
Abstract
Drug resistance is the main culprit of failure in cancer therapy that may lead to cancer relapse. This resistance mostly originates from rare, but impactful presence of cancer stem cells (CSCs). Ability to self-renewal and differentiation into heterogeneous cancer cells, and harboring morphologically and phenotypically distinct cells are prominent features of CSCs. Also, CSCs substantially contribute to metastatic dissemination. They possess several mechanisms that help them to survive even after exposure to chemotherapy drugs. Although chemotherapy is able to destroy the bulk of tumor cells, CSCs are left almost intact, and make tumor entity resistant to treatment. Eradication of a tumor mass needs complete removal of tumor cells as well as CSCs. Therefore, it is important to elucidate key features underlying drug resistance raised by CSCs in order to apply effective treatment strategies. However, the challenging point that threatens safety and specificity of chemotherapy is the common characteristics between CSCs and normal peers such as signaling pathways and markers. In the present study, we tried to present a comprehensive appraisal on CSCs, mechanisms of their drug resistance, and recent therapeutic methods targeting this type of noxious cells.
Similar content being viewed by others


Introduction
Cancer treatment has reached promising breakthroughs during the last decades [1]. Despite all these progresses, chemoresistance has been remained as the main hurdle to achieve success in eliminating cancer cells [2]. Chemoresistance, which means non-optimal respond to chemical drugs, limits drug efficacy [3]. Indeed, chemoresistance is associated with transformation of tumor cells into a more aggressive and/or metastatic forms, [4, 5] and it is considered as the main reason of death in cancer patients [6]. About nine out of ten cancer deaths are due to spreading cancer cells from the primary tumor mass to local and remote tissues (metastasis) [7].
Stem cells (SCs) maintain tissue homeostasis using the unique property of self-renewal [8]. Drug resistance and cancer relapse are occurred due to the presence of a type of stem cells called cancer stem cells (CSCs) within a tumor with the ability to generate heterogeneous lineages of cancer cells based on their self-renewal and differentiation potential [9]. Following chemotherapy, the density of CSCs within the tumor is enriched because CSCs are able to survive and proliferate even after eradication of the majority of cancer cells [10]. Recent findings revealed that chemotherapy induces reprogramming or differentiation of normal cancer cells toward generation of CSC-like cells [11]. Another challenge in cancer therapy is the presence of heterogenous cells in the tumor. These cells have different morphology and proliferative index, are genetically variable, and dissimilar in responding to chemotherapy agents [12]. Multidrug resistance, which means resistance to a broad spectrum of agents, is frequently seen for tumor eradication at the clinical level [13]. Drug resistance could be represented either intrinsically, inherent resistance to drugs, or acquired, which emerges after exposure of tumor cells to chemicals [14]. CSCs have mechanisms that show endogenous resistance at a much higher degree than normal tumor cells [15]. There is an intense need to have a clear picture of the features and mechanisms of resistance employed by CSCs [9].
CSCs
In the nineteenth century, Conheim declared that dormant embryonic stem cells become active, and start to proliferate after certain stimulations leading to the formation of large masses of tumors [16]. This is for the first time that such a tumor-developing role has been assigned to stem cells. Later in 1994, Lapidot and coworkers isolated CSCs from peripheral blood of patients with leukemia. Implantation of the isolated cells into mice generates human leukemia [17]. In the following years, identification of CSCs in breast tumors and other solid tumors such as brain cancer, colorectal cancer, and liver cancer were reported [18]. High number of CSCs increases proliferation capacity, the risk of poor clinical outcome, and genetic instability.
Although CSCs are a small population of cells in the cancerous entity [19], CSCs clusters harbor a huge potential of metastasis, 25 to 50 folds higher than CSCs alone [20]. They justify their appellation due to their similar characteristics with normal stem cells including ability to self-renew, differentiation, and expression of surface stemness markers [21].
The origin of CSCs is not well-understood as they possess a dynamic state. Interestingly, a non-CSCs population could produce CSCs, and a population with high density of CSCs could generate non-CSCs [22]. CSC plasticity within a tumor is largely influenced by some factors such as context and environment [23]. These cells may originate from normal stem cells that become tumorigenic because of genetic or environmental changes. An alternative theory is that the differentiated cells transform into cancer cells with stem-like properties [24]. Others believe that epigenetic plasticity that is often represented in epithelial-mesenchymal-transition (EMT) is involved in the generation of CSCs. In EMT, epithelial characteristics like cell–cell adhesion is lost, and mesenchymal traits like increased motility and invasiveness are gained [25]. Cancer cells show stem cell-like properties such as invasion to neighboring tissues and resistance to therapeutics upon EMT [26]. These alterations are mainly determined epigenetically through methylation of DNA, modifications of histone, and differential genes expression. Notably, EMT is not a biphasic process, and dynamic transitional states such as hybrid epithelial/mesenchymal state could be deployed by the cells [27]. Lack of precision about the origin of CSCs makes many researchers to use alternative terms like cancer stem-like cells or tumor initiating cells [2].
Mechanisms of drug resistance by CSCs
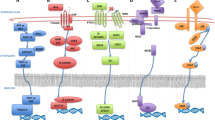
Different intrinsic and extrinsic factors regulate CSCs functions. Intrinsic regulators are genetic, epigenetic, and metabolic effectors while extrinsic regulators are microenvironment and the immune system [28]. Signaling pathways like those in normal stem cells are also found in CSCs though deregulated in some cases [29]. Discovery of CSCs has revolutionized the understanding of how tumors are formed, and it shed light into new avenues of therapy and prognosis. The presence of CSCs now explains the heterogeneous nature of many tumors [30]. Having efficient diagnostic and therapeutic methods in the battle against cancer needs in-depth perception of the mechanisms of resistance used by CSCs. CSCs use several ways to resist against chemical agents in cancer therapy (Fig. 1).

Different mechanisms used by CSCs to resist against chemotherapy
Signaling pathways
CSCs and other stem cells like that of embryonic ones have common characteristics particularly developmental signaling pathways. These highly conserved pathways controls self-renewal of the stem cells [31]. Activation of such pathways expand CSCs yielding resistance to therapy [32]. Wnt/β-catenin, Hedgehog, Notch, Janus kinase-signal transducer and activator of transcription (JAK-STAT), Nuclear factor erythroid 2-related factor 2 (NRF2), and Hippo-YAP/TAZ play critical roles in CSCs [2]. However, they are not tightly regulated in these cells. This dysregulation is one of the underlying reasons of distinction in proliferation, metastasis, and resistance to treatment between cancer and normal stem cells [33]. For instance, deregulated Notch signaling stimulates self-renewal in CSCs in breast cancer and oral squamous cell carcinoma. Furthermore, interconnection of signaling pathways with each other affects the function of the downstream effectors S. For instance, Notch pathway is influenced by signal transfer between Wnt and Hedgehog pathways [34]. Other signaling pathways like Transforming growth factor beta (TGFβ), phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt), and epidermal growth factor receptor (EGFR) along with transcriptional regulators such as SRY-Box Transcription Factor 2 (SOX2), cellular Myc (c-Myc), NANOG, and Octamer-binding transcription factor 4 (OCT4) are active in CSCs to maintain self-renewal and differentiation properties [35]. It was shown that Wnt, Notch, Hedgehog, and Yes-associated protein 1/ Transcriptional coactivator with PDZ-binding motif (YAP/TAZ) signaling pathways contribute substantially to metastasis [36].
Wnt pathway
Wnt pathway regulates cell proliferation, survival, and cell fate as well as embryonic development. Asymmetrical cell division, cell polarity, and cell migration are all under the control of Wnt pathway. This pathway supports somatic stem cells in a variety of tissues [12], and in combination with Notch pathway regulates the expression of the main markers of stemness like c-Myc [37]. Wnt pathway is abnormally activated in many cancers [38]. For instance, nearly 90% of patients with colon malignancy show dysregulated high Wnt signaling in their CSCs [39]. Overactivation of Wnt/B-catenin is a hint for discrimination of tumor cells and differentiated cells [40].
Resistance to combination therapy of IFN-α/5-FU results from activation of Wnt/β-catenin signaling [41]. Endogenous activation of Wnt/β-catenin signaling in OV6+ cells makes them resistant to standard chemotherapy [42]. Nuclear β-catenin translocation and transactivation of Wnt genes like multidrug resistance gene 1, an important player in chemoresistance, activates Wnt/β-catenin pathway in neuroblastoma cancer cells due to overexpression of frizzled-1 Wnt receptor resulting in attenuation of sensitivity to chemotherapy [43]. In ovarian cancer, c-Kit, which is a stem cell factor receptor, activates Wnt/β-catenin and ATP-binding cassette G2 pathway producing chemoresistance [44]. CD44+/CD133+ CSCs are considerably associated with high expression of Wnt pathway in patients with colorectal cancer [45]. Wnt signaling was associated with CSC-related metastasis in breast cancer. Proteins of Wnt signaling like LEF1, cyclin D1, β-catenin, and TCF-4 are highly expressed in breast CSCs in comparison with non-stem cancer cells. In this regard, CSC population as well as stemness-mediated genes including CD44, ALDH1, and Sca-1 were downregulated upon Wnt1 knockdown [46] (43 as 2).
Notch pathway
Proliferation, differentiation, apoptosis, and intercellular communication of normal stem cells are controlled by Notch signaling pathway. This pathway also regulates survival and self-renewal of CSCs. Upregulated Notch components were seen in CSCs of pancreas tumor [12]. Notch-1 pathway activates nuclear factor kappa light chain enhancer of activated B cells (NF-kB) followed by stimulation of CSC to proliferate via downregulation of some anti-apoptotic proteins like survivin and B-cell lymphoma 2 [47]. Activation of Notch-1 signaling leads to EMT phenotype in those lung cancer cells that are resistant to gefitinib [48, 49]. In glioma, CD133+ CSCs contribute to the activation of genes involved in Notch and Hedgehog pathways, and gives resistance against temozolomide treatment [50]. Chemical drugs such as oxaliplatin increases γ-secretase activity and instigates Notch-1 receptor and its upcoming target Hes-1 in colon cancer. Hence, inhibitors of γ-secretase is a good alternative to sensitize colon cancer cells to chemotherapy [51]. Tumorsphere formation by CSCs was increased or decreased by activation or knock down of Notch target gene (Hes1), respectively [52]. Maintenance of CSCs and their resistance to platinum are regulated by Notch signaling pathway, particularly Notch3, in ovarian cancer [53]. Cisplatin therapy activates Notch signaling, and enriches CD133+ cells in lung adenocarcinoma [54].
According to a study on 115 samples of breast tumor tissues, Notch activity were significantly associated with ALDH1 expression [55]. Notch signaling was associated with overactivation of iota, a protein kinase C, that plays role in survival of stem-like cells in glioblastoma. Deactivation of iota causes proliferation reduction and apoptosis of glioblastoma CSCs [56].
Hedgehog pathway
This pathway controls several cellular and molecular processes during embryogenesis as well as development and homeostasis of adult tissues. Hedgehog pathway also regulates CSCs in different cancer types [12]. In fact, self-renewal property is maintained in CSCs relying on overexpression of the Hedgehog pathway [8]. It was shown that Hedgehog inhibitors decrease proliferation, survival, self-renewal, and clonogenicity of CSCs in glioma. Furthermore, expression of stemness genes like NANOG, SOX2, and OCT4 are decreased upon inhibition of Hedgehog pathway [57]. Regarding Hedgehog signaling, the expression of SMO (a G protein–coupled receptor protein) is inversely related to the CSCs activity in chronic myeloid leukemia in a way that SMO knockout leads to CSCs enhancement and disease progression [58]. Hedgehog ligand in breast cancer cells reprograms cancer fibroblasts to activate FGF5 expression and fibrillary collagen production in order to support niche giving resistance to therapy [59].
PTEN/PI3K/Akt pathway
This pathway controls many important physiological and pathological processes like cell proliferation, angiogenesis, metabolism, differentiation, and survival. Mutations of phosphatase and tensin homolog (PTEN) are evident in more than half of the glioblastoma multiforme specimens [12]. Interestingly, stemness genes (OCT4, SOX2, and NANOG) are expressed in PTEN-knock out neural stem cells. These cells retain differentiation capability, and produce different cell lineages. They also acquire neoplastic phenotype including increased growth, resistant to death, and elevated migration potential along with in vivo invasiveness [12].
One of the functions of PI3K/Akt pathway is regulation of ABCG2 activity through its homing in the plasma membrane. The side population phenotype of glioma cancer stem-like cells is promoted thereafter due to PTEN loss [60]. With regard to the role of Notch pathway in resistance of glioma stem cells, γ-secretase inhibitors cause glioma stem cells to be more vulnerable to therapy because of PI3K/Akt activation and overexpression of truncated apoptotic isoform of Mcl-1 [61]. Stroma-derived factor 1a and CXCR4, as its cognate receptor, involve in the migration of hematopoietic cells to the bone marrow (125, 126 as 1). Binding of this ligand to the receptor plays an important role in resistance of leukemic cells to apoptosis after chemotherapy [62]. Therefore, AMD3100, as an inhibitor of CXCR4, prevents Akt phosphorylation and induction of PARP cleavage totally increasing the sensitivity of cancer cells to chemotherapy in leukemia [63]. As mobilization of hematopoietic stem cells depends on CXCR4, inhibition of CXCR4 makes multiple myeloma cells vulnerable to chemotherapy by disintegrating adhesion of myeloma cells to the stromal cells in the bone marrow [64]. In a PI3K/Akt manner, PD-L1 regulates stemness markers including OCT-4, NANOG, and BMI1 in breast cancer [65].
JAK/STAT pathway
Expression of different cytokines and growth factors to control proliferation, growth, and immune functions are regulated by JAK/STAT pathway. It is also involved in hematopoiesis, neurogenesis, and maintenance of self-renewal in embryonic stem cell. For example, STAT3 induces angiogenesis, immunosuppression, and tumor invasion, which all substantiate in progression of gliomagenesis [12]. IL-6/JAK2/STAT3 pathway are more active in breast CSCs compared with other cancer cells, and thereby, number of CSCs as well as growth of xenograft are decreased following JAK2 inhibition [66]. JAK/STAT signaling was active in CSCs of acute myeloid leukemia, and JAK2 inhibition hampers the growth of leukemic stem cells [67]. Self-renewal and tumorigenicity of glioblastoma stem-like cells were activated by FOXM1-PDGFA-STAT3 signaling [68].
NF-kB pathway
NF-kB is implicated in the regulation of innate and adaptive immunity. It is a fundamental mediator in inflammatory responses. NF-Kb regulates cell survival, activation, and differentiation of cells. Activation of NF-kB upregulates the expression of Interleukin 3 (IL-3) and granulocyte–macrophage colony-stimulating factor (GM-CSF), which in turn provokes the proliferation and survival of stem cells in leukemia [12]. In breast and lung cancer cells, NF-kB signaling activates LIN28/TCF4 interaction continued by promoting stemness and metastasis [69]. Breast cancer cells and CSCs highly express IL-8 after chemotherapy leading to the formation of an inflammatory loop between NF-kB and STAT3 signaling pathways [70]. Antagonizing Toll-like receptor-7 lowers the growth rate of CSCs in hepatocellular carcinoma through TLR7-IKK-NF-kB-IL6 signaling pathway [71].
Hippo-YAP/TAZ signaling
During normal organ development, this pathway controls cell fate and differentiation of progenitor cells [72]. Dedifferentiation and expansion of stem or progenitor cells are among the other functions of Hippo-YAP pathway [73]. Activation of YAP/TAZ signaling leads to the dedifferentiation of cancer cells to gain CSCs characteristics like self-renewal and chemoresistance [72]. The impact of YAP on the promoters of mammary stem cells results in the induction of breast CSCs [74]. Direct upregulation of SOX9 is performed by YAP that is known as a strong inducer of CSC properties [75]. Also, SOX2 activates YAP which maintain CSCs in osteosarcoma and glioblastoma [76]. Altogether, huge body of evidence confirm that YAP/TAZ significantly involve in the maintenance and progression of CSCs.
Microenvironment
The microenvironment, in which division and differentiation of stem cells happen, controls stem cell functions through intercellular communication between stem cells and differentiated peers. Also, interactions of the cell with extracellular matrix, paracrine communication, hormone signaling, the effects of growth factors and mediators, and physicochemical characteristics of the microenvironment affect its components [29, 77]. These delicately acting signals protect and regulate normal stem cells. This is the case for CSCs as well. CSCs are exposed to a variety of growth factors and cytokines released by different types of stromal cells [78]. Plasticity of CSCs represents in reversible phenotypic changes like induction of expression of some markers upon microenvironment-related stimuli [79]. CSCs are also capable to recruit components of tumor microenvironment [80].
The microenvironment promotes CSCs survival in two ways: activation of specific molecular signaling pathways, and formation of a physical barrier to prevent penetration of therapeutic agents [8]. The cells present in the nearby microenvironment of CSCs instigate some signaling pathways like Notch and Wnt leading to metastasis, evasion from anoikis, and alteration of divisional property [9]. Other than helping CSCs to expedite their divisional dynamics by providing inherent properties of self-renewal and differentiation capabilities, microenvironment maintains CSCs in a primitive development state [2].
It is concluded that microenvironment is an influential factor that establishes a balance between self-renewal and differentiation of CSCs, possibly directing them toward proliferation, invasion, and metastasis [81]. Endothelial cells, cancer-associated fibroblasts, mesenchymal stromal/stem cells, inflammatory cells, and extracellular matrix constitute the tumor microenvironment. This pool of cells and mediators aids CSCs to grow and proliferate [79].
Cancer-associated fibroblasts
Fibroblast is the most prevalent component of tumor microenvironment especially in certain cancers like breast tumors. These cells are known as cancer-associated fibroblasts (CAFs) Coculture of breast cancer cells with CAFs increases resistance to tamoxifen up to 4.4 folds [8]. CAFs secrete a variety of growth factors, cytokines, and chemokines. Hepatocyte growth factor, as one of the secreting elements, activates MET receptor protecting CSCs against apoptosis following cetuximab therapy in colorectal cancer [82]. TGF-β is the most important mediator secreted by CAFs that promotes EMT and drug resistance [8]. It was reported that following paclitaxel treatment in breast cancer, TGF-β signaling and IL-8 expression are augmented, and thereby, population of CSC is enriched leading to tumor recurrence [83].
CAFs increase secretion of some cytokines and chemokines, support self-renewal and invasiveness of CSCs, and ultimately provide chemoresistance [84]. Exosomes that secreted from CAFs help resistance to 5-fluorouracil in colon cancer [85]. Some other mediators like Neuregulin 1 (NRG1) are secreted by CAFs, which provoke stemness and activate NF-KB signaling. Type I collagen is also secreted by CAFs, and decreases drug influx [8]. On the other hand, CSCs have the ability to differentiate into CAFs-like cells leading to tumor maintenance and survival [86]. Chemicals that specifically target CAFs show their therapeutic benefits in the treatment of breast cancer [87].
Immune cells
CSCs directly attenuate immune surveillance facilitating tumor growth [88]. There are many reports regarding the contribution of inflammatory cells like monocytes and macrophages to the tumor microenvironment [89]. Macrophages, dendritic cells, myeloid-derived suppressor cells, and other components of the innate immunity regulate CSCs and tumor growth [90]. Macrophages in the tumor microenvironment are called tumor-associated macrophages (TAMs) or M2 macrophages. CSCs release proinflammatory cytokines and chemokines in order to recruit macrophages [8]. Once reaching to the tumor niche, macrophages transform into TAMs. Some cytokines like CCL2 promote infiltration of TAMs into the tumors, in primary or even metastatic regions. Interaction of TAMs with tumor cells is mediated through a variety of factors such as TGF-β and TNF-α that stimulate EMT process [8]. Cancer cells of pancreatic adenocarcinoma secrete colony-stimulating factor 1. This is followed by attraction and stimulation of its receptor on TAMs [91]. TAMs increase the expression of cytidine deaminase that catabolizes gemcitabine. TAMs also activate certain signaling pathways in CSCs such as STAT3 and Hedgehog via secretion of some cytokines like milk fat globule epidermal growth factor 8 and Interleukin-6 (IL-6) totally providing chemoresistance. Activation of STAT3 by TAMs inhibits antitumor responses raised by CD8+ T lymphocytes, and provokes characteristics of CSC in pancreatic tumor cells [92]. Thus, TAMs are potent targets for downregulation of tumor-initiating cells. Another player among immune components is dendritic cells that makes a bridge between innate immunity and adaptive immunity. These cells are capable of inducing chemoresistance and tumorigenesis [12].
Mesenchymal stem cells
One type of adult stem cells is mesenchymal stem cells (MSCs). Under normal conditions, MSCs act as immunomodulators, and differentiate into specialized cells supporting hemostasis. In the tumor microenvironment, MSCs activate NF-KB signaling that induces CSC phonotype and chemoresistance [29]. It was shown that abnormal DNA hypermethylation of the two tumor suppressor genes (HIC1 and RassF1A) change MSCs to CSCs causing resistance to cisplatin [93]. Breast CSCs secrete IL-6 that recruits MSCs, which in turn leads to CXCL7 release. This cytokine accelerates tumor growth and chemoresistance in animal models. Also, it was shown that IL-6 activates STAT-3 signaling that enriches CSC population, and results in trastuzumab resistance in breast cancer. CSCs are able to differentiate into endothelial cells, and endothelial cells differentiate into MSCs suggesting that CSCs could generate MSCs [8]. Based on higher expression of integrin-linked kinase in mesenchymal cells compared with epithelial cells, the former possess elevated expression of genes involved in metastasis and invasion, and accordingly, they become resistant to some chemotherapy drugs like erlotinib, gefitinib, and cetuximab [94]. Trastuzimab resistance was gained by PTEN downregulation and c-Src activation following coculturing of breast cancer cells and MSCs [95].
Extracellular matrix
Extracellular matrix (ECM) is constituted from a population of molecules that mostly produced by fibroblasts. Cancer cells attach to the ECM in order to form a tumor. The high stiffness of ECM in solid tumors is like a physical barrier that protect CSCs to be reached by therapeutic drugs. Furthermore, proteins of the ECM interact with membrane proteins of CSCs. This interaction activates signaling pathways involved in stemness, proliferation, and eventually chemoresistance [8].
Endothelial cells and oxygen content
Oxygen and nutrients are delivered to the tumor microenvironment by blood vessels. It was shown that this vasculature plays an important role in maintaining the properties of CSCs like self-renewal. Endothelial cells of the vascular system release several growth factors such as EGF, which induces EMT process and CSC characteristics in many tumors [8]. Irregular shape of blood vessels in the tumor microenvironment hampers drug delivery to the CSCs [96]. CSCs themselves show a complex interaction with the endothelial cells. CSCs recruit, and also able to directly differentiate into endothelial cells. It was shown that hypoxia and glucose shortage are inducers of CSCs differentiation into endothelial cells. So, maintenance and development of self-renewal in CSCs are influenced by the hypoxic condition [8]. Interestingly, CSCs have a tendency to be in a close proximity of the hypoxic regions within the tumors [97]. In fact, certain features of CSCs are expressed in the hypoxic conditions [98].
Hypoxia provokes drug resistance via activation of stem-related pathways and promotion of quiescence. Activation of hypoxia-inducible factor (HIF)-1α leads to the expression of many effectors involving in EMT process like Wnt, Hedgehog, and Notch pathways besides upregulation of some stemness markers [99]. Hypoxia contributes to CSCs enrichment via hyperactivation of VEGF, IL-6, and stemness-related genes like NANOG, OCT4, and EZH2, as shown in pancreatic tumor cells [100]. Increasing the expression of insulin-like growth factor 1 due to hypoxia through HIF1α and activation of IGF1 receptor induce resistance to gefitinib in lung CSCs [101].
Cellular responses to hypoxia are mainly regulated by HIF-1α, which is degraded at high oxygen levels. In low oxygen tension, it becomes activated, moves to the nucleus, undergoes dimerization with HIF-1β, and eventually, stimulates the expression of response elements in 60 genes that are important in angiogenesis, oxygen delivery, and activation of survival pathways. HIF-1α decreases reactive oxygen species (ROS) as well. This function results in drug resistance and induction of quiescence in CSCs [8]. Cellular growth is retarded and quiescence is induced in the tumor cells in the conditions of low oxygen and nutrients deprivation [80].
ATP-binding cassette transporters
Protecting normal stem cells is a vital task because these cells are biologically necessary to support the pool of cells in different tissues. Therefore, stem cells are equipped with several mechanisms to avoid apoptosis or senescence. Meanwhile, CSCs use the same mechanisms to repel anti-cancer therapeutic drugs like cisplatin, paclitaxel, docetaxel, and cetuximab [102]. There are protein in the cell membrane called ATP-binding cassette (ABC transporters) that efficiently translocate molecules across the membrane. Rapid transportation of chemotherapeutic agents from inside to the extracellular space causes multidrug resistance in CSCs [103]. Some believe that this may be the most powerful resistance strategy that is used by CSCs [104].A great number of these proteins are expressed on the cell surface of CSCs. Of the important ones are ABCB1, ABCG2, ABCB5, and ABCC1. Staining a population of tumor cells with Hoechst 33342 dye and Rhodamine 123 dye helps to identify overexpressed ABC transporters, and consequently, determine the location of CSCs within a tumor [8]. Notably, stemness markers like c-Myc are involved in CSCs chemoresistance by increasing the expression of ABC transporters. CD44, another stemness marker, activates chemoresistance through anti-apoptotic proteins and ABC transporters [2].
ABCG2/ABCB1 transporters are highly expressed in hematopoietic stem cells [105]. Coexpression of ABCG2 and CD133 is a hint for identification of tumor-initiating cells in melanoma cancer. Progression of melanoma is clinically correlated with ABCB5 expression. Rather than other stem cell markers, simultaneous expression of ABCB1, ABCB5, and ABCC2 were seen in a subpopulation of melanoma cells [106]. Side population cells in glioma that efflux Hoechst 33342 dye and express ABCG2 transporter through PTEN/PI3K/Akt signaling are strongly tumorigenic, and show resistance against temozolomide [60].
Dormancy
In CSCs, cell death mechanisms are controlled via modulation of cell cycle. This strategy, which also contributes to EMT, creates resistance. While conventional cancer therapy mainly targets cancer cells relying on their rapid proliferation, slow cycling-cancer cells are not affected [12]. CSCs maintain themselves in a quiescent state resulting in an inherent resistance to such treatments [107], and are found in melanoma, glioblastoma, squamous cell carcinoma, and bladder cancer [108].
Such cells promote long-term tumor growth, and generate a cell population with highly proliferative potential eventually leads to cancer relapse [109]. Furthermore, the imposed injuries caused by chemotherapy drugs recruit quiescent cells during the interval between treatment cycles resulting in repopulation of tumor cells [110].
Genetically distinct subclones produced by heterogeneous population within a tumor have diverse functions [111]. Moreover, previously dormant or slow-proliferating cancer cells are activated after chemotherapy. Totally these situations reduce the treatment efficacy and increase the risk of cancer relapse [111]. Cancer relapse several years after initial treatment reveals that CSCs could survive through the dormant state during treatment course and reactivate afterward [112].
Ferroptosis
Ferroptosis is a form of non-apoptotic cell death. It differs from apoptosis, necrosis, and autophagy as ferroptosis is an important type of cell death in cancer cells. It occurs as a result of imbalance between lipid hydroperoxides (possibly due to overproduction of lipid ROS) and specific detoxification enzymes. Lipid metabolism, ROS production, and iron addiction are different between cancer cells and normal peers. CSCs, similar to other cells, metabolize lipid, though in a higher degree, in order to provide enough energy for their different functions. In CSCs, cytoplasmic organelles called lipid droplets are generated for lipid storage. Lipids are protected in these organelles from peroxidation, and this causes resistance against ferroptosis. Lipid desaturation is the other protective mechanism against lipid peroxidation in CSCs. Conversion of saturated fatty acids to mono-unsaturated fatty acids (MUFAs) prevents ferroptosis because MUFAs reduce ROS and poly-unsaturated fatty acids (PUFA)-containing phospholipids. Lipid desaturation is regulated by stearoyl-CoA desaturase 1. This enzyme along with lipid droplets are implicated in stemness of CSCs [2].
CSCs are unique with respect to iron metabolism due to their unusual expression of some proteins involved in iron import, export, and storage [2]. For instance, ferritin as the iron storage protein, is upregulated in CSCs, and consequently, CSCs have higher iron level. This intracellular storage of iron augments stem-related characteristics like proliferation and maintenance [2]. It was shown that low level of iron is associated with downregulation of EMT markers like E-cadherin in breast cancer cells [113].
Autophagy
Autophagy is a fundamental determinant for CSCs aggressiveness. In fact, autophagy is a bidirectional road that promotes survival or death by either sustaining the cell viability or activating a phagosome-lysosome-dependent feature. Cellular context determines direction of the cell toward each pathway. Autophagy is able to alter the microenvironment in favor of CSCs by making an imbalance between CSCs and normal cells. Autophagy also instigates ferroptosis to induce cell death via degradation of ferritin [2]. In the case of hypoxia and nutrient deprivation in the microenvironment, autophagy is increased [114].
Reactive oxygen species
One of the main reasons of chemoresistance in CSCs is the low content of ROS [115]. Scavenging system in CSCs is strong enough to keep the intracellular ROS at a level similar to that of normal stem cells [116]. Breast CSCs demonstrate enhanced expression of scavenging genes like superoxide dismutase, glutathione peroxidase, and catalase [116,117,118]. Activation of oncogenic transcription factors like c-Myc increases ROS levels followed by NRF2 activation. NRF2 is an important transcription factor that upregulates genes involved in detoxification and antioxidant activity such as NADPH quinone oxidoreductase (NQO-1), glutathione (GSH), and glutathione peroxidase (GPX) [2]. Indeed, NRF2 activates the expression of efflux transporters and anti-apoptotic proteins like BCL-2 [119]. Also, it was shown that NRF2 contributes considerably to iron homeostasis [120]. In this regard, NRF2 could be considered as a promising target in CSCs therapy [2].
High NRF2 level in CSCs is related to the high expression of some markers like CD44 and ALDH [121]. Enhanced expression of CD44 translates into higher GSH synthesis, and thereby, stronger shield against ROS [122]. Some cancer cells like those in oral cavity have high contents of antioxidant enzymes such as SOD2 and peroxiredoxin 3. This leads to low levels of ROS representing in resistance to cisplatin [123]. Another protecting mechanism of CSCs is oxidation of intracellular aldehyde by ALDH in order to prevent ROS-induced cell injury [124]. In normal condition, ALDH has diverse functions such as acetaldehyde oxidation, cellular detoxification, and regulation of stem cell tasks [117]. ALDH activity is considered as a selective marker for identification of CSCs in many types of tumors [125]. The main isoform of ALDH is ALDH1. This isoform has a detoxifying function by concurrent reduction of ROS and generation of antioxidant compounds like NADP. ALDH1 also keeps cells safe against alkylating agents like paclitaxel [126]. It was shown that ALDH1A1 and ALDH3A1 are overexpressed in the cancer cells [117]. Also, ALDH1A1 augments the activation of DNA repair mechanism in such cells [127]. Elevated expression of ALDH1 is a predictor of poor response and prognosis in esophageal cancer [128]. CSCs that express ALDH1A1 are significantly resistant to gefitinib, cisplatin, etoposide, and fluorouracil in the lung tissues [129].
Activation of DNA damage responses increases the number of CSCs to about 2–4 folds [8]. In glioma, activation of DNA damage checkpoints is more effective in CD133+ cells after radiation compared with CD133− peers [15]. DNA repair mechanisms are enhanced in glioblastoma stem cells compared with progenitor cells [130]. So, glioblastoma stem cells profoundly become sensitive by inhibition of PARP and ATR [131]. Overexpression of polymerase η gives CSCs resistance to cisplatin in ovarian cancer cells, and sensitivity of CSCs to cisplatin is increased by activating mir-93, which regulates the expression of polymerase η [132].
However, response to DNA damage has opposing consequences. Normal stem cells use this strategy in order to provide optimal functions in healthy tissues while leads to CSCs survival and resistance. High levels of replication stress are tolerated by CSCs via this mechanism. Even, CSCs resist against chemical therapeutics that are specifically designed to damage DNA [131].
Other mechanisms
There are other mechanisms that give CSCs resilience [2]. Epigenetic changes including elevation of the euchromatin mark H3-lys4 trimethylation, reduction of the heterochromatin mark H3-lys9 dimethylation, and increasing transcriptional mark H3-lys36 trimethylation are happened during TGF-β mediated EMT. These alterations, particularly the first one, implicate in chemoresistance [133]. EZH2, a PcG member and a subunit of polycomb repressor complex 2 that provokes gene silencing by histone trimethylation, participates in pancreatic cancer chemoresistance via turning off the p27 tumor suppressor gene [134]. In chronic myelogenous leukemia, quiescent stem cells that are resistant to imatinib mesylate are effectively eliminated after treatment with inhibitors of histone deacetylase [135].
Binding of NANOG to the promoter region decreases K14 and K27 histone H3 acetylation. This event activates histone deacetylase 1, and represses cell cycle inhibitors of CDKN2D and CDKN1B inducing stem-like features. Also, silencing of E3 ubiquitin-ligase TRIM17 and NOXA upregulate MCL-1 facilitating immune resistance and chemoresistance [136]. Complete hypermethylation of the hMLH1 promoter leads to loss of DNA mismatch repair gene [137]. This arrests cell death and cell cycle after chemotherapy-induced DNA damage and consequently, leads to low rate of survival in ovarian and breast cancer patients [138].
MicroRNAs are non-coding RNAs that have roles in chemoresistance, EMT activation, and acquisition of CSCs characteristics [139]. Repression of PI3K signaling by mir-126 increases self-renewal and decreases differentiation ability in leukemic stem cells through keeping them in a quiescent state and restricting their entry to the cell cycle [140]. By targeting PTEN, mir-10b regulates expression of stem cell markers in breast cancer lines [141].
CSCs produce different proinflammatory signals like Interferon (IFN) regulatory factor-5, a transcription factor that specifically induces chemoresistance via induction of macrophage colony-stimulating factor (M-CSF) [142]. Significant expression of IFN regulatory factor-5 by CSCs is assumed to be necessary for tumorigenic activities in myeloid cells [142]. In some cases, CSCs use inflammatory mediators to resist against therapeutic drugs. For instance, breast CSCs use IL-6 inflammatory loop to resist against trastuzumab [143]. Telomere shortening causes chromosome instability, cell fusion, and senescence. Telomerase, an enzyme that adds repeats to the telomeres end, is active in tumor cells, and give them self-renewal. Telomerase activity was found in nearly 90% of breast carcinoma cells while it was completely absent in breast normal cells [8].
Therapeutic strategies
Conventional chemotherapy eliminates bulk of proliferating cancer cells, but CSCs survive and move to a higher level of invasiveness and chemoresistance. On the other hand, chemotherapy provokes heterogeneity in both normal and cancer cells that consequently attenuates treatment efficiency. Higher expression of CSC biomarkers in the tumor is associated with poor prognosis [2]. Different therapeutic approaches have been designed to sensitize Achilles’ hill of the tumor, CSCs. However, these advancements though promising are not desirable possibly because they are not fine-tuned for eradication of CSCs [2]. Hence, the way toward finding effective cancer treatment is still in its infancy, and needs more developments for future treatment plans (Table 1). Here, we describe some of the important and undergoing strategies that specifically target mechanisms used by CSCs for resistance (Fig. 2).

Specific therapeutic strategies for elimination of cancer stem cells
Blocking the signaling pathways
Blocking vital pathways of CSCs like Notch, Wnt, and Hedgehog that are necessary to their self-renewal would be a favorable approach [35]. Some small molecules such as evodiamine, IGC-001, and acridine derivatives are developed to deactivate CSCs through Wnt inhibition [144]. Salinomycine and its derivative, ironomycin, are inhibitors of Wnt/β-catenin pathway that also interfere with ABC drug transporters [8]. It was shown that Notch and Hedgehog signaling pathways are inhibited by Honokiol and cyclopamine, respectively [8]. γ-secretase inhibitors are among the most important class of Notch blockers [117]. Vismodegib is an inhibitor of Hedgehog pathway that was approved by FDA [8]. JAK/STAT pathway is downregulated by molecules such as EC-70124 and napabucasin [144]. One of the therapeutic strategies against CSCs could be delivered through targeting EMT pathway [145]. Three general target groups have been designed in this regard. These include regulators of EMT extracellular inducers and extracellular matrix components, certain transcription factors that promote EMT transcriptome and downstream effectors, and regulators of EMT-related transcription factors and epigenetic effectors [146].
Restricting the microenvironment
Surprisingly, deactivation of CSCs solely does not guarantee tumor eradication. Tumor microenvironment, which harbors different cells and multiple factors, enhances survival, plasticity, and drug resistance of CSCs. Therefore, targeting tumor microenvironment increases CSCs sensitivity [80]. To substantiate, differentiation of non-CSCs into CSCs is done through reprogramming processes like EMT [8]. Components of tumor microenvironment like CAFs or TAMs release EMT inducers [8]. ECM and related proteins are valuable targets in the era of CSCs eradication. Destruction of hyaluronic acid and subsequent reduction in stroma reduce interstitial pressure while increasing the expansion of the vasculature; these totally augment the accessibility of chemotherapy drugs to CSCs [147]. CSCs also use their microenvironment to escape from immune surveillance. In this regard, components of microenvironment like CAFs are suitable targets in order to make CSCs available for immune system [148]. Also, innate immune cells release protumorigenic factors during chronic inflammation. It is important especially in those cancers (e.g. colon cancer) that inflammation plays a significant role. Hence, anti-inflammatory anti-cyclooxygenase-2 considerably decreases the risk of colon cancer [149].
Inhibiting ABC transporters
Making ABC transporters dysfunctional in tandem with restoration of apoptosis pathways sensitizes CSCs to chemotherapy [150]. Metformin showed promising results for attenuating resistance via inhibition of an ABC transporter [8].
Targeting dormant cells
Induction of activation in dormant CSCs to activate and commence the cell cycling increases their vulnerability to certain therapies. Meanwhile, efforts are undergoing to specifically identify and target dormant CSCs [151].
Inducing ferroptosis
Activation of ferroptosis offers a safe treatment because cancer cells are more susceptible to ferroptotic death in comparison with normal cells. Ironomycin, ebselen, and other pyrazole and benzylisothiourea-containing agents as well as artemisinin derivatives are among the many ferroptosis inducing drugs that specifically target CSCs. Combinatorial therapy of ferroptosis inducing agents like erastin with chemotherapeutic agents like cisplatin, doxorubicin, and temozolomide increases treatment efficiency synergistically. However, different types of cancer cells show varying degrees of vulnerability to this type of death. Some cancer types like that of the breast, colon, and lung are more resistant to ferroptosis than some others like renal cell carcinoma and diffuse large B cell lymphomas. This variety may be partly explained by different degrees of aggressiveness of the tumors [2].
Enhancing antioxidant repertoire and altering iron metabolism
One less studied fact about CSCs is overexpression of antioxidant and detoxifying genes as well as aberrant metabolism of iron and lipid [152]. Some researchers suggested that targeting iron metabolism in CSCs is a good approach for cancer therapy. Salinomycin sequesters iron in lysosomes, degrades ferritin, and promotes the Fenton reaction totally produces lipid peroxides leading to ferroptosis [153].
Working on surface markers
Surface markers of CSCs are potential targets [154]. For instance, CD44 and CD133, surface biomarkers of CSCs, are targeted by H90 and oxytetracycline [155]. However, lack of specific markers in CSCs hinders the efforts to reach favorable outcomes [8]. Unfortunately, surface markers of CSCs in different tumor types are not similar [154]. Another challenging issue is that these markers are also expressed by normal cells [154].
Knockdown of ALDH
Targeting ALDH is an alternative choiceto eliminate CSCs [8]. CSCs become vulnerable to cyclophosphamide after knockdown of two isoforms of ALDH [156]. High intracellular level of retinoic acid decreases the expression of ALDH. Therefore, all-trans retinoic acid is a potent therapeutic agent in acute promyelocytic leukemia. Combination of all-trans retinoic acid with retinoids suppress ALDH activation in a synergistic manner [117].
Immnunotherapy
Immunotherapy is considered as a breakthrough in cancer therapy that specifically targets CSCs within a tumor entity. In this type of treatment, several components of the immune system including natural killer (NK) cells and γδT cells of the innate immunity, antibodies of acquired humoral immunity, CSC-based dendritic cells, and CSC-primed cytotoxic T lymphocytes (CTLs) of acquired cellular immunity recognize and kill CSCs-associated molecules. Finding specific targets such as appropriate antigens like markers of CSCs, antigens involved in the interactions of CSC with microenvironment, cytokines, and immune checkpoint, and genetic alterations in CSCs are of eminent importance in immunotherapy [157].
Modifying epigenetic changes
Epigenetic changes like histone acetylation and DNA methylation affect the regulation of drug resistance in CSCs [9]. CSCs are associated with aberrant expression of histone deacetylase (HDAC) [158]. Inhibition of HDAC and targeting histones per se are assumed as the therapeutic strategies against CSCs [84]. HDAC inhibitors like vorinostat suppress CSCs both in in vitro and in vivo experiments of different tumor cells. DNA methylation inhibits tumor suppressor genes promoting chemoresistance. For instance, hypermethylation of promoter region of insulin-like growth factor binding protein-3 downregulates its expression, and consequently, modifies cell growth, transformation, and survival toward enhancing chemoresistance [9].
Other strategies
While a growing body of evidence revealed that miRNA and other long noncoding RNAs substantially contribute to the regulation of vital CSCs characteristics like self-renewal, asymmetric cell division, tumor initiation, drug resistance, and tumor relapse [159], targeting such miRNAs is considered in CSCs-based therapy. Also, CSCs possess a strict dependence on the biogenesis of mitochondrion, which is inhibited by doxycycline. In this way, doxycycline sensitizes CSCs to paclitaxel as well. Doxycycline reduces metastasis in breast cancer along with attenuating the tumor burden in pancreas cancer [8]. Of other strategies is the development of some drugs like atovaquone and artesunate that inhibit oxygen consumption and induce mitochondrial dysfunction in CSCs [160].
Limitations
Although these novel strategies seem efficient especially in combination with conventional therapeutics, challenges still hamper the desirable goal. Some of the novel drugs may be toxic due to lack of specificity as there are similarities in markers and pathways between CSCs and normal stem cells [33]. Another important issue is the heterogeneity within a tumor. This causes different cells to express different markers with different dysregulated pathways [148]. Hence, designing a single agent that efficiently destroys CSCs despite all of these diversities is a laborious task if possible at all. Therefore, seeking the unique characteristic of CSCs, which are absent in normal stem cells as well as other vital contributors, is the gate of success toward elimination of cancer cells.
Conclusion
Major therapeutic advancements have been reached in the battle against cancer. However, certain properties of the tumors that are mainly derived from CSCs challenge optimal treatment. Targeting this population of cancer cells is of utmost essence. To reach this goal, obtaining an in-depth understanding of the mechanisms that make CSCs alive despite anticancer therapy is very critical. Implementation of the strategies that discriminate between CSCs and other normal peers advance this road forward significantly.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Abbreviations
- CSCs:
-
Cancer stem cells
- SCs:
-
Stem cells
- ALDH1:
-
Aldehyde dehydrogenase 1
- EMT:
-
Epithelial-mesenchymal-transition
- JAK-STAT:
-
Janus kinase-signal transducer and activator of transcription
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- PTEN:
-
Phosphatase and tensin homolog
- CAFs:
-
Cancer-associated fibroblasts
- TAMs:
-
Tumor-associated macrophages
- MSCs:
-
Mesenchymal stem cells
- ECM:
-
Extracellular matrix
- HIF:
-
Hypoxia-inducible factor
- ROS:
-
Reactive oxygen species
- ABC transporters:
-
ATP-binding cassette
- MUFAs:
-
Mono-unsaturated fatty acids
- PUFA:
-
Poly-unsaturated fatty acids
- NQO-1:
-
NADPH quinone oxidoreductase
- GSH:
-
Glutathione
- GPX:
-
Glutathione peroxidase
- NK:
-
Natural killer
- CTLs:
-
Cytotoxic T lymphocytes
- HDAC:
-
Histone deacetylase
- YAP/TAZ:
-
Yes-associated protein 1/Transcriptional coactivator with PDZ-binding motif
- c-Myc:
-
Cellular Myc
- SOX2:
-
SRY-Box Transcription Factor 2
- OCT4:
-
Octamer-binding transcription factor 4
- TGFβ:
-
Transforming growth factor beta
- PI3K/Akt:
-
Phosphatidylinositol 3-kinase/protein kinase B
- EGFR:
-
Epidermal growth factor receptor
- NF-kB:
-
Nuclear factor kappa light chain enhancer of activated B cells
- IL-3:
-
Interleukin 3
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- NRG1:
-
Neuregulin 1
- IL-6:
-
Interleukin-6
- IFN:
-
Interferon
- M-CSF.:
-
Macrophage colony-stimulating factor
References
Zhang H, Chen J. Current status and future directions of cancer immunotherapy. J Cancer. 2018;9(10):1773–81.
Elgendy SM, Alyammahi SK, Alhamad DW, Abdin SM, Omar HA. Ferroptosis: an emerging approach for targeting cancer stem cells and drug resistance. Crit Rev Oncol Hematol. 2020;155: 103095.
Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–54.
Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Mazeedi M, et al. The role of tumor microenvironment in chemoresistance: to survive, keep your enemies closer. Int J Mol Sci. 2017;18(7):1586.
Fesler A, Guo S, Liu H, Wu N, Ju J. Overcoming chemoresistance in cancer stem cells with the help of microRNAs in colorectal cancer. Epigenomics. 2017;9(6):793–6.
Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299–309.
Qian CN, Mei Y, Zhang J. Cancer metastasis: issues and challenges. Chin J Cancer. 2017;36(1):38.
Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug resistance driven by cancer stem cells and their niche. Int J Mol Sci. 2017;18(12):2574.
Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, et al. Cancer Stem Cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018:5416923.
Alison MR, Lim SM, Nicholson LJ. Cancer stem cells: problems for therapy? J Pathol. 2011;223(2):147–61.
Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol. 2016;11:47–76.
Makena MR, Ranjan A, Thirumala V, Reddy AP. Cancer stem cells: road to therapeutic resistance and strategies to overcome resistance. Biochim Biophys Acta Mol Basis Dis. 2020;1866(4): 165339.
Efferth T, Konkimalla VB, Wang YF, Sauerbrey A, Meinhardt S, Zintl F, et al. Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin Cancer Res. 2008;14(8):2405–12.
Schmidt F, Efferth T. Tumor heterogeneity, single-cell sequencing, and drug resistance. Pharmaceuticals (Basel). 2016;9(2):33.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60.
Capp JP. Cancer stem cells: from historical roots to a new perspective. J Oncol. 2019;2019:5189232.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–8.
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–5.
Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. 2017;7(3):339–48.
Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–22.
Zhao W, Li Y, Zhang X. Stemness-related markers in cancer. Cancer Transl Med. 2017;3(3):87–95.
Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. 2015;17(3):260–71.
Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–94.
Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012;44(12):2144–51.
Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18(2):128–34.
Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–73.
Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556(7702):463–8.
Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203–17.
Carnero A, Garcia-Mayea Y, Mir C, Lorente J, Rubio IT, LLeonart ME. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat Rev. 2016;49:25–36.
Wang K, Wu X, Wang J, Huang J. Cancer stem cell theory: therapeutic implications for nanomedicine. Int J Nanomed. 2013;8:899–908.
Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445–64.
Kurth I, Peitzsch C, Baumann M, Dubrovska A. The role of cancer stem cells in tumor radioresistance. Cancer Stem Cells. 2014. https://doi.org/10.1002/9781118356203.ch35.
Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol. 2015;42(Suppl 1):S3-17.
Takebe N, Warren RQ, Ivy SP. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011;13(3):211.
Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512.
Nwabo Kamdje AH, Takam Kamga P, Tagne Simo R, Vecchio L, Seke Etet PF, Muller JM, et al. Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog. Cancer Biol Med. 2017;14(2):109–20.
Yan Y, Liu F, Han L, Zhao L, Chen J, Olopade OI, et al. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J Exp Clin Cancer Res. 2018;37(1):256.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–73.
Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468–76.
Radulescu S, Ridgway RA, Cordero J, Athineos D, Salgueiro P, Poulsom R, et al. Acute WNT signalling activation perturbs differentiation within the adult stomach and rapidly leads to tumour formation. Oncogene. 2013;32(16):2048–57.
Noda T, Nagano H, Takemasa I, Yoshioka S, Murakami M, Wada H, et al. Activation of Wnt/β-catenin signalling pathway induces chemoresistance to interferon-α/5-fluorouracil combination therapy for hepatocellular carcinoma. Br J Cancer. 2009;100(10):1647–58.
Yang W, Yan H-X, Chen L, Liu Q, He Y-Q, Yu L-X, et al. Wnt/β-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68(11):4287–95.
Flahaut M, Meier R, Coulon A, Nardou K, Niggli F, Martinet D, et al. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/β-catenin pathway. Oncogene. 2009;28(23):2245–56.
Chau W, Ip C, Mak A, Lai H, Wong AJO. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/β-catenin–ATP-binding cassette G2 signaling. Oncogene. 2013;32(22):2767–81.
Zhang S-S, Huang Z-W, Li L-X, Fu J-J, Xiao B. Identification of CD200+ colorectal cancer stem cells and their gene expression profile. Oncol Rep. 2016;36(4):2252–60.
Jang G-B, Kim J-Y, Cho S-D, Park K-S, Jung J-Y, Lee H-Y, et al. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. 2015;5(1):1–15.
Fulda S, Pervaiz S. Apoptosis signaling in cancer stem cells. Int J Biochem Cell Biol. 2010;42(1):31–8.
Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, Oyama T. Epithelial− mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010;30(7):2513–7.
Xie M, He C, Wei S. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. Cancer Res Clin. 2015;27:298–304.
Ulasov IV, Nandi S, Dey M, Sonabend AM, Lesniak MS. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133+ glioma stem cells to temozolomide therapy. Mol Med. 2011;17(1):103–12.
Meng RD, Shelton CC, Li Y-M, Qin L-X, Notterman D, Paty PB, et al. γ-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009;69(2):573–82.
Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, et al. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS ONE. 2014;9(3): e91983.
McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS, et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci USA. 2012;109(43):E2939–48.
Liu Y-P, Yang C-J, Huang M-S, Yeh C-T, Wu AT, Lee Y-C, et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013;73(1):406–16.
Zhong Y, Shen S, Zhou Y, Mao F, Lin Y, Guan J, et al. NOTCH1 is a poor prognostic factor for breast cancer and is associated with breast cancer stem cells. OTT. 2016;9:6865.
Phillips E, Lang V, Bohlen J, Bethke F, Puccio L, Tichy D, et al. Targeting atypical protein kinase C iota reduces viability in glioblastoma stem-like cells via a notch signaling mechanism. Int J Cancer. 2016;139(8):1776–87.
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17(2):165–72.
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–9.
Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan C-L, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nature. 2018;9(1):1–18.
Bleau A-M, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–35.
Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28(1):17–28.
Zeng Z, Samudio IJ, Munsell M, An J, Huang Z, Estey E, et al. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther. 2006;5(12):3113–21.
Azab AK, Runnels JM, Pitsillides C, Moreau A-S, Azab F, Leleu X, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113(18):4341–51.
Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells. 2010;28(6):1019–29.
Almozyan S, Colak D, Mansour F, Alaiya A, Al-Harazi O, Qattan A, et al. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int J Cancer. 2017;141(7):1402–12.
Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+ CD24–stem cell–like breast cancer cells in human tumors. J Clin Investig. 2011;121(7):2723–35.
Cook AM, Li L, Ho Y, Lin A, Li L, Stein A, et al. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood. 2014;123(18):2826–37.
Gong A, Wei P, Zhang S, Yao J, Yuan Y, Zhou A, et al. FoxM1 drives a feed-forward STAT3-activation signaling loop that promotes the self-renewal and tumorigenicity of glioblastoma stem-like cells. Cancer Res. 2015;75(11):2337–48.
Chen C, Cao F, Bai L, Liu Y, Xie J, Wang W, et al. IKKβ enforces a LIN28B/TCF7L2 positive feedback loop that promotes cancer cell stemness and metastasis. Cancer Res. 2015;75(8):1725–35.
Jia D, Li L, Andrew S, Allan D, Li X, Lee J, et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017;8(7):2932.
Ren X, Wang F, Ji B, Gao C. TLR7 agonist induced repression of hepatocellular carcinoma via the TLR7-IKK-NF-κB-IL6 signaling pathway. Oncol Lett. 2016;11(5):2965–70.
Park JH, Shin JE, Park HW. The role of hippo pathway in cancer stem cell biology. Mol Cells. 2018;41(2):83.
Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157(6):1324–38.
Kim T, Yang S-J, Hwang D, Song J, Kim M, Kyum Kim S, et al. A basal-like breast cancer-specific role for SRF–IL6 in YAP-induced cancer stemness. Nat Commun. 2015;6(1):1–15.
Song S, Ajani JA, Honjo S, Maru DM, Chen Q, Scott AW, et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014;74(15):4170–82.
Basu-Roy U, Bayin NS, Rattanakorn K, Han E, Placantonakis DG, Mansukhani A, et al. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat Commun. 2015;6(1):1–14.
Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124(6):1111–5.
Liu H, Zhang W, Jia Y, Yu Q, Grau GE, Peng L, et al. Single-cell clones of liver cancer stem cells have the potential of differentiating into different types of tumor cells. Cell Death Dis. 2013;4(10): e857.
Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–38.
Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 2017;50(3):117–25.
Daverey A, Drain AP, Kidambi S. Physical intimacy of breast cancer cells with mesenchymal stem cells elicits trastuzumab resistance through Src activation. Sci Rep. 2015;5:13744.
Luraghi P, Reato G, Cipriano E, Sassi F, Orzan F, Bigatto V, et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 2014;74(6):1857–69.
Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, et al. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Investig. 2013;123(3):1348–58.
Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J Exp Med. 2013;210(13):2851–72.
Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE. 2015;10(5): e0125625.
Nair N, Calle AS, Zahra MH, Prieto-Vila M, Oo AKK, Hurley L, et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci Rep. 2017;7(1):6838.
Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9(1):2897.
Silver DJ, Sinyuk M, Vogelbaum MA, Ahluwalia MS, Lathia JD. The intersection of cancer, cancer stem cells, and the immune system: therapeutic opportunities. Neuro Oncol. 2016;18(2):153–9.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29.
Sultan M, Coyle KM, Vidovic D, Thomas ML, Gujar S, Marcato P. Hide-and-seek: the interplay between cancer stem cells and the immune system. Carcinogenesis. 2017;38(2):107–18.
Amit M, Gil ZJO. Macrophages increase the resistance of pancreatic adenocarcinoma cells to gemcitabine by upregulating cytidine deaminase. Oncoimmunology. 2013;2(12): e27231.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–41.
Teng IW, Hou PC, Lee KD, Chu PY, Yeh KT, Jin VX, et al. Targeted methylation of two tumor suppressor genes is sufficient to transform mesenchymal stem cells into cancer stem/initiating cells. Cancer Res. 2011;71(13):4653–63.
Fuchs BC, Fujii T, Dorfman JD, Goodwin JM, Zhu AX, Lanuti M, et al. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008;68(7):2391–9.
Daverey A, Drain AP, Kidambi S. Physical intimacy of breast cancer cells with mesenchymal stem cells elicits trastuzumab resistance through Src activation. Sci Rep. 2015;5(1):1–13.
Fukumura D, Jain RK. Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. J Cell Biochem. 2007;101(4):937–49.
Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, Yeger H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells. 2008;26(7):1818–30.
Liang D, Ma Y, Liu J, Trope CG, Holm R, Nesland JM, et al. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer. 2012;12:201.
Liu L, Salnikov AV, Bauer N, Aleksandrowicz E, Labsch S, Nwaeburu C, et al. Triptolide reverses hypoxia-induced epithelial-mesenchymal transition and stem-like features in pancreatic cancer by NF-κB downregulation. Int J Cancer. 2014;134(10):2489–503.
Bao B, Ali S, Ahmad A, Azmi AS, Li Y, Banerjee S, et al. Hypoxia-induced aggressiveness of pancreatic cancer cells is due to increased expression of VEGF, IL-6 and miR-21, which can be attenuated by CDF treatment. PLoS ONE. 2012;7(12): e50165.
Murakami A, Takahashi F, Nurwidya F, Kobayashi I, Minakata K, Hashimoto M, et al. Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. Sci Rep. 2014;9(1):e86459.
da Silva SD, Hier M, Mlynarek A, Kowalski LP, Alaoui-Jamali MA. Recurrent oral cancer: current and emerging therapeutic approaches. Front Pharmacol. 2012;3:149.
Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18(7):452–64.
Alisi A, Cho WC, Locatelli F, Fruci D. Multidrug resistance and cancer stem cells in neuroblastoma and hepatoblastoma. Int J Mol Sci. 2013;14(12):24706–25.
Scharenberg CW, Harkey MA, Torok-Storb B. The Journal of the American Society of Hematology. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99(2):507–12.
Welte Y, Adjaye J, Lehrach HR, Regenbrecht CRA. Cancer stem cells in solid tumors: elusive or illusive? Cell Commun Signal. 2010;8(1):1–10.
Chen W, Dong J, Haiech J, Kilhoffer MC, Zeniou M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016;2016:1740936.
Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol Mech Dis. 2016;11:47–76.
Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–6.
Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517(7533):209–13.
Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AM, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339(6119):543–8.
Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112(13):4793–807.
Guo W, Zhang S, Chen Y, Zhang D, Yuan L, Cong H, et al. An important role of the hepcidin-ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim Biophys Sin (Shanghai). 2015;47(9):703–15.
Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S, et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol. 2012;227(3):325–35.
Bystrom LM, Guzman ML, Rivella S. Iron and reactive oxygen species: friends or foes of cancer cells? Antioxid Redox Signal. 2014;20(12):1917–24.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3.
Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol. 2015;31:16–27.
Krause M, Dubrovska A, Linge A, Baumann M. Cancer stem cells: radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Deliv Rev. 2017;109:63–73.
Jia Y, Chen J, Zhu H, Jia ZH, Cui MH. Aberrantly elevated redox sensing factor Nrf2 promotes cancer stem cell survival via enhanced transcriptional regulation of ABCG2 and Bcl-2/Bmi-1 genes. Oncol Rep. 2015;34(5):2296–304.
Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23: 101107.
Ryoo IG, Choi BH, Ku SK, Kwak MK. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: implications for cancer stem cell resistance. Redox Biol. 2018;17:246–58.
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–400.
Chang CW, Chen YS, Chou SH, Han CL, Chen YJ, Yang CC, et al. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 2014;74(21):6291–305.
Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M, et al. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014;74(13):3579–90.
Nakahata K, Uehara S, Nishikawa S, Kawatsu M, Zenitani M, Oue T, et al. Aldehyde Dehydrogenase 1 (ALDH1) is a potential marker for cancer stem cells in embryonal rhabdomyosarcoma. PLoS ONE. 2015;10(4): e0125454.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Clark DW, Palle K. Aldehyde dehydrogenases in cancer stem cells: potential as therapeutic targets. Ann Transl Med. 2016;4(24):518.
Ajani J, Wang X, Song S, Suzuki A, Taketa T, Sudo K, et al. ALDH-1 expression levels predict response or resistance to preoperative chemoradiation in resectable esophageal cancer patients. Mol Oncol. 2014;8(1):142–9.
Huang C-P, Tsai M-F, Chang T-H, Tang W-C, Chen S-Y, Lai H-H, et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013;328(1):144–51.
Yuan M, Eberhart CG, Kai MJO. RNA binding protein RBM14 promotes radio-resistance in glioblastoma by regulating DNA repair and cell differentiation. Oncotarget. 2014;5(9):2820.
Vitale I, Manic G, De Maria R, Kroemer G, Galluzzi L. DNA damage in stem cells. Mol Cell. 2017;66(3):306–19.
Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, et al. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc Natl Acad Sci. 2015;112(14):4411–6.
McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011;18(8):867–74.
Ougolkov AV, Bilim VN, Billadeau DD. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin Cancer Res. 2008;14(21):6790–6.
Zhang B, Strauss AC, Chu S, Li M, Ho Y, Shiang K-D, et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell. 2010;17(5):427–42.
Song K-H, Choi CH, Lee H-J, Oh SJ, Woo SR, Hong S-O, et al. HDAC1 upregulation by NANOG promotes multidrug resistance and a stem-like phenotype in immune edited tumor cells. Cancer Res. 2017;77(18):5039–53.
Strathdee G, MacKean M, Illand M, Brown R. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene. 1999;18(14):2335–41.
Gifford G, Paul J, Vasey PA, Kaye SB, Brown R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res. 2004;10(13):4420–6.
Ma C, Ding YC, Yu W, Wang Q, Meng B, Huang T. MicroRNA-200c overexpression plays an inhibitory role in human pancreatic cancer stem cells by regulating epithelial-mesenchymal transition. Minerva Med. 2015;106(4):193–202.
Lechman ER, Gentner B, Ng SW, Schoof EM, van Galen P, Kennedy JA, et al. miR-126 regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell. 2016;29(2):214–28.
Bahena-Ocampo I, Espinosa M, Ceballos-Cancino G, Lizarraga F, Campos-Arroyo D, Schwarz A, et al. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016;17(5):648–58.
Yamashina T, Baghdadi M, Yoneda A, Kinoshita I, Suzu S, Dosaka-Akita H, et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res. 2014;74(10):2698–709.
Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47(4):570–84.
Müller S, Caneque T, Acevedo V, Rodriguez R. Targeting cancer stem cells with small molecules. Isr J Chem. 2017;57(3–4):239–50.
Du B, Shim JS. Targeting epithelial–mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21(7):965.
Malek R, Wang H, Taparra K, Tran PT. Therapeutic targeting of epithelial plasticity programs: focus on the epithelial–mesenchymal transition. Cells Tissues Organs. 2017;203(2):114–27.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–29.
Sun HR, Wang S, Yan SC, Zhang Y, Nelson PJ, Jia HL, et al. Therapeutic strategies targeting cancer stem cells and their microenvironment. Front Oncol. 2019;9:1104.
Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol. 1997;39:1–20.
Talukdar S, Emdad L, Das SK, Sarkar D, Fisher PB. Evolving strategies for therapeutically targeting cancer stem cells. Adv Cancer Res. 2016;131:159–91.
Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. 2011;17(15):4936–41.
Begicevic RR, Arfuso F, Falasca M. Bioactive lipids in cancer stem cells. World J Stem Cells. 2019;11(9):693–704.
Mai TT, Hamaï A, Hienzsch A, Cañeque T, Müller S, Wicinski J, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9(10):1025–33.
Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu-Economescu M. Therapies targeting cancer stem cells: current trends and future challenges. World J Stem Cells. 2015;7(9):1185–201.
Song Y, Kim IK, Choi I, Kim SH, Seo HR. Oxytetracycline have the therapeutic efficiency in CD133(+) HCC population through suppression CD133 expression by decreasing of protein stability of CD133. Sci Rep. 2018;8(1):16100.
Moreb JS, Mohuczy D, Ostmark B, Zucali JR. RNAi-mediated knockdown of aldehyde dehydrogenase class-1A1 and class-3A1 is specific and reveals that each contributes equally to the resistance against 4-hydroperoxycyclophosphamide. Cancer Chemother Pharmacol. 2007;59(1):127–36.
Dammeijer F, Lievense LA, Kaijen-Lambers ME, van Nimwegen M, Bezemer K, Hegmans JP, et al. Depletion of tumor-associated macrophages with a CSF-1R kinase inhibitor enhances antitumor immunity and survival induced by DC immunotherapy. Cancer Immunol Res. 2017;5(7):535–46.
Witt AE, Lee CW, Lee TI, Azzam DJ, Wang B, Caslini C, et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene. 2017;36(12):1707–20.
Deng J, Yang M, Jiang R, An N, Wang X, Liu B. Long Non-Coding RNA HOTAIR regulates the proliferation, self-renewal capacity, tumor formation and migration of the cancer stem-like cell (CSC) subpopulation enriched from breast cancer cells. PLoS ONE. 2017;12(1): e0170860.
Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, et al. Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget. 2016;7(23):34084–99.
Acknowledgements
I would like to express my sincere thanks to Shiraz Institute for Cancer Research for the invaluable guidance and support in completing this project.
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
IR-J conceived of the presented idea. IR-J and HR developed the theory. IR-J and MR wrote the manuscript. HR and IR-J edited the final version of the manuscript. All authors discussed the results and contributed to the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rezayatmand, H., Razmkhah, M. & Razeghian-Jahromi, I. Drug resistance in cancer therapy: the Pandora's Box of cancer stem cells. Stem Cell Res Ther 13, 181 (2022). https://doi.org/10.1186/s13287-022-02856-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-022-02856-6