
Abstract
Background
Human dental pulp stem cells (hDPSCs) have received widespread attention in the fields of tissue engineering and regenerative medicine. Although amphiregulin (AREG) has been shown to play a vital function in the biological processes of various cell types, its effects on DPSCs remain largely unknown. The aim of this study was to explore the specific role of AREG as a biologically active factor in the regeneration of dental pulp tissue.
Methods
The growth of hDPSCs, together with their proliferation and apoptosis, in response to AREG was examined by CCK-8 assay and flow cytometry. We explored the effects of AREG on osteo/odontogenic differentiation in vitro and investigated the regeneration and mineralization of hDPSCs in response to AREG in vivo. The effects of AREG gain- and loss-of-function on DPSC differentiation were investigated following transfection using overexpression plasmids and shRNA, respectively. The involvement of the mitogen-activated protein kinase (MAPK) or phosphatidylinositol 3-kinase (PI3K)/Akt pathways in the mineralization process and the expression of odontoblastic marker proteins after AREG induction were investigated by using Alizarin Red S staining and Western blotting, respectively.
Results
AREG (0.01–0.1 µg/mL) treatment of hDPSCs from 1 to 7 days increased hDPSCs growth and affected apoptosis minimally compared with negative controls. AREG exposure significantly promoted hDPSC differentiation, shown by increased mineralized nodule formation and the expression of odontoblastic marker protein expression. In vivo micro-CT imaging and quantitative analysis showed significantly greater formation of highly mineralized tissue in the 0.1 μg/mL AREG exposure group in DPSC/NF-gelatin-scaffold composites. AREG also promoted extracellular matrix production, with collagen fiber, mineralized matrix, and calcium salt deposition on the composites, as shown by H&E, Masson, and Von Kossa staining. Furthermore, AREG overexpression boosted hDPSC differentiation while AREG silencing inhibited it. During the differentiation of hDPSCs, AREG treatment led to phosphorylation of extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and PI3K/Akt. Notably, a specific inhibitor of ERK, JNK, and PI3K/Akt signaling markedly reduced AREG-induced differentiation, as well as levels of phosphorylated ERK and JNK in hDPSCs.
Conclusions
The data indicated that AREG promoted odontoblastic differentiation and facilitated regeneration and mineralization processes in hDPSCs.
Similar content being viewed by others


Background
Mesenchymal stem cells (MSCs) are multifunctional cells present in various tissues, including bone marrow, adipose tissue, Wharton’s jelly, umbilical cord blood, and peripheral blood, that can be used for cell therapy and tissue/organ regeneration [1, 2]. Dental pulp stem cells (DPSCs) isolated from human dental pulp are specific mesenchymal stem cells that can be induced to differentiate into odontoblasts like cells in vitro or to form dentine-/pulp-like tissue in vivo [3,4,5,6]. DPSCs are relatively easy to obtain from waste tooth tissue and exhibit relatively high levels of proliferation, clonal potential, and mineralization compared with bone marrow mesenchymal stem cells (BMMSCs) [7]. Consequently, DPSCs have received widespread attention in the fields of tissue engineering and regenerative medicine. Despite this, signal transduction mechanisms involved in the oriented differentiation of DPSC are not yet fully understood.
Growth factor signaling plays a pivotal regulatory role in both cell growth and differentiation. Amphiregulin (AREG) is a member of the epidermal growth factor (EGF) family and is expressed by a variety of epithelial and mesenchymal cell types during development; it is also associated with homeostasis [8, 9]. According to previous reports, AREG is transcribed as a 1.4-kb mRNA composed of six exons that encode for a transmembrane polarized glycoprotein precursor (Pro-AREG) comprised of 252 aminoacids [8]. As a membrane-anchored precursor protein, it is mainly engaged in juxtacrine signaling on adjacent cells. There are three different levels of regulation of AREG functionality and these include: (1) at the transcriptional level through the binding of activators and repressors to different regions on the AREG promoter, (2) at the post-transcriptional level through modulation of mRNA stability, and (3) at the post-translational level through covalent modifications and the regulation of AREG shedding [8]. Recently, AREG has been implicated in the regulation of a wide range of biological processes, including cell growth, proliferation, neurogenesis, cell migration, and bone formation, by binding to the EGF receptor (EGFR) on the cell membrane [10]. Interestingly, AREG can regulate squamous cell differentiation and neuronal differentiation from stem/progenitor cell sources [11, 12]. AREG has also been found to accumulate in multiple myeloma-derived exosomes and is involved in regulating osteoclast differentiation through a circuitous mechanism in osteoblasts [13]. However, despite these demonstrations of AREG’s role in the differentiation of a variety of cell types, its effects on the growth and differentiation of DPSCs remain largely unknown.
AREG was initially identified in culture supernatants from the human breast cancer cell line MCF-7 [14] and was found to bind to EGFR. Subsequently, it has been shown to activate a variety of downstream intracellular signaling pathways, including Ras/MAPK, PI3K/AKT, mTOR, and STAT [15, 16]. These signal transduction cascades regulate gene expression and initiate diverse cellular responses, including proliferation, survival, invasion, differentiation, and angiogenesis. AREG can promote migratory activity and doxorubicin resistance through activation of the MAPK pathway [17]. Furthermore, AREG-induced growth can also be regulated partially through the MAPK and PI3K-Akt/PKB pathways [18]. Currently, the role of AREG in activating downstream signaling in the regeneration of dental pulp tissue is not known. Therefore, our study aimed to explore the effects of AREG on the growth, differentiation, and regeneration processes of DPSCs.
Methods
Reagents
Amphiregulin (AREG) (262-AR-100) was obtained from R&D Systems (R&D, CA, USA). The specific inhibitors of ERK kinase, p38 kinase, JNK kinase, and AKT kinase are U0126, SB203580, SP600125, and LY294002, respectively, were all obtained from Cell Signaling Technology (Danvers, MA, USA). AREG lentivirus kits were purchased from Hanbio Biotechnology (Hanbio, China).
DPSC culture
Freshly extracted sound teeth were collected from patients (18–22-years-old), under the approval of the Ethics Committee of the Fourth Military Medical University (FMMU), Xi’an, China with informed consent. The hDPSCs were isolated, cultured, and characterized as previously described [19, 20]. Briefly, the pulp tissue was dissected and digested with 3 mg/mL type I collagenase and 4 mg/mL dispase (Sigma-Aldrich, St Louis, MO, USA) for 45–60 min at 37℃. Single-cell suspensions were cultured in 60-mm culture dishes and maintained in a-minimum essential medium (α-MEM; Invitrogen, Carlsbad, CA, USA) with 10% (v/v) fetal bovine serum (Gibco-BRL, Grand Island, NY, USA), 100 units/mL penicillin-G, and 100 mg/mL streptomycin (Invitrogen) in a humidified atmosphere with 5% CO2 at 37℃. Single-cell clones of DPSCs were isolated and passaged as previously described [21]. DPSCs were grown in a 5% CO2 incubator at 37 °C, and cultures at passages 3–5 were used for all studies.
Multi-lineage differentiation in vitro
Cells were cultured in 6-well plates in osteo/odontogenic induction medium containing 50 mg/mL ascorbic acid, 10 mM β-glycerophosphate, and 10 nM dexamethasone (all from Sigma-Aldrich) with 10% FBS for 14 days. The cells were washed twice in PBS and fixed with 4% polyoxymethylene (Sigma-Aldrich, St Louis, MO, USA) for 15 min. The cells were then stained with Alizarin Red S (Sigma-Aldrich, St Louis, MO, USA). For adipogenic induction, the cells were cultured in adipogenic induction medium containing 1 mM IBMX, 0.4 mM indomethacin, 0.2 μM dexamethasone, 0.02 mg/mL insulin, 100 μg/mL streptomycin, 100 U/mL penicillin, and 10% FBS in α-MEM medium) for 21 days. Cultures were stained with 0.3% (w/v) Oil-Red O (Sigma-Aldrich)/60% isopropanol reagent for 60 min. Finally, cultures were washed three times in water prior to analysis.
Flow cytometric analysis of stem cell surface markers
DPSCs (2 × 105 cells) were digested by trypsinization and harvested. Cells (1 × 106/100 µL) were resuspended and incubated with fluorescein isothiocyanate (FITC)-coupled anti-human monoclonal antibodies against CD105-FITC, CD34-PE, CD45-PE, CD90-PE, CD29-PE, and CD146-PE (1: 100 dilution; BD Biosciences, San Jose, CA, USA) in PBS with 3% FBS for 1 h in the dark at room temperature. Finally, the samples were analyzed on a flow cytometer (FACS Calibur; BD Biosciences, Franklin Lakes, NJ, USA) using CellQuest PROTM software (BD Biosciences).
Flow cytometric analysis of proliferation and apoptosis in hDPSCs
Third-passage DPSCs (3 × 106 cells/dish) were inoculated in 60 mm dishes. The experimental groups were treated with 0.01 μg/mL and 0.1 μg/mL AREG for 16 h, respectively. The control group received no AREG treatment. After removal from the dishes, cells were washed three times in pre-cooled phosphate-buffered saline (PBS) and were fixed in 75% ice-cold ethanol. A flow cytometer (BD Biosciences, San Jose, CA, USA) was used to monitor the changes in the G0/G1, S, and G2/M phases of the cell cycle and to calculate the cell proliferation index (PI = G2/M + S). For the measurement of apoptosis, the AREG-treated DPSCs were harvested and washed as above, and 500 μl of cells was diluted with 1 × Annexin V Binding Buffer working solution according to the instructions of the apoptosis kit (KeyGENBioTECH, China). Then, 5 μl Annexin V-APC and 5μL 7-ADD staining solution were added to the cells and the number of apoptotic cells was detected by FCM.
Cell counting kit-8 assay (CCK-8)
Five thousand DPSCs per well were seeded and cultured in 96-well plates with a range of concentrations of AREG (0-1 μg/mL). In a separate experiment, DPSCs were incubated with 20 µL CCK-8 reagents (KeyGENBioTECH, China) at 37 °C for 2 h for 1, 3, 5, and 7 days. Finally, absorbances at 450 nm were read in a microplate reader (ModelELX 808; Bio-Tek, Winooski, VT, USA).
Alizarin Red S staining and quantitation
DPSCs (4 × 105 cells/well) were seeded into 6-well plates and cultured in either control medium or osteo/odontogenic induction medium with AREG for 14 days. At specified times, the cells were fixed in 1 ml of 4% paraformaldehyde for 30 min at room temperature. After three washes with distilled water, 0.1 g/ml Alizarin Red S (ARS) (Sigma-Aldrich) was used to stain the cultures for 10 min at room temperature. The unbound stain was removed by washing with deionized water until the discarded liquid appeared colorless. Five hundred microliters of water were added to each dish to ensure the cells remained hydrated. Cells were observed under an Olympus inverted microscope (Tokyo, Japan). For quantitation, alizarin red S stain dissolved in 10% cetylpyridinium chloride (CPC) (Sigma-Aldrich) was added to the cells and the absorbance at 450 nm was read in the microplate reader.
In vivo studies
All animal surgical procedures were approved by the Animal Care Committee and the Institutional Review Board (IRB) for Human Subjects Research of the Fourth Military Medical University. The high-stiffness three-dimensional (3D) nanofibrous gelatin (NF-gelatin) scaffolds were a kind gift from Prof. Tiejun Qu [22]. Initially, the NF-gelatin scaffolds were placed in 70% alcohol for half an hour and then washed three times in sterile PBS to remove residual ethanol. Before the seeding of human DPSCs (5 × 105), the scaffolds were soaked in α-MEM containing 10% FBS. The cell-scaffold composites were cultured in α-MEM supplemented with 10% FBS for 24 h on an orbital shaker (Orbi-shaker™, Benchmark, USA) in an incubator with 5% CO2 at 37℃. Subsequently, the cell-scaffold composites were stimulated with 0.1 μg/mL AREG in osteo/odontogenic induction medium for 7 days. Controls received no AREG exposure. The medium was changed every other day. After 7 days, the cell-scaffold composites were implanted subcutaneously on the dorsal surfaces of immune-compromised nude mice (nu/nu, 6–8 weeks old). After 4 weeks, the mice were euthanized by an anesthetic overdose and the tissue growths were then harvested. Tissue samples were immediately fixed in 4% paraformaldehyde overnight. The samples (n = 4 in each group) were scanned and analyzed using a micro-CT (eXplore Locus SP micro-CT; GE Healthcare, USA) as previously described [23] and the 3D micro-architectural properties of specimens were evaluated using analysis software (MicroView; GE Healthcare). After decalcification in 17% ethylenediamine tetra-acetic acid, hematoxylin–eosin (H&E), Masson’s trichrome, and von Kossa staining were used for histological observation.
Overexpression and knockdown of AREG in DPSCs
AREG-green fluorescent protein lentivirus kits and their respective control kits were purchased from Hanbio (China). DPSCs were transfected with AREG lentivirus according to the manufacturer's instructions. In brief, third-passage DPSCs were inoculated in 12-well plates at 0.5 × 105/mL and infected for 4 h, after which 0.5 mL fresh complete medium was added. After a further 24-h infection period, the medium was replaced with 1 mL fresh medium and after a further 72 h, the transfection rate was examined under a fluorescence microscope (DMI8, Leica, German). Puromycin was used to select stably transfected cells with the puromycin concentration determined in a preliminary experiment. For simplicity, AREG-overexpressing and AREG-silenced cells were referred to as AREG (+) and AREG (−), respectively. The effects on the oriented differentiation of DPSCs were examined by ARS and Western blot analysis as above and described above and below.
Western blot analysis
DPSCs were cultured in serum-free medium for 24 h and were treated with 0.1 µg/mL AREG for 0, 30, 60, and 90 min in the presence or absence of the specific inhibitors U0126 (25 µM), SP600125 (25 μM), SB203580 (25 μM), and LY294002 (10 μM). Protein extraction from the cells, SDS-PAGE, and blotting was performed as previously described [17]. After incubating with secondary antibody (1:4000; Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h, the protein bands were imaged by using an enhanced chemiluminescence system (Amersham, Piscataway, NJ, USA).
Statistical analysis
All experiments were repeated separately in triplicate or quintuplicate. Data are expressed as means ± SD. For statistical processing, we used SPSS software 16.0 (version 16.0; SPSS, Chicago, IL, USA). Inter-group differences were compared by the ONE test. P < 0.05 was considered to be statistically significant.
Results
Culture and characterization of hDPSCs
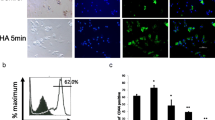
Fibroblast-like clonal cells were obtained from the dental pulp tissue by limiting dilution and colony cloning (Fig. 1A a, b). Putative stem cells derived from the clonal cells (Fig. 1A c) were characterized by multiple lineage differentiation tests and flow cytometry. If cultured under inductive conditions, the cells formed mineral nodules and lipid droplets, shown by Alizarin Red S and Oil Red O staining (Fig. 1B a, b). Flow cytometry analysis showed that CD90, CD105, CD29, CD146, and STRO-1 were highly expressed in isolated cultured DPSCs (Fig. 1C d–h). In contrast, the hematopoietic cell markers CD34 and CD45 (Fig. 1C b, c) were detected at minimal levels in the cultured stem cells. Analysis of morphology, colony formation, immunophenotype, and the ability to differentiate into multiple lineages indicated that mesenchymal stem cells had been isolated.

Culture and characterization of DPSCs. A (a) DPSCs isolated and cultured from pulp tissue samples, (b) clonal DPSCs, (c) were cultured for 2 weeks. B (a) Mineralized nodule formation in osteo/odontogenic induction medium demonstrated by Alizarin Red S staining, (b) lipid droplet formation in adipogenic induction medium using Oil Red O staining. C Flow cytometry of molecular surface antigen markers in DPSCs. (a) Negative control for DPSCs. Representative profiles are shown for the expression of the immunophenotypic markers CD34 (b), CD45 (c), CD90 (d), CD105 (e), CD29 (f), CD146 (g) and STRO-1 (h) in DPSCs
Effects of AREG on growth of hDPSCs
The effects of AREG on hDPSC numbers after 1-, 3-, 5-, and 7-days incubation were assessed using the CCK-8 assay (Fig. 2A). The data showed that cell numbers in the 0.01 μg/mL and 0.1 μg/mL AREG-treated groups were significantly increased (P < 0.05). In contrast, cell numbers in the 1 μg/mL AREG-treated group were reduced. Cell cycle analysis showed a minimal increase in the proliferation index in the 0.01 μg/mL and 0.1 μg/mL AREG exposure groups (PI = G2/M + S), whereas treatment with 1 μg/mL AREG decreased the proliferation index compared with the control group (CTRL) (Fig. 2B). In addition, FCM analysis showed that AREG did not significantly affect apoptosis of DPSCs compared with the CTRL group (Fig. 2C).

Effects of AREG on the growth of DPSCs. A The CCK-8 assay was used to detect cell proliferation in different AREG-treated groups (0.01–1 mg/mL) and untreated DPSCs at 1, 3, 5 and 7 days (n = 5, *P < 0.05). B The cell cycle proliferation index (PI = G2/M + S) in AREG-treated and control groups analyzed by flow cytometry. C Flow cytometry analysis of AREG-treated and control groups
Effects of AREG on odontogenic differentiation of hDPSCs in vitro
After two weeks of osteo/odontogenic induction, it was apparent that AREG concentrations between 0.01 and 0.1 µg/mL promoted mineralized nodule formation in a dose-dependent manner, although a significant decrease was apparent at 1 µg/mL AREG exposure (Fig. 3A, B). Consequently, 0.1 µg/mL AREG exposure was selected as the optimal concentration for the following studies to investigate the influence of AREG on DPSCs. When the expression of odontoblastic marker proteins was examined by Western blotting, it was found that the levels of DSPP, BSP, RUNX2, and OCN were noticeably up-regulated in the AREG-treated groups compared with the controls by day 3. Levels were markedly elevated on days 7 and 14 (P < 0.05) (Fig. 3C, D). Taken together, these results indicated that AREG could stimulate DPSC differentiation.

AREG enhances the oriented differentiation potential of DPSCs. A Alizarin Red S staining of DPSCs after 14 days’ culture with a range of AREG concentrations in mineralizing medium. B Quantitative analysis of Alizarin Red S staining (n = 5, **P < 0.05). C Western blot analysis showing expression of the odontoblastic markers DSPP, RUNX2, BSP, and OCN. The full-length gels and blots are included in Additional file 1: Fig. S1A. D ImageJ software analysis of the gray level of the panel. Data are expressed as means ± SDs, n = 3, *P < 0.05, **P < 0.01
Effects of AREG on regeneration and mineralization of hDPSCs in vivo
To analyze the regenerative capability in response to AREG, the cell-scaffold composites were subcutaneously implanted into nude mice. Subsequently, the specimens were scanned and images were reconstructed using micro-CT analysis (Fig. 4A, B). Micro-CT images showed the formation of more highly mineralized tissue in the 0.1 μg/mL AREG exposure group in the composites (Fig. 4A a, b). Additionally, quantitative analysis also indicated that the bone volume fraction, trabecular thickness, and trabecular number were superior in the AREG group compared with the control group, although the trabecular separation was decreased (Fig. 4B a–d). Combined, these findings supported the ability of AREG in enhancing the odonto/osteogenic potential of DPSCs.

The effect of AREG on the regeneration and mineralization of hDPSCs in vivo. A Micro-CT images of the DPSC/S-scaffold construct in nude mice after subcutaneous implantation for 4 weeks. A (a) and (b) show the DPSC/NF-gelatin-scaffold composites of the control and AREG group, respectively. B Quantitative analysis of micro-CT images (a) BV/TV (Bone Volume to Tissue Volume); (b) Tb.Th. (Trabecular Thickness); (c) Tb.N. (Trabecular Number); (d) Tb.Sp. (Trabecular Separation). C Histological staining of the DPSC/S-scaffold construct after subcutaneous implantation in nude mice for 4 weeks. (a, d, g) H&E staining; (b, e, h) Masson staining; (c, f, i) Von Kossa staining. *P < 0.05 represents a significant change compared with the control
The results of the H&E and Masson staining showed that the cell-scaffold composites stimulated by 0.1 μg/mL AREG produced greater DPSC penetration into the pores of scaffold, as well as showing greater extracellular matrix secretion and collagen fiber wrapping in the NF-gelatin scaffold, compared with the control group (Fig. 4C a, b, d, e, g, h). Von Kossa staining confirmed the presence of mineralization and further demonstrated greater deposition of mineralized matrix and calcium salts in the AREG exposure group (Fig. 4C c, f, i). These data confirmed that AREG promoted the regeneration and mineralization capability of hDPSCs in vivo.
Effects of AREG overexpression and knockdown on hDPSCs differentiation
To further investigate AREG function during DPSC differentiation, cells were transfected with the AREG vector to induce AREG overexpression. Western blotting demonstrated increased expression of the odontoblastic markers at 3, 7, and 14 days after induction, compared with the control group (Fig. 5C, D). Furthermore, overexpression of AREG promoted mineralized nodule formation in DPSCs as detected by ARS staining and quantitation (Fig. 5A, B). These data supported the ability of AREG to promote differentiation of DPSCs.

The effects of AREG overexpression/knockdown on the differentiation of DPSCs. A Alizarin Red S staining showing mineralized nodule formation in the AREG ( +) and AREG (–) groups. B Quantitative analysis of A (n = 5, **P < 0.05). C Protein expression of odontoblastic markers DSPP, RUNX2, BSP, and OCN shown by Western blotting in the AREG ( +) group. The full-length gels and blots are included in Additional file 1: Fig. S1B. D ImageJ analysis of the gray level of the panel. E Protein expression of odontoblastic markers DSPP, RUNX2, BSP, and OCN shown by Western blotting in the AREG ( −) group. The full-length gels and blots are included in Additional file 1: Fig. S1C. F ImageJ was used to analyze the gray level of the panel. Data represent means ± SDs, n = 3, *P < 0.05, **P < 0.01
To investigate whether AREG up-regulation was necessary for DPSC differentiation, AREG expression was silenced by shRNA transfection in DPSCs. Silencing was confirmed by qRT-PCR at 72 h after transfection following puromycin selection. Protein expression of DPSC odontoblast markers was decreased in response to AREG inhibition, as shown by Western blotting (Fig. 5E, F). ARS staining and quantitation showed that AREG silencing attenuated mineralized nodule formation in DPSCs (Fig. 5A, B). These results indicated that reduced AREG expression resulted in the suppression of odontogenic differentiation.
Involvement of MAPK signaling in AREG-induced differentiation of hDPSCs
Treatment with AREG increased the protein expression level of p-ERK after 60 min stimulation. AREG stimulation also resulted in phosphorylation of JNK in DPSCs (Fig. 6A, B). However, AREG showed minimal effects on phosphorylated p38 levels (Fig. 6A, B). Notably, incubation with the ERK, JNK, and p38 MAPK inhibitors markedly antagonized the effect of AREG on phosphorylated ERK and JNK in hDPSCs (Fig. 6C, D). These inhibitors also reduced the expression of the mineralization markers assayed by day 14, and mineralized nodule formation in DPSCs (Fig. 6E–H). Taken together, these data indicated that the ERK MAPK and JNK MAPK pathways are involved in AREG-induced differentiation of hDPSCs.

AREG promotes differentiation of DPSCs by activation of the ERK, JNK, and AKT pathways. A Protein levels of ERK and p-ERK, JNK and p-JNK, p38 and p-p38, and ATK and p-AKT treated with AREG at different time points (0, 30, 60, and 90 min) shown by Western blotting in DPSCs. The full-length gels and blots are included in Additional file 1: Fig. S2A. B Quantitative analysis of the gray intensity of A. C Treatment of DPSCs with specific ERK, JNK, or PI3K inhibitors (U0126, SP600125, and LY294002, respectively). Protein levels of ERK and p-ERK, JNK and p-JNK, p-AKT/AKT are shown by Western blotting at the indicated times. The full-length gels and blots are included in Additional file 1: Fig. S2B. D Quantitative analysis of the ratio of p-ERK/ERK, p-JNK/JNK and p-AKT/AKT from C. E Alizarin Red S staining showing mineralized nodule formation in DPSCs treated with the specific inhibitors. F Quantitative analysis of Alizarin Red S staining (n = 5, *P < 0.05). G Western blots showing protein expression of odontoblastic markers (DSPP, RUNX2, BSP, and OCN) in different groups at day 14. The full-length gels and blots are included in Additional file 1: Fig. S2C. H Quantitative analysis of data presented in G. Data represent means ± SDs, n = 3, *P < 0.05, **P < 0.01
Involvement of PI3K/AKT signaling in AREG-induced differentiation of hDPSCs
AREG phosphorylated AKT in DPSCs in a time-dependent manner (Fig. 6A, B); this effect was inhibited by the PI3K pathway inhibitor LY294002 (Fig. 6C, D). Furthermore, Western blotting showed that the levels of DSPP, BSP, RUNX2, and OCN were markedly reduced in cells treated with AREG + LY204002 (P < 0.05) (Fig. 6G, H). The PI3K pathway inhibitor LY294002 also markedly antagonized mineralized nodule formation and the expression of mineralization markers in DPSCs (Fig. 4E–H). These findings indicate that the PI3K/AKT pathways are involved in the AREG-induced differentiation of hDPSCs.
Discussion
DPSCs provide a cellular reservoir which have significant applications in several tissue engineering applications. As with other somatic stem cells, a range of studies have demonstrated the potential of DPSCs in regenerative therapies, based on their multipotent differentiation capability [24]. Importantly, their cell growth and differentiation is regulated by action of several stimulatory growth factors and numerous studies have now demonstrated the importance of the EGF family in various development processes [25, 26]. EGF is known to exert its biological actions through its binding to EGFR on cell surfaces and AREG is also able to interact with the same receptor to activate downstream signaling pathways [10]. In the present study, results indicated that AREG was able to promote odontoblastic differentiation in hDPSCs through the activation of the ERK MAPK, JNK MAPK, and PI3K/Akt pathways, and facilitated the regeneration and mineralization of hDPSCs. This understanding now has the potential to be applied for the development of novel strategies for the repair and regeneration of damaged pulp tissue.
A previous study demonstrated that AREG was able to inhibit or induce cell growth and proliferation in a range of cell types [14]. Indeed, AREG was shown to induce a potent proliferative response in colon carcinoma cells [27] and an increased proliferation in airway epithelial and smooth muscle cells [28]. AREG plays a vital function in skin wound healing by stimulating keratinocyte proliferation [29,30,31,32], and it has been reported that overexpression of AREG induces self-sufficient growth and survival in lung, liver, colon, breast, and pancreatic carcinoma cells [10, 33,34,35,36,37]. In the present study, we observed that AREG exerted a biphasic effect. Data demonstrated that the numbers of hDPSCs treated with 0.01μg/mL and 0.1μg/mL AREG were significantly increased after 3, 5, and 7 days of treatment while exposure to the higher dose of 1μg/mL AREG reduced cell numbers by day 7. The analysis of cell cycle phases demonstrated that the 0.01 μg/mL and 0.1 μg/mL AREG treatment groups had a marginally higher proliferation index (PI = G2/M+S), in comparison with both the control group and the 1μg/mL AREG-treatment group. Notably, flow cytometry analysis showed that treatment with 0.01-1µg/ml AREG had minimal impact on apoptosis in hDPSCs. These results are largely consistent with those of current studies [29,30,31,32].
AREG is potentially suited for use in tissue repair and regeneration applications as it is not only able to enhance proliferation but can also stimulate differentiation. These data are consistent with previous findings as AREG has been reported to induce the differentiation of neuronal PC12 cells [38] and it has also been shown to be more effective for human mammary epithelial differentiation than other EGFR ligands [39]. Similar results have been reported for human mammary myoepithelial cells [40]. In our study, AREG was shown to induce mineralized nodule formation in a dose-dependent manner, although the high dose of 1 µg/mL AREG induced a marked decrease. In addition, AREG also increased DSPP, BSP, RUNX2, and OCN expression. Together, these data indicated that AREG induced odontogenic differentiation of DPSCs in vitro.
The biomimetic NF-gelatin scaffold used here provided an excellent resource for bone tissue engineering studies due to its physical architecture and chemical composition which is similar to that of natural bone ECM. Constructs exhibited excellent biocompatibility, mechanical stability, and enhancement of osteogenic differentiation [22, 23, 41]. To clarify the role of AREG in regeneration and mineralization in vivo, the DPSC/NF-gelatin-scaffold composites with or without AREG treatment were subcutaneously implanted in nude mice for 4 weeks. Micro-CT analysis was initially used to detect the content, density, and distribution of both bone tissue and mineralized hard tissue formed. The quantitative analysis using Micro CT showed that BV/TV (Bone Volume to Tissue Volume), Tb.Th. (Trabecular Thickness), and Tb.N. (Trabecular Number) were markedly increased in the AREG-stimulated group, although the trabecular separation was decreased. These findings indicate that AREG supplementation produced more newly mineralized tissue together with increased numbers and thicker bone trabeculae, and that the trabecular structure was more compact. Importantly, AREG supplementation also promoted the formation of the mineralized tissue in vivo. Furthermore, both H&E and Masson staining showed increased DPSC penetration into the pores of the scaffold, with relatively large amounts of ECM and collagen fiber deposition and formation in the AREG group. Von Kossa staining indicated that biomineralization occurred in all the composites, with greater mineralized matrix and calcium salt deposition on the composites in the AREG treated group. This finding was consistent with the micro CT quantitative analysis data. All the results demonstrated that AREG facilitated the formation of a dentin-like matrix in DPSC/NF-gelatin-scaffold composites. Furthermore, AREG overexpression experiments showed a similar promotion of odontogenic differentiation, whereas the knockdown experiments inhibited this process. These results indicated that AREG was necessary for odontoblastic differentiation of DPSCs and could facilitate the regeneration and mineralization of hDPSCs. Although not yet determined, it is possible that AREG may contribute substantially to injured pulp repair and regeneration.
AREG plays a vital role in biological processes through its interaction with EGFR and tyrosine phosphorylation of downstream proteins, activating the two major intracellular pathways of MAPK and PI3K/Akt signaling [42, 43]. Interestingly, a previous study has shown that AREG interacts with EGFR, activating PI3K/Akt, and subsequently inducing the NF-κB transcription factor interaction with the MMP-13 promoter, inducing cartilage destruction in osteoarthritis [44]. MAPK signaling has also been observed to participate in AREG-induced morphological effects in MDCK cells [45]. Indeed, AREG stimulation was found to be required for the differentiation of K5 + K19-hMECs through activation of ERK and MAPK but not Akt signaling [39]. Moreover, inhibition of ERK1/2 blocked AREG-induced myoepithelial differentiation [40]. In the present study, ERK MAPK, JNK MAPK, and AKT were phosphorylated in response to AREG stimulation, an action that was markedly antagonized by specific inhibitors of these proteins. In addition, the ERK MAPK, JNK MAPK, and PI3K/Akt inhibitors also significantly reduced mineralized nodule formation and expression of protein mineralization markers. These data support the involvement of ERK MAPK, JNK MAPK, and PI3K/AKT pathways in the AREG-induced differentiation of hDPSCs.
Conclusions
In conclusion, our in vitro and in vivo data demonstrated that AREG was necessary for odontoblastic differentiation of DPSCs and promoted their regeneration and mineralization potential. Furthermore, the investigation of the roles of the MAPK and PI3K/AKT pathways in AREG-induced growth and osteo/odontogenic differentiation in hDPSCs implicated ERK MAPK, JNK MAPK, and PI3K/AKT signaling in AREG-mediated differentiation of hDPSCs. Further investigation of these mechanisms may provide novel targets and treatment modalities for use in the repair of damaged and diseased pulp and the regeneration of dental tissues.
Availability of data and materials
The authors confirm the availability of all data generated or analyzed in this manuscript.
Abbreviations
- AREG:
-
Amphiregulin
- MAPK:
-
Mitogen-activated protein kinase
- PI3K:
-
Phosphatidylinositol 3-kinase
- ERK:
-
Extracellular signal-regulated kinase
- JNK:
-
C-Jun N-terminal kinase
- EGF:
-
Epidermal growth factor
- DSPP:
-
Dentin sialophosphoprotein
- RUNX2:
-
Runt-related transcription factor 2
- OCN:
-
Osteocalcin
- BSP:
-
Bone sialoprotein
- NF-gelatin:
-
Nanofibrous gelatin
- BV/TV:
-
Bone volume to tissue volume
- Tb.Th.:
-
Trabecular thickness
- Tb.N.:
-
Trabecular number
- Tb.Sp.:
-
Trabecular separation
References
Augello A, Kurth TB, De Bari C. Mesenchymal stem cells: a perspective from in vitro cultures to in vivo migration and niches. Eur Cell Mater. 2010;20:121–33. https://doi.org/10.22203/ecm.v020a11.
Kolios G, Moodley Y. Introduction to stem cells and regenerative medicine. Respiration. 2013;85(1):3–10. https://doi.org/10.1159/000345615.
Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, et al. Stem cell properties of human dental pulp stem cells. J Dent Res. 2002;81(8):531–5. https://doi.org/10.1177/154405910208100806.
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A. 2000;97(25):13625–30. https://doi.org/10.1073/pnas.240309797.
d’Aquino R, Graziano A, Sampaolesi M, Laino G, Pirozzi G, De Rosa A, et al. Human postnatal dental pulp cells co-differentiate into osteoblasts and endotheliocytes: a pivotal synergy leading to adult bone tissue formation. Cell Death Differ. 2007;14(6):1162–71. https://doi.org/10.1038/sj.cdd.4402121.
Yu J, He H, Tang C, Zhang G, Li Y, Wang R, et al. Differentiation potential of STRO-1+ dental pulp stem cells changes during cell passaging. BMC Cell Biol. 2010;11:32. https://doi.org/10.1186/1471-2121-11-32.
Alge DL, Zhou D, Adams LL, Wyss BK, Shadday MD, Woods EJ, et al. Donor-matched comparison of dental pulp stem cells and bone marrow-derived mesenchymal stem cells in a rat model. J Tissue Eng Regen Med. 2010;4(1):73–81. https://doi.org/10.1002/term.220.
Berasain C, Avila MA. Amphiregulin. Semin Cell Dev Biol. 2014;28:31–41. https://doi.org/10.1016/j.semcdb.2014.01.005.
Berasain C, García-Trevijano ER, Castillo J, Erroba E, Lee DC, Prieto J, et al. Amphiregulin: an early trigger of liver regeneration in mice. Gastroenterology. 2005;128(2):424–32. https://doi.org/10.1053/j.gastro.2004.11.006.
Willmarth NE, Ethier SP. Autocrine and juxtacrine effects of amphiregulin on the proliferative, invasive, and migratory properties of normal and neoplastic human mammary epithelial cells. J Biol Chem. 2006;281(49):37728–37. https://doi.org/10.1074/jbc.M606532200.
Zuo WL, Yang J, Gomi K, Chao I, Crystal RG, Shaykhiev R. EGF-amphiregulin interplay in airway stem/progenitor cells links the pathogenesis of smoking-induced lesions in the human airway epithelium. Stem Cells. 2017;35(3):824–37. https://doi.org/10.1002/stem.2512.
Takenouchi T, Kadosaka M, Shin SY, Munekata E. Biological actions of the epidermal growth factors-like domain peptides of mouse schwannoma-derived growth factor and human amphiregulin. J Pept Res. 1999;53(2):120–5. https://doi.org/10.1034/j.1399-3011.1999.00004.x.
Raimondo S, Saieva L, Vicario E, Pucci M, Toscani D, Manno M, et al. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J Hematol Oncol. 2019;12(1):2. https://doi.org/10.1186/s13045-018-0689-y.
Shoyab M, McDonald VL, Bradley JG, Todaro GJ. Amphiregulin: a bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc Natl Acad Sci U S A. 1988;85(17):6528–32. https://doi.org/10.1073/pnas.85.17.6528.
Rosa V, Zhang Z, Grande RH, Nör JE. Dental pulp tissue engineering in full-length human root canals. J Dent Res. 2013;92(11):970–5. https://doi.org/10.1177/0022034513505772.
Huang GT, Gronthos S, Shi S. Mesenchymal stem cells derived from dental tissues vs. those from other sources: their biology and role in regenerative medicine. J Dent Res. 2009;88(9):792–806. https://doi.org/10.1177/0022034509340867.
Chen JC, Huang C, Lee IN, Wu YP, Tang CH. Amphiregulin enhances cell migration and resistance to doxorubicin in chondrosarcoma cells through the MAPK pathway. Mol Carcinog. 2018;57(12):1816–24. https://doi.org/10.1002/mc.22899.
Kato M, Inazu T, Kawai Y, Masamura K, Yoshida M, Tanaka N, et al. Amphiregulin is a potent mitogen for the vascular smooth muscle cell line, A7r5. BiochemBiophys Res Commun. 2003;301(4):1109–15. https://doi.org/10.1016/s0006-291x(03)00093-7.
Luo Z, Kohli MR, Yu Q, Kim S, Qu T, He WX. Biodentine induces human dental pulp stem cell differentiation through mitogen-activated protein kinase and calcium-/calmodulin-dependent protein kinase II pathways. J Endod. 2014;40(7):937–42. https://doi.org/10.1016/j.joen.2013.11.022.
He W, Wang Z, Luo Z, Yu Q, Jiang Y, Zhang Y, Zhou Z, et al. LPS promote the odontoblastic differentiation of human dental pulp stem cells via MAPK signaling pathway. J Cell Physiol. 2015;230(3):554–61. https://doi.org/10.1002/jcp.24732.
He W, Qu T, Yu Q, Wang Z, Lv H, Zhang J, et al. LPS induces IL-8 expression through TLR4, MyD88, NF-kappaB and MAPK pathways in human dental pulp stem cells. Int Endod J. 2013;46(2):128–36. https://doi.org/10.1111/j.1365-2591.2012.02096.x.
Qu T, Jing J, Ren Y, Ma C, Feng J, Yu Q, et al. Complete pulpodentin complex regeneration by modulating the stiffness of biomimetic matrix. Acta Biomater. 2015;16:60–70. https://doi.org/10.1016/j.actbio.2015.01.029.
He X, Jiang W, Luo Z, Qu T, Wang Z, Liu N, et al. IFN-γ regulates human dental pulp stem cells behavior via NF-κB and MAPK signaling. Sci Rep. 2017;7:40681. https://doi.org/10.1038/srep40681.
Mori G, Brunetti G, Oranger A, Carbone C, Ballini A, Lo Muzio L, et al. Dental pulp stem cells: osteogenic differentiation and gene expression. Ann N Y Acad Sci. 2011;1237:47–52. https://doi.org/10.1111/j.1749-6632.2011.06234.x.
Shirasawa S, Sugiyama S, Baba I, Inokuchi J, Sekine S, Ogino K, et al. Dermatitis due to epiregulin deficiency and a critical role of epiregulin in immune-related responses of keratinocyte and macrophage. Proc Natl Acad Sci U S A. 2004;101(38):13921–6. https://doi.org/10.1073/pnas.0404217101.
Pastore S, Mascia F, Mariani V, Girolomoni G. The epidermal growth factor receptor system in skin repair and inflammation. J Invest Dermatol. 2008;128(6):1365–74. https://doi.org/10.1038/sj.jid.5701184.
Solic N, Davies DE. Differential effects of EGF and amphiregulin on adhesion molecule expression and migration of colon carcinoma cells. Exp Cell Res. 1997;234(2):465–76. https://doi.org/10.1006/excr.1997.3635.
Hirota N, Risse PA, Novali M, McGovern T, Al-Alwan L, McCuaig S, et al. Histamine may induce airway remodeling through release of epidermal growth factor receptor ligands from bronchial epithelial cells. FASEB J. 2012;26(4):1704–16. https://doi.org/10.1096/fj.11-197061.
Liu B, Xia X, Zhu F, Park E, Carbajal S, Kiguchi K, et al. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell. 2008;14(3):212–25. https://doi.org/10.1016/j.ccr.2008.07.017.
Stoll SW, Johnson JL, Bhasin A, Johnston A, Gudjonsson JE, Rittié L, et al. Metalloproteinase-mediated, context-dependent function of amphiregulin and HB-EGF in human keratinocytes and skin. J Invest Dermatol. 2010;130(1):295–304. https://doi.org/10.1038/jid.2009.211.
Stoll SW, Johnson JL, Li Y, Rittié L, Elder JT. Amphiregulin carboxy-terminal domain is required for autocrine keratinocyte growth. J Invest Dermatol. 2010;130(8):2031–40. https://doi.org/10.1038/jid.2010.98.
Cook PW, Mattox PA, Keeble WW, Pittelkow MR, Plowman GD, Shoyab M, et al. A heparin sulfate-regulated human keratinocyte autocrine factor is similar or identical to amphiregulin. Mol Cell Biol. 1991;11(5):2547–57. https://doi.org/10.1128/mcb.11.5.2547-2557.1991.
Hurbin A, Dubrez L, Coll JL, Favrot MC. Inhibition of apoptosis by amphiregulin via an insulin-like growth factor-1 receptor-dependent pathway in non-small cell lung cancer cell lines. J Biol Chem. 2002;277(51):49127–33. https://doi.org/10.1074/jbc.M207584200.
Shao J, Lee SB, Guo H, Evers BM, Sheng H. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res. 2003;63(17):5218–23. https://doi.org/10.1002/yea.320080508.
Castillo J, Erroba E, Perugorría MJ, Santamaría M, Lee David C, Prieto J, et al. Amphiregulin contributes to the transformed phenotype of human hepatocellular carcinoma cells. Cancer Res. 2006;66(12):6129–38. https://doi.org/10.1158/0008-5472.CAN-06-0404.
Johnson GR, Saeki T, Gordon AW, Shoyab M, Salomon DS, Stromberg K. Autocrine action of amphiregulin in a colon carcinoma cell line and immunocytochemical localization of amphiregulin in human colon. J Cell Biol. 1992;118(3):741–51. https://doi.org/10.1083/jcb.118.3.741.
Funatomi H, Itakura J, Ishiwata T, Pastan I, Thompson SA, Johnson GR, et al. Amphiregulin antisense oligonucleotide inhibits the growth of T3M4 human pancreatic cancer cells and sensitizes the cells to EGF receptor-targeted therapy. Int J Cancer. 1997;72(3):512–7. https://doi.org/10.1002/(sici)1097-0215(19970729)72:3%3c512::aid-ijc21%3e3.0.co;2-c.
Kimura H, Schubert D. Schwannoma-derived growth factor promotes the neuronal differentiation and survival of PC12 cells. J Cell Biol. 1992;116(3):777–83. https://doi.org/10.1083/jcb.116.3.777.
Mukhopadhyay C, Zhao X, Maroni D, Band V, Naramura M. Distinct effects of EGFR ligands on human mammary epithelial cell differentiation. PLoS ONE. 2013;8(10):e75907. https://doi.org/10.1371/journal.pone.0075907.
Pasic L, Eisinger-Mathason TS, Velayudhan BT, Moskaluk CA, Brenin DR, Macara IG, et al. Sustained activation of the HER1-ERK1/2-RSK signaling pathway controls myoepithelial cell fate in human mammary tissue. Genes Dev. 2011;25(15):1641–53. https://doi.org/10.1101/gad.2025611.
Liu X, Smith LA, Hu J, Ma PX. Biomimetic nanofibrous gelatin/apatite composite scaffolds for bone tissue engineering. Biomaterials. 2009;30(12):2252–8. https://doi.org/10.1016/j.biomaterials.2008.12.068.
Toulany M, Baumann M, Rodemann HP. Stimulated PI3K-AKT signaling mediated through ligand or radiation-induced EGFR depends indirectly, but not directly, on constitutive K-Ras activity. Mol Cancer Res. 2007;5(8):863–72. https://doi.org/10.1158/1541-7786.MCR-06-0297.
Shankaran H, Wiley HS. Oscillatory dynamics of the extracellular signal-regulated kinase pathway. Curr Opin Genet Dev. 2010;20(6):650–5. https://doi.org/10.1016/j.gde.2010.08.002.
Chen YT, Hou CH, Hou SM, Liu JF. The effects of amphiregulin induced MMP-13 production in human osteoarthritis synovial fibroblast. Mediators Inflamm. 2014;2014:759028. https://doi.org/10.1155/2014/759028.
Chung E, Graves-Deal R, Franklin JL, Coffey RJ. Differential effects of amphiregulin and TGF-alpha on the morphology of MDCK cells. Exp Cell Res. 2005;309(1):149–60. https://doi.org/10.1016/j.yexcr.2005.05.012.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation of China (Grant Nos. 81771060 and 81970932), Natural Science Basic Research Plan in Shaanxi Province of China (Program No. 2022JZ-42), and the Science and Technology Program of Guizhou Province (ZK[2022] -Normal 638).
Author information
Authors and Affiliations
Contributions
JL contributed to data acquisition, analysis, and interpretation and drafted and critically revised the manuscript; ZW contributed to design, drafted the manuscript, and critically revised the manuscript; JW contributed to design, drafted the manuscript, and performed data acquisition; QG, YF, ZD, and MW contributed to data acquisition and analysis; YB and XL contributed to analysis and data interpretation. PC contributed to design and drafting and critically revised the manuscript; JW and WH contributed to conception, design, data interpretation, and critical revisions of the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study had approval from the Ethics Committee of the Fourth Military Medical University (FMMU), Xi’an, China, with informed consent. All animal surgical procedures were approved by the Animal Care Committee and the Institutional Review Board (IRB) for Human Subjects Research of the Fourth Military Medical University.
Consent for publication
Not applicable.
Competing interests
The authors declared no potential conflicts of competing interest with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure 3
. The picture shows the full length of the strip, and the strips circled in the red box are the strips used in the article. A corresponds to Figure C in Fig 3. B corresponds to Figure C in Fig 5. C corresponds to Figure E in Fig 5. Supplementary Figure 5. The picture shows the full length of the strip, and the strips circled in the red box are the strips used in the article. A corresponds to Figure A in Fig 6. B corresponds to Figure C in Fig 6. C corresponds to Figure G in Fig 6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, J., Wang, Z., Wang, J. et al. Amphiregulin regulates odontogenic differentiation of dental pulp stem cells by activation of mitogen-activated protein kinase and the phosphatidylinositol 3-kinase signaling pathways. Stem Cell Res Ther 13, 304 (2022). https://doi.org/10.1186/s13287-022-02971-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-022-02971-4