino S, Ueha S, Nakajima T, Hashimoto S, Yamazaki S, et al. Mesenchymal–epithelial interactome analysis reveals essential factors required for fibroblast-free alveolosphere formation. iScience. 2019;11:318–33.
Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359(6380):1118–23.
Li J, Wang Z, Chu Q, Jiang K, Li J, Tang N. The strength of mechanical forces determines the differentiation of alveolar epithelial cells. Dev Cell. 2018;44(3):297–312.
Liu M, Tanswell AK, Post M. Mechanical force-induced signal transduction in lung cells. Am J Physiol. 1999;277(4):L667–83.
Joshi R, Batie MR, Fan Q, Varisco BM. Mouse lung organoid responses to reduced, increased, and cyclic stretch. Am J Physiol Lung Cell Mol Physiol. 2022;322(1):L162–73.
Sanchez-Esteban J, Cicchiello LA, Wang Y, Tsai SW, Williams LK, Torday JS, et al. Mechanical stretch promotes alveolar epithelial type II cell differentiation. J Appl Physiol. 2001;91(2):589–95.
Sanchez-Esteban J, Wang Y, Gruppuso PA, Rubin LP. Mechanical stretch induces fetal type II cell differentiation via an epidermal growth factor receptor–extracellular-regulated protein kinase signaling pathway. Am J Respir Cell Mol Biol. 2004;30(1):76–83.
Nakamura T, Liu M, Mourgeon E, Slutsky A, Post M. Mechanical strain and dexamethasone selectively increase surfactant protein C and tropoelastin gene expression. Am J Physiol Lung Cell Mol Physiol. 2000;278(5):L974–80.
Nguyen TM, van der Merwe J, Elowsson Rendin L, Larsson-Callerfelt AK, Deprest J, Westergren-Thorsson G, et al. Stretch increases alveolar type 1 cell number in fetal lungs through ROCK-Yap/Taz pathway. Am J Physiol Lung Cell Mol Physiol. 2021;321(5):L814–26.
Acknowledgements
We like to thank Dr. Andras Nagy, University of Toronto, and the Center for Commercialization of Regenerative Medicine, Toronto, for their generous gifts of CA1 ES and NCRM1 iPS cells, respectively. The hSFTPC promoter construct was a generous gift of Dr. Jeffrey Whistsett, Cincinatti, OH. Claudia Bilodeau was supported by a RESTRACOMP scholarship award from the Hospital for Sick Children.
Funding
This research was supported by Canadian Institutes of Health Research (FND-143309 to MP).
Author information
Authors and Affiliations
Contributions
OG and CB: Conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing. JW, DL, MA, LB, and AP: Design, Collection and/or assembly of data. SLL: manuscript writing. MP: Conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent of participants
This study does not involve animal experiments or human participants. Use of human pluripotent cell lines was approved by the Stem Cell Oversight Committee of The Canadian Institute of Health Research (Patient-specific alveolar type II (ATII) cells from surfactant protein-B deficient induced pluripotent stem cells) in March 2014. The Research Ethical Board of the Hospital for Sick Children confirmed no need for additional ethical approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Artificial intelligence
The authors declare that artificial intelligence is not used in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Goltsis, O., Bilodeau, C., Wang, J. et al. Influence of mesenchymal and biophysical components on distal lung organoid differentiation. Stem Cell Res Ther 15, 273 (2024). https://doi.org/10.1186/s13287-024-03890-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-024-03890-2