
Abstract
Recent advances in single-cell RNA sequencing (scRNA-seq) and epithelium lineage labeling have yielded identification of multiple abnormal epithelial progenitor populations during alveolar type 2 (ATII) cell differentiation into alveolar type 1 (ATI) cells during regenerative lung post-fibrotic injury. These abnormal cells include basaloid/basal-like cells, ATII transition cells, and persistent epithelial progenitors (PEPs). These cells occurred and accumulated during the regeneration of distal airway and alveoli in response to both chronic and acute pulmonary injury. Among the alveolar epithelial progenitors, PEPs express a distinct Krt8+ phenotype that is rarely found in intact alveoli. However, post-injury, the Krt8+ phenotype is seen in dysplastic epithelial cells. Fully understanding the characteristics and functions of these newly found, injury-induced abnormal behavioral epithelial progenitors and the signaling pathways regulating their phenotype could potentially point the way to unique therapeutic targets for fibrosing lung diseases. This review summarizes recent advances in understanding these epithelial progenitors as they relate to uncovering regenerative mechanisms.
Similar content being viewed by others


Introduction
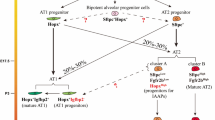
In normal lung, epithelial cells are the key components for both environmental barrier and gas exchange function [1]. Diverse populations of epithelial cells populate upper, mid, and lower airways. In the trachea and proximal conducting airways, the predominate epithelial cells are secretory, goblet, ciliated, and club cells (Fig. 1). In the terminal bronchiolar epithelium, neuroendocrine and basal cells (BCs) are present, along with BASCs. In the alveoli space, thin, flat ATI cells and cuboidal ATII cells are the two primary epithelial populations [2]. Depending on the location within the lung and the severity of injury, different progenitor populations respond to restore the damaged epithelium. Club cells are mainly found in the bronchioles, and they self-renew, give rise to new ciliated cells [3], and contribute to proximal airway repair. BCs have extensive proliferative potential, self-renewal capacity, and the capability to differentiate into club cells and ciliated cells [4]. In the lower airways and alveoli, epithelial cell turnover is slow compared to other epithelial linings (e.g., intestinal and epidermal). Bronchioalveolar stem cells (BASCs) are progenitor cells that originate in the bronchioalveolar duct junctions (BADJs) and migrate down into the alveolar space [5]. BASCs, which co-express Scgb1a1 and Sftpc, contribute descendants to both bronchioles and alveoli [5] and, as demonstrated by lineage tracing, include club cells, ciliated cells, ATII and ATI cells [6, 7]. ATII cells are responsible for both self-renewal and differentiation into ATI cells [8].

Airway progenitors migrated from the proximal airway to the distal airway. In the healthy airway, basal cells differentiate into ciliated, goblet, tuft, and club cells. In pathogenic states, basal-like and basaloid cells populate the airway. BASCs differentiate into club and ATII cells in the middle airway. ATII cells differentiate into AEP, ATII proliferating, ATII primed, cell cycle arrest, and ATI cells. In pathological alveolar states, ATIIs differentiate into PEPs (DATPs, PATs, Krt8 + , and ADI). This figure is created with BioRender.com
In the injured lung, ATII cells serve the critical role of replacing damaged and abnormal alveolar epithelial cells [9, 10]. Recent studies have identified ATII transition types (or PEPs) that occur as ATII cells differentiate into ATI cells. During homeostasis and normal repair, PEPs do not accumulate or contribute significantly to the total number of alveolar epithelial cells, but rather efficiently complete their transfer into ATI cells. However, under certain disease conditions, such as idiopathic pulmonary fibrosis (IPF), PEPs accumulate and do not effective repopulate ATI cells [10,11,12], a defect that would contribute markedly to impaired respiration.
In this review, we will discuss these diverse epithelial progenitor lineages, the signaling pathways involved in controlling the fate of these progenitors, and the diseases and genetic predispositions in which alterations in their behavior contribute to maladaptive injury repair mechanisms. We also discuss current research approaches that are used to study these epithelial progenitors.
Progenitors in alveolar niche and ATII transition cells/PEPs during alveolar regeneration
The alveolar space comprises over 70% of the lung and is the basic ventilatory unit involved in regulating gas exchange. In the adult lung, there are regional progenitor cell populations. BASCs are a specific type of progenitor that expresses both club cell marker SCGB1A1 and ATII cell marker SFTPC and are located at the BADJ [5]. BASCs contribute to both bronchiolar repair and alveolar repair and can give rise to club, ciliated, ATII, and, in turn, ATI cells. ATII cells are mainly account for alveolar repair (Fig. 1). Recent scRNA-seq and lineage tracing studies has suggested that BASCs can be segregated into two subpopulations (BASC-1 and BASC-2) [6], indicating that BACSs are a heterogeneous population. The BASC-1 subpopulation is enriched with Cyp2t2 and Scgb1a1 gene expression, while BASC-2 subpopulation is highly expressed with H2-Ab1, CD74, and H2-Aa genes. As Scgb1a1 is a club cell maker and CD74 labels ATII cells [13, 14], the sharing of gene expression signatures with club and ATII cells suggests the differentiation potential of the subpopulations of BASCs. BASC-1 might correspond to bronchiolar repair by differentiating into club cells, and BASC-2 may differentiate into ATII cells for alveolar repair, but these assumptions warrant further investigation. Using a Sftpc+ ATII depletion mouse model, it was concluded that there are more Sftpc+ ATII cells derived from alveolar ATII cells than from BASCs [15]. However, a recent study found that mouse Scgb1a1 derived Sftpc+ BASCs (also termed secretory ATII cells, sAT2 cells) are capable of long-term self-renewal, more so than residual AT2 cells in in vitro organoids [16]. It remains unknown in lung fibrosis if ATII cell depletion occurs within fibrotic area or if BASCs or the remaining ATII cells are the major source for regeneration of ATII cells in the alveoli.
Besides BASCs, studies have shown that club cells [3], BCs, integrin α6β4 positive cells, H2-K1 high, and Uroplakin3a (Upk3a) expressing airway epithelial progenitors can migrate to the alveoli and give rise to ATII cells under conditions of severe injury that involve broad epithelial denudation [17,18,19,20,21]. An abnormal cluster of Fstl1+ secretory cells were found during defective club cells (or secretory cells) transition to ATII cells [16]. These cells were marked by Id3, Porcn, and Cdkn1c and blocked the fate conversion from secretory to ATII cells when lacking sustained Notch activity. The assumption that these above-mentioned airway progenitor cells regenerate ATII cells is partially based on the structurally contiguous of airway and alveoli. It is assumed that the severity of the injury determines the degree that ATII cells derived from airway progenitors can be replenished. Consistently, in other organs, ductal epithelial cells can contribute to functional unit progenitor cell regeneration, such as β cell in pancreas [22] and hepatocytes in liver [23].
A majority of the ATII cells utilize self-renewal to maintain the alveoli population during homeostasis of slow cellular cycling and in injury conditions of ATII cell loss [15, 24]. Alternatively, a population of facultative ATII progenitor cells (AEPs), which is Wnt-responsive and labeled by Transmembrane 4 L Six Family Member 1 (TM4SF1), were suggested to participate in alveolar regeneration [25]. AEPs also show distinct genetic profiles from ATIIs in lung development, including the import gene Nkx2.1. In addition, AEPs were shown to have ~ 40% of their chromatin open (via scATAC seq) compared to ATII cells.
Besides their self-renewal function, ATII cells can differentiate into ATI cells. Originally, it was thought that ATII cells are the only genitor of ATI cells. Except for the minor contribution of alveolar epithelial from bipotential progenitors [26, 27], there is no evidence that other airway progenitor cells can differentiate into ATI cells without initially differentiating into ATII cells. However, there is emerging evidence that ATII transition cells or PEPs occur during the differentiation of ATII cells to ATI cells, which were regarded as potential key characteristics of fibrotic injury. The ATII transition cells, previously described in nitrogen dioxide-induced lung injury [28], was recently found in pseudomonas aeruginosa injured and post-pneumonectomy ATII lineage labeled mouse lungs [29, 30]. These transition epithelial progenitors accumulated following injury in persistent Notch activation condition, expressed low levels of ATII and ATI cell markers, and were associated with retarded differentiation of ATII cells into ATI cells [29].
Furthermore, a subset of distinct ATII transition cells or PEPs population, damage-associated transient progenitors (DATPs), were characterized by gene expression of Krt8+, Cldn4, and Lgals3, and were extremely rare at steady state but arose during alveolar regeneration after single-dose bleomycin injury. DATPs accumulated following inflammatory signal IL-1β stimulation, blocked ATI differentiation, and impaired alveolar regeneration [10]. The DATPs subpopulation was also found in COVID19 patient lungs and were associated with a progressive decrease in ATI cell abundance [31]. In addition, a pre-alveolar type-1 transitional cell state (PATS) were observed during the differentiation of ATII cells into ATI cells. These PATS occurred in mouse lungs post-single-dose bleomycin injury and presented in the fibrotic regions of the idiopathic pulmonary fibrosis (IPF) lungs [12]. By using Sftpc ATII and Krt19 lineage labeled mice, PATS that displayed a stretched morphology were shown to derive from ATII and Krt19 lineage cells post-bleomycin injury [30].
Lastly, a unique Krt8+ alveolar differentiation intermediate (ADI) population was found during alveolar regeneration post-single-dose bleomycin lung injury, was derived from ATII cells, and might give rise to ATI cells as demonstrated by scRNA velocity analyses [11]. These abundant Krt8+ ADI belong to ATII transition cells or PEPs population and had stretched morphology (partially spread, but not cuboidal), features p53 and NFkB activation, and displayed transcriptional features of epithelial–mesenchymal transition (EMT) and cellular senescence. The Krt8+ ADI cells were also found to be persistently present in human fibrotic lungs and appeared in acute respiratory distress syndrome (ARDS) caused by influenza-A and pneumococcal infection as well as interstitial lung disease patients [11]. These reports showed, regardless of the different naming of PEPs that they occurred in different injury/disease settings with the similarities resulting from abnormal differentiation of ATII cells. It remains unclear whether, and under what conditions, the various PEPs might have the potential of completing terminal differentiation into ATI cells. Figure 2 summarizes the markers found in the previously discusses PEPs.

Venn diagram shows the types of published PEPs and signature marker genes. Genes labeled in bold represent overlapping markers. All genes are presented with the standard nomenclature for homo sapiens even if work was one in model organisms
There are potentially two types of ATI cells in adult mouse lungs, as have been shown in post-pneumonectomy (PNX) alveolar regeneration [9]. Among this population of ATI1 cells, Hopx+/Igfbp2+ ATI cells are considered as terminal ATI cells while Hopx+/Igfbp2− ATI cells are not. It is not fully understood whether these PEPs have the capacity to fully differentiate into terminal and functional ATI cells and what are the underlining mechanisms that drive the process.
There are other subclusters of ATII cells that have been shown by recent scRNA-seq analysis. For example, activated ATII cells marked by injury-induced genes (Lcn2 and Il33) and Mki67 + proliferation clusters were found concurrently with Krt8+ ADI cells in bleomycin injured mouse lung [11]. In another study, ATII cells were segregated into proliferating, cell cycle arrest (senescence) and transdifferentiating (intermediate) ATII subpopulations in mouse lung 7 days post-LPS injury [32]. Primed ATII cells and cycling ATII cells were found concurrently with DATPs [10]. Further, stepwise differentiation trajectories were suggested from primed ATII cells to DATPs and then to ATI cells by scRNA velocity analysis. These studies started to explore the subpopulation of ATII cells and the interrelationship of the ATII subpopulations. It is hoped that the subpopulation of ATII cells and the differentiation checkpoints could be dissected in a deeper degree by multiple methods, especially in the human lung.
ATII self-renewal has been shown to be influenced by the mesenchymal cells in the alveolar niche/microenvironment [33, 34]. Pdgfra+ cells can facilitate the self-renewal and differentiation of ATII cells in vitro [15]; it is reported that Axin2+/Pdgfra+ cells supported ATII cell regeneration through functional signaling pathways, including interleukin-6 (IL6)/Stat3, Bmp, and Fgf pathways [15, 35, 36]. In addition, Lgr5-expressing mesenchymal cells promoted ATII cell differentiation [35]. Furthermore, fibroblasts provide short-range Wnt signals to neighboring ATII stem cells and maintain their stemness [24]. Hedgehog activation in lung mesenchymal fibroblasts led to reduced ATII regeneration and can be rescued by HGF in vitro [37]. Profibrotic mesenchymal cells retarded ATII cell growth and were associated with suppressed growth hormone receptor (GHR) expression in secreted vesicles [38]. These reports demonstrated that the impairment of ATII renewal in the chronically injured lungs could be due to the loss of supportive paracrine signaling from mesenchymal cells. The changes that occur in secreted supportive factors from mesenchymal cells during injury subsequently impact ATII renewal and make the disease more severe. It will be interesting to investigate whether niche supporting factors can influence the occurrence and differentiation of the PEPs.
Involvement of airway progenitor cells for alveolar remodeling
BCs are speculated to be the major airway stem/progenitor cells and have been shown to differentiate into airway club cells, ciliated cells, tuft cells, and secretory goblet cells [39]. In cases of severe injury with massive depletion of ATII cells, BCs can migrate to lower airway or alveoli, become distal basal-like cells, and promote alveoli regeneration by differentiating into ATII and ATI cells [18, 21, 40]. BC expansion is gradually recognized as a common feature of epithelial remodeling, as originally discovered in influenza-induced mouse injury models as BC “pod” [40], and later in chronic lung diseases [41] and IPF [42] as BC hyperplasia, which might resemble a precancerous state [43]. BCs have traditionally been thought to line the airway basement membrane, to express markers including KRT5, TP63, and NGFR, and to be cuboidal in shape [44].
Recently, a type of basal-like cells predominantly accumulated at the edge of myofibroblast foci in IPF lungs, termed basaloid cells [45], might contribute to the airway epithelial-lined cysts known as “honeycombs” in the distal parenchyma. These “honeycombs” might contain this basaloid subtype of BCs that are secretory or goblet-like [46]. These basaloid cells are transcriptionally distinct from other epithelial cell types subclustered by scRNA-seq, and they express markers including TP63, KRT17, LAMB3, and LAMC2, but not KRT5 and KRT15. They also feature EMT and senescence-related genes. These basaloid/basal-like cells might be able to seal the injured wound area by using their produced laminin [47], and these cells might produce serials of extracellular matrix and function as a sticking type of cell that pulls the mesenchymal cells and other epithelial progenitor cells together for sealing the injured wound [48].
Another assumption is that these basal-like/basaloid cells might be able to provide a basal membrane to help ATII progenitors reside prior to self-renew and/or further differentiate. A trial of transplantation of Sox9+ cells (including basal-like cells) into injured lung claimed to be able to improve the recipients’ (bronchiectasis patients) lung function [49]. These transplanted Sox9+ basal-like cells seemed to be incorporated into the bronchiolar region and gave rise to club cells and ATI cells in immunodeficient non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice post-bleomycin injury. Sox9 was expressed in the tip of the distal epithelium and has been shown to regulate extracellular matrix (ECM) and cell movement. Loss of Sox9 leads to defects in laminin deposition [50]. It is worth noting that these isolated Sox9+ basal-like cells have the capacity to proliferate massively in plastic culture and can be easily passaged. The massive proliferation feature might suggest they have acquired features of mesenchymal cells, which corresponds with the EMT feature noticed by scRNA-seq in IPF basaloid cells [45].
Senescence-related gene expression in the fresh sorted basaloid cells was found by scRNA-seq. It is assumed that at the stage in returning to pulmonary homeostasis, these senescent basaloid cells might be detected and be cleared by immune scavenger cells, such as macrophages [51]. Apparently in the IPF lungs, these senescent basaloid cells are persistent and pathogenically halt the regeneration of functional alveoli [52]. It is noticeable that the accumulation of basaloid cells were accompanied by elevated regulatory T cells [45], and regulatory T cells were found to contribute to tumor development and progression [53]. From this aspect, it is possible that accumulation of basaloid cells in the EMT in the early tumor microenvironment resembles a precancerous state.
A specific regional mesenchymal lineage Gli1 + cells, located in airway epithelium niche, exhibit properties of mesenchymal stem cells (MSCs) [54]. Gli1 + MSCs were shown to promote metaplastic differentiation of airway progenitors into Krt5 + basal cells [55]. This study provides a functional link between the Gli1 + MSC niche population and metaplastic basal cell transition of airway epithelial progenitors in fibrotic zones in mouse lungs [55]. It is not fully clear whether other mesenchymal cell populations play roles in the basal cell niche.
Diverse airway and alveolar epithelial progenitors as new features of disease pathogenesis
These ATII transition cells/PEPs have been observed across chronic and acute fibrosis-related pulmonary diseases. The cellular characteristics and phenotypes of the abnormal epithelial progenitors varies by disease pathology. The PEPs we described above were mainly found in patients with IPF or in bleomycin-induced lung fibrosis mouse models and were considered as new features of IPF pathology. We will describe below the epithelial progenitors found in other fibrosis-related pulmonary diseases.
Chronic obstructive pulmonary disease (COPD)
COPD is a progressive condition of chronic bronchitis, pulmonary obstruction, and emphysema. Fibrosis of small airways and respiratory vasculature has been established as a feature of COPD. There were aberrant basaloid cells found in COPD lungs, but these were far less prevalent than in the IPF lungs [52]. In another report, basal-like extra vivo clones, expressing markers p63 and KRT5, were derived from distal epithelial progenitors obtained from lung tissues of COPD patients [56]. The pathological implications from these basaloid cells and basal-like clones suggests a possible link to the etiology and progression of the disease.
Coronavirus disease 2019 (COVID19) infection
The leading cause of death from the COVID-19 virus is respiratory failure coupled with ARDS, and post-covid pulmonary fibrosis has been reported [57,58,59]. Studies of the recent COVID-19 lung cell populations found that hospitalized COVID-19 positive patients showed a significant decrease in total epithelial cells and global impairment of epithelial cell regeneration in the proximal and distal lung [31]. A population of PEPs with the KRT8/CLDN4/CDKN1A phenotype previously identified as DATPs were more frequent in the lungs of COVID-19 patients versus controls [10, 31]. The elevated levels of Il1β secreted from macrophages/monocytes in COVID-19 patients was speculated as the reason why this inflammation activated PEP, DATPs, were identified in high numbers. Whether PEP occurrences could be a driving force of the unremitting post-covid pulmonary fibrosis warrants further investigation.
Influenza infection
Lung fibrosis was developed in some cases post-H1N1 influenza viral infection [60]. Chronic lung disease patients with frequent exacerbations of influenza infection subsequently developed fibrotic interstitial lung disease [61]. Following H1N1 influenza viral infection, an expansion of distal airway stem cells (DASC) expressing BC markers Trp63 and Krt5 assembled into nascent alveoli at sites of interstitial lung inflammation. Selective ablation of the Krt5+ DASCs in vivo prevented this remodeling, leading to pre-fibrotic lesions and deficient oxygen exchange [21]. These basal-like DASCs have the potential to differentiate into ATI cells, ATII cells, and bronchiolar secretory cells, which is supported by the evidence that transplantation of DASCs diminished the structural changes in the infected lungs [21]. Similarly, another study suggested that p63+/Krt5+ basal-like lineage-negative epithelial progenitors (LNEPs) cells were activated after influenza or bleomycin injury in mice, and these Scgb1a1−/integrin α6β4+ LNEPs proliferated and migrated to injured alveoli, exhibiting differentiation potential towards mature ATII cells, and potentially contributing to the micro-honeycombing structure that characterizes progressive fibrotic lung disease [19, 40]. More recently, a population of ectopic solitary chemosensory cells (SCCs), expressing tuft cell marker DCLK1 and derived from p63+/Krt5+ BCs by lineage labeling, appeared in the areas of remodeled alveoli post-A/H1N1/PR/8 influenza viral infection [62]. These influenza activated basal-like cells, and activated BCs seems to have the ability of differentiation towards alveolar epithelial cells, but it is uncertain at which disease stage these cells will start to differentiate into the alveolar epithelial cells.
Bronchopulmonary dysplasia (BPD)
BPD is a chronic lung disease that usually occurs in premature infants. The pathologic findings of BPD includes airway fibrosis and interstitial fibrosis [63]. Alteration of the composition of alveolar epithelial cells were found in hyperoxia injured mouse models that represented the BPD phenotype [64]. This recent scRNA-seq analysis for hyperoxia mouse lung revealed an ATII lyz1+ population as a subpopulation of ATII cells. lyz1, an encoding lysozyme with decreased secretion in BPD [65], was found to be a marker of the activated ATII subpopulation [11] and enriched in ATII late (a stage right before becoming a mature ATII cell) in another scRNA-seq study for embryonic mouse lung [27]. The similarity of marker for ATII transition cells or PEPs found in different diseases or different cell stages implicates the immature or defect condition of ATII cells in pathology of certain diseases.
Recent findings on signaling pathways involved in epithelial progenitors
The occurrence and differentiation of diverse types of epithelial progenitors involve similarly prevalent signaling pathways under different injury conditions. Here we highlight the WNT/β-catenin, TGFβ, Notch, cellular senescence, and inflammatory-related signaling pathways.
WNT/β-catenin signaling
WNT/β-catenin signaling pathway is essential for lung development and homeostasis of progenitor ATII function [24, 25], and activation of WNT/β-catenin signaling facilitates the repair and regeneration of the lung after injury [34, 66]. Recently, it was reported that WNT/β-catenin signaling can be induced by blockage of LTβR signaling in ATII cells, which leads to enhanced lung alveolar regeneration in chronic smoking-induced lung diseases such as chronic obstructive pulmonary disease (COPD) [67]. Furthermore, WNT/β-catenin signaling induced by hypoxia was reported to promote the ATII reconstruction and blockade the Krt5+ basal-like activation from LNEPs for alveolar regeneration in influenza injured mouse lungs [19]. In another influenza study, facultative Wnt-responsive AEPs were shown to contribute to functional alveolar epithelial regeneration by generating both ATII and ATI cells after injury [25]. Furthermore, in an in vitro study, Wnt activation was shown to increase ATII self-renewal, while Wnt inhibition enhanced ATI differentiation in both human and mouse ATII organoids [25]. These observations indicated the beneficial roles of WNT/β-catenin signaling for ATII cell mediated alveolar regeneration in different conditions post-injury. In regard to PEPs, chronic activation of WNT/β-catenin signaling (7 day WNT3A treatment in vitro), but not WNT5A or acute WNT3A (24 h treatment) stimulation, has been found to induce cellular senescence and Krt8 expression (PEP features) in primary murine ATII cells in vitro [68]. These data suggest chronic WNT3A signaling might enhance the production of PEPs from ATII cells in vitro by activation of the canonical WNT/β-catenin pathway. It is assumed that WNT/β-catenin signaling spatiotemporally regulates the ATII self-renewal and affects ATI differentiation, while chronic activation of canonical WNT/β-catenin signaling might promote ATII-to-PEP transition.
TGFβ signaling pathway
TGFβ1 has been long recognized as a growth factor that is elevated in pro-fibrotic conditions. TGFβ1 was enriched in aberrant basaloid cells lined by myofibroblast foci in IPF lung tissue, which might be activated locally through their integrin repertoire [52, 69]. A TGFB1hi CTHRC1+ mesenchymal population were found adjacent to KRT17+/KRT5− basaloid cells (also termed ABIs) [70]. Suppression of TGFβ induced BCs reentry into the cell cycle and initiates epithelial regeneration via enhancing Id2 expression [71]. Activation of TGFβ signaling was found in IPF HTII-280+ ATII cells by scRNA-seq analysis [72], and TGFβ signaling might be responsible for halting ATII self-renewal and inducing ATII differentiation into ATI cells in LPS-induced acute lung injury. TGFβ signaling was highly upregulated in the cell cycle arrested ATII subpopulation and relatively downregulated in ATII transdifferentiating cells detected by scRNA-seq analysis [32]; this suggests TGFβ signaling is required for ATII cell cycle arrest and ATII differentiation into ATI cells.
Another report suggested that TGFβ signaling is highly activated during early differentiation of ATII into ATI cells, and subsequent TGFβ deactivation promotes late differentiation. They also found that Krt8 and Krt18 were upregulated during ATII differentiation into ATI cells in vitro, and inhibition of TGFβ signaling attenuates Krt8/Krt18 upregulation [73], suggesting that TGFβ signaling might contribute to ATII transition cells/PEPs formation, thus recapitulating a failure of TGFβ signaling deactivation that may result in the persistence of ATII transition cells and PEPs in IPF [12, 73]. Indeed, another study found that TGFβ signaling was upregulated in a presumably ATII transition cell subpopulation accumulated in PNX-treated Cdc42-null mouse lungs and was responsible for mechanical tension driven periphery-to-center progression of lung fibrosis in a spatially regulated manner [30]. These studies elaborated on the pivotal role of TGFβ signaling in basaloid cells, BCs, and in ATII transition cells/PEPs during ATII differentiation into ATI cells. Specifically, early activation of TGFβ enhances PEP marker expression in ATII cells, and subsequent suppression of TGFβ promotes differentiation into ATI cells. However, it seems obvious to speculate but still no experimental verification has determined whether it is the myofibroblast secreted TGFβ that activates the integrin receptors on ATII cells that stimulates ATII-to-PEP transition with subsequent PEP accumulation.
Notch signaling pathway
Airway homeostasis can be easily disrupted by endogenous and exogenous insults, such as chronic micro-wound scarring, acute hypoxemic injury, pseudomonas-induced acute lung injury, and H1N1 infection in the lung. Elevated Notch signaling was found in collapsed alveoli post-hypoxemia and H1N1 infection, as well as in fibrotic lungs [19, 29, 42]. It is reported that Notch signaling initiates the expansion of LNEPs to basal-like Krt5+ cells in an alveolar remodeling following H1N1(PR8) influenza infection in a HIF1A dependent manner. Further, Wnt activity antagonized Notch signaling to favor LNEPs differentiation into ATII cells for recovery [19, 29]. In addition, Notch inhibition skews the differentiation of human secretory cells (CC10+KDR+HTII280−) into ATII cells instead of ciliated cells and BCs in culture conditions [16]. Recent scRNA-seq mapping of epithelial cell types for normal and IPF human airways suggested that BC subpopulations constitute a hierarchy regulated by Notch signaling. Within this hierarchy, Notch receptor, Notch2, maintains undifferentiated in BCs and restricts basal-to-ciliated differentiation, and Notch3 restrains BC secretory differentiation [42]. Furthermore, deletion of Dlk1 (a non-canonical Notch ligand) in ATII cells induced persistent Notch activation and led to accumulation of ATII transition cells following pseudomonas injury. These findings suggested the cell fate selection feature of Notch signaling in the subpopulations of basal and ATII cells. Suppression of Notch facilitates ATII cell recovery, while persistent Notch activation promotes ATII transition cells or PEPs. It will be interesting to investigate whether this phenomenon is also representative of PEPs in the fibrotic lung.
Cellular senescence pathways
Another common signaling pathway found in PEPs is cellular senescence. PEPs in fibrotic lung, including Basaloid cells, DATPs, PATs, and Krt8+ ADI cells, all display highly expressed genes related to senescence [10,11,12, 52]; this suggests that senescence not only is a feature for fibrotic fibroblast [74, 75], but represent a PEP stage halting full ATII differentiation into ATI cells in fibrotic lungs. These PEP senescence-related genes include CDKN1A, CDKN2A, CCND1, CCND2, MDM2, and GDF15 [52]. In the same line, loss of Sin3a in ATII cells has been shown to initiate a program of p53-dependent cellular senescence, ATII cell depletion, and spontaneous, progressive pulmonary fibrosis [76], but whether Sin3a-null ATII cells represent PEPs is currently unclear. In another study, senescent marker CDKN2A was overlapping with the basal epithelial markers Krt5 and Krt17, which localize to bronchiolized epithelial structures in fibrotic regions of IPF and systemic sclerosis-associated interstitial lung disease (SSc-ILD) lung tissue [77]. Whether senescence can be a reversible transient state accompanying tissue regeneration in fibrotic lung is currently unclear. It is challenging and the tools to lineage label senescent cells [78] are being developed to presumably able to resolve this issue.
Inflammatory-related signaling pathways
Recent studies have provided evidence that indicate inflammatory signals trigger ATII cells response to injury. IL6 has been shown to promote ATII cell self-renewal in an innate immune receptor TLR4 dependent manner at an early stage of fibrotic lung injury [79]. It is reported that macrophage derived IL1β activated a subpopulation of primed ATII cells expressing IL1R1 for differentiation into DATPs, while chronic IL1β halted DATPs transition into functional ATI cells, which results in impaired alveolar regeneration in fibrotic lung [10]. The same group did follow-up research and found that IL1β signaling through IL1R1 on ciliated cells via paracrine Notch ligands, JAG1 and JAG2, is essential for secretory cell differentiation of ATII cells [16]. These studies suggest that inflammatory signals are required for ATII cell mediated alveolar regeneration but sustained inflammatory response can cause defects for generating functional alveolar niche following injury. These inflammatory signals can function directly on the effected ATII cells, or indirectly on upstream epithelial cells, thus executing a sequential effect. A recent study that transplanted human ATII cells into bleomycin-induced fibrotic lungs of NOD scid gamma immunodeficient (NSG) mice revealed ATII-to-basaloid/basal cell transition [70]. It is intriguing to see whether, in fibrotic human lung where inflammatory signals present, these inflammatory signals are required for ATII-to-basal cell transition.
Up-to-date approaches for researching epithelial progenitors
Single-cell RNA (scRNA)
ScRNA-seq from multiple companies and across several technology platforms has been utilized to understand epithelial progenitors. The expansion of single-cell sequencing into MULTI-seq and spatial RNA-seq has been further used to determine the epithelial progenitor subpopulations and their gene expression characteristics [27, 45, 64, 80]. A stem cell progenitor hierarchy in the distal airway has been well documented in the previous literature [1, 15, 33, 72]. Additionally, scRNA-seq has allowed for greater probing of heterogeneous airway and alveolar epithelial progenitor types and with the combination of cell lineage label produced convincing evidence for determining several types of PEPs [10, 12, 52].
Lineage tracing mouse tools
Lineage-traced mouse tools for lung epithelial cells are well described elsewhere [81]. Here we updated the recent used epithelial cell type-specific mouse lines that have been used in studying the PEPs. There are many diverse mouse lineage tracing tools, and we have included short summaries of the most recent, novel, and pertinent to airway and alveolar epithelial cells (Table 1), and we have specifically organized these by PEP lineages, including DATP, PATS, and BC lineages (Table 2).
Epithelial progenitor transplantation for treating clinical fibrotic lung diseases
It is thought that replacing the pathologic epithelial cells by exogenous progenitors is a promising treatment for fibrotic pulmonary diseases. Investigators have transplanted ATII cells, fresh isolated from healthy rat or differentiated from induced pluripotent stem cells (iPSCs) for bleomycin-induced mouse lung fibrosis, and found that transplanted ATII cells can be engrafted, reduce collagen deposition in the lung, and abrogate pulmonary fibrosis [87,88,89]. Adult rat lung spheroid cells containing mixed epithelial progenitors were reported to attenuate rat lung fibrotic progression [90]. Furthermore, ATII cells isolated from deceased organ donors were transplanted intratracheally in progressive IPF patients and were well tolerated [91]. More recently, human SOX9+ BCs expanded from bronchoscopic brushing were reported to give rise to alveolar and bronchiolar epithelium post-transplantation into bleomycin injured mouse lung and were found to enhance the recipients’ lung functions. Transplantation of autologous SOX9+ BCs enhanced lung tissue repair and lung function in two patients with bronchiectasis [49]. More mechanistic research is needed to validate the potential of epithelial progenitor transplantation in vivo in patients with lung fibrosis and other lung diseases. Long-term follow-up should be conducted to evaluate the tumorigenic potential of the exogenous or endogenous lung epithelial progenitors.
Conclusions, current challenges, and prospects
The existence of epithelial progenitors in the distal airway with regenerative properties (both migration into and proliferation in the alveolar space) is established [5, 92]. Wnt/β-catenin, Notch, TGFβ, and senescence pathways have been shown to regulate ATII transition cells/PEPs under diverse causes and driver of lower airway injury [19, 32, 40, 68, 72, 76]. ScRNA-seq in combination with lineage tracing has helped to show that there are distinct ATII transition cells/PEPs, and while Krt expression is crucial, what combination of Krt genes (Krt8, Krt14, and Krt17 have all be shown to be present in vitro transition states and utilized in vivo models) defines these ATII transition cells/PEPs and at what cellular checkpoints is unsettled. The dominant immunological, senescent, or hypoxic signaling pathways present in the alveolar microenvironment may determine what specific ATII transition cells/PEPs drives regeneration and repair in the alveoli. Further lineage-based studies to determine how these transition cells/PEPs originate may clarify these states.
Efforts to integrate the many single-cell seq datasets into a functional database has been an ongoing project in IPF research. Differentiating between PEP phenotypes and defining transition states makes utilizing multiple datasets from different research groups a challenge. An initiative to standardize single-cell terminology or single-cell taxonomy could aid in the creation of this database. Further experimentation may provide a more accurate framework for how to categorize transition state cell populations in IPF.
Availability of data and materials
Not applicable.
Abbreviations
- ScRNA-seq:
-
Single-cell RNA sequencing
- ATII:
-
Alveolar type 2 cell
- ATI:
-
Alveolar type 1 cell
- PEPs:
-
Persistent epithelial progenitors
- BASCs:
-
Bronchioalveolar stem cells
- BADJs:
-
Bronchioalveolar duct junctions
- Upk3a:
-
Uroplakin3a
- TM4SF1:
-
Transmembrane 4 L six family member 1
- DATPs:
-
Damage-associated transient progenitors
- PATS:
-
Pre-alveolar type-1 transitional cell state
- IPF:
-
Idiopathic pulmonary fibrosis
- ADI:
-
Alveolar differentiation intermediate
- ARDS:
-
Acute respiratory distress syndrome
- PNX:
-
Pneumonectomy
- IL6:
-
Interleukin-6
- GHR:
-
Growth hormone receptor
- NOD/SCID:
-
Non-obese diabetic/severe combined immunodeficiency
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial–mesenchymal transition
- COPD:
-
Chronic obstructive pulmonary disease
- LNEPs:
-
Lineage-negative epithelial progenitors
- AEPs:
-
ATII epithelial progenitors
- SSc-ILD:
-
Systemic sclerosis-associated interstitial lung disease
- COVID19:
-
Coronavirus disease 2019
- DASC:
-
Distal airway stem cells
- SCCs:
-
Solitary chemosensory cells
- BPD:
-
Bronchopulmonary dysplasia
- NEBs:
-
Neuroepithelial bodies
- IS:
-
Interlobar serous
- iPSCs:
-
Induced pluripotent stem cells
- BCs:
-
Basal cells
References
Snyder JC, Teisanu RM, Stripp BR. Endogenous lung stem cells and contribution to disease. J Pathol. 2009;217(2):254–64. https://doi.org/10.1002/path.2473.
Whitsett JA, Kalin TV, Xu Y, Kalinichenko VV. Building and regenerating the lung cell by cell. Physiol Rev. 2019;99(1):513–54. https://doi.org/10.1152/physrev.00001.2018.
Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BL. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell. 2009;4(6):525–34. https://doi.org/10.1016/j.stem.2009.04.002.
Dean CH, Snelgrove RJ. New rules for club development: new insights into human small airway epithelial club cell ontogeny and function. Am J Respir Crit Care Med. 2018;198(11):1355–6. https://doi.org/10.1164/rccm.201805-0925ED.
Kim CFB, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–35. https://doi.org/10.1016/j.cell.2005.03.032.
Liu Q, Liu K, Cui G, Huang X, Yao S, Guo W, Qin Z, Li Y, Yang R, Pu W, Zhang L, He L, Zhao H, Yu W, Tang M, Tian X, Cai D, Nie Y, Hu S, Ren T, Qiao Z, Huang H, Zeng YA, Jing N, Peng G, Ji H, Zhou B. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat Genet. 2019;51(4):728–38. https://doi.org/10.1038/s41588-019-0346-6.
Liu K, Tang M, Jin H, Liu Q, He L, Zhu H, Liu X, Han X, Li Y, Zhang L, Tang J, Pu W, Lv Z, Wang H, Ji H, Zhou B. Triple-cell lineage tracing by a dual reporter on a single allele. J Biol Chem. 2020;295(3):690–700. https://doi.org/10.1074/jbc.RA119.011349.
Logan CY, Desai TJ. Keeping it together: pulmonary alveoli are maintained by a hierarchy of cellular programs. BioEssays. 2015;37(9):1028–37. https://doi.org/10.1002/bies.201500031.
Wang Y, Tang Z, Huang H, Li J, Wang Z, Yu Y, Zhang C, Li J, Dai H, Wang F, Cai T, Tang N. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc Natl Acad Sci U S A. 2018;115(10):2407–12. https://doi.org/10.1073/pnas.1719474115.
Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, Lee JH. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell. 2020;27(3):366-82e7. https://doi.org/10.1016/j.stem.2020.06.020.
Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, Ogar P, Sengupta A, Kukhtevich I, Schneider R, Zhao Z, Voss C, Stoeger T, Neumann JHL, Hilgendorff A, Behr J, O’Reilly M, Lehmann M, Burgstaller G, Konigshoff M, Chapman HA, Theis FJ, Schiller HB. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun. 2020;11(1):3559. https://doi.org/10.1038/s41467-020-17358-3.
Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, Banovich NE, Kropski JA, Tata PR. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol. 2020;22(8):934–46. https://doi.org/10.1038/s41556-020-0542-8.
Lee DF, Salguero FJ, Grainger D, Francis RJ, MacLellan-Gibson K, Chambers MA. Isolation and characterisation of alveolar type II pneumocytes from adult bovine lung. Sci Rep. 2018;8(1):11927. https://doi.org/10.1038/s41598-018-30234-x.
Marsh LM, Cakarova L, Kwapiszewska G, von Wulffen W, Herold S, Seeger W, Lohmeyer J. Surface expression of CD74 by type II alveolar epithelial cells: a potential mechanism for macrophage migration inhibitory factor-induced epithelial repair. Am J Physiol Lung Cell Mol Physiol. 2009;296(3):L442–52. https://doi.org/10.1152/ajplung.00525.2007.
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025–36. https://doi.org/10.1172/JCI68782.
Choi J, Jang YJ, Dabrowska C, Iich E, Evans KV, Hall H, Janes SM, Simons BD, Koo BK, Kim J, Lee JH. Release of Notch activity coordinated by IL-1beta signalling confers differentiation plasticity of airway progenitors via Fosl2 during alveolar regeneration. Nat Cell Biol. 2021;23(9):953–66. https://doi.org/10.1038/s41556-021-00742-6.
Guha A, Deshpande A, Jain A, Sebastiani P, Cardoso WV. Uroplakin 3a(+) cells are a distinctive population of epithelial progenitors that contribute to airway maintenance and post-injury repair. Cell Rep. 2017;19(2):246–54. https://doi.org/10.1016/j.celrep.2017.03.051.
Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA. Distinct airway epithelial stem cells hide among club cells but mobilize to promote alveolar regeneration. Cell Stem Cell. 2020;26(3):346-58e4. https://doi.org/10.1016/j.stem.2019.12.014.
Xi Y, Kim T, Brumwell AN, Driver IH, Wei Y, Tan V, Jackson JR, Xu J, Lee DK, Gotts JE, Matthay MA, Shannon JM, Chapman HA, Vaughan AE. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat Cell Biol. 2017;19(8):904–14. https://doi.org/10.1038/ncb3580.
Yang Y, Riccio P, Schotsaert M, Mori M, Lu J, Lee DK, Garcia-Sastre A, Xu J, Cardoso WV. Spatial-temporal lineage restrictions of embryonic p63(+) progenitors establish distinct stem cell pools in adult airways. Dev Cell. 2018;44(6):752-61e4. https://doi.org/10.1016/j.devcel.2018.03.001.
Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, Crum CP, Xian W, McKeon F. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature. 2015;517(7536):616–20. https://doi.org/10.1038/nature13903.
Dirice E, De Jesus DF, Kahraman S, Basile G, Ng RW, El Ouaamari A, Teo AKK, Bhatt S, Hu J, Kulkarni RN. Human duct cells contribute to beta cell compensation in insulin resistance. JCI Insight. 2019. https://doi.org/10.1172/jci.insight.99576.
Minnis-Lyons SE, Ferreira-Gonzalez S, Aleksieva N, Man TY, Gadd VL, Williams MJ, Guest RV, Lu WY, Dwyer BJ, Jamieson T, Nixon C, Van Hul N, Lemaigre FP, McCafferty J, Leclercq IA, Sansom OJ, Boulter L, Forbes SJ. Notch-IGF1 signaling during liver regeneration drives biliary epithelial cell expansion and inhibits hepatocyte differentiation. Sci Signal. 2021. https://doi.org/10.1126/scisignal.aay9185.
Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359(6380):1118–23. https://doi.org/10.1126/science.aam6603.
Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, Morrisey EE. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555(7695):251–5. https://doi.org/10.1038/nature25786.
Frank DB, Penkala IJ, Zepp JA, Sivakumar A, Linares-Saldana R, Zacharias WJ, Stolz KG, Pankin J, Lu M, Wang Q, Babu A, Li L, Zhou S, Morley MP, Jain R, Morrisey EE. Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc Natl Acad Sci U S A. 2019;116(10):4362–71. https://doi.org/10.1073/pnas.1813952116.
Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509(7500):371–5. https://doi.org/10.1038/nature13173.
Evans MJ, Cabral LJ, Stephens RJ, Freeman G. Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp Mol Pathol. 1975;22(1):142–50. https://doi.org/10.1016/0014-4800(75)90059-3.
Finn J, Sottoriva K, Pajcini KV, Kitajewski JK, Chen C, Zhang W, Malik AB, Liu Y. Dlk1-mediated temporal regulation of notch signaling is required for differentiation of alveolar type II to type I cells during repair. Cell Rep. 2019;26(11):2942–54. https://doi.org/10.1016/j.celrep.2019.02.046.
Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, Shi M, Zhao X, Yuan J, Li J, Yang X, Bin E, Wei D, Zhang H, Zhang J, Yang C, Cai T, Dai H, Chen J, Tang N. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell. 2020;180(1):107-21e17. https://doi.org/10.1016/j.cell.2019.11.027.
Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, Katsyv I, Rendeiro AF, Amin AD, Schapiro D, Frangieh CJ, Luoma AM, Filliol A, Fang Y, Ravichandran H, Clausi MG, Alba GA, Rogava M, Chen SW, Ho P, Montoro DT, Kornberg AE, Han AS, Bakhoum MF, Anandasabapathy N, Suarez-Farinas M, Bakhoum SF, Bram Y, Borczuk A, Guo XV, Lefkowitch JH, Marboe C, Lagana SM, Del Portillo A, Zorn E, Markowitz GS, Schwabe RF, Schwartz RE, Elemento O, Saqi A, Hibshoosh H, Que J, Izar B. A molecular single-cell lung atlas of lethal COVID-19. Nature. 2021. https://doi.org/10.1038/s41586-021-03569-1.
Riemondy KA, Jansing NL, Jiang P, Redente EF, Gillen AE, Fu R, Miller AJ, Spence JR, Gerber AN, Hesselberth JR, Zemans RL. Single cell RNA sequencing identifies TGFbeta as a key regenerative cue following LPS-induced lung injury. JCI Insight. 2019. https://doi.org/10.1172/jci.insight.123637.
Lee JH, Bhang DH, Beede A, Huang TL, Stripp BR, Bloch KD, Wagers AJ, Tseng YH, Ryeom S, Kim CF. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell. 2014;156(3):440–55. https://doi.org/10.1016/j.cell.2013.12.039.
Uhl FE, Vierkotten S, Wagner DE, Burgstaller G, Costa R, Koch I, Lindner M, Meiners S, Eickelberg O, Konigshoff M. Preclinical validation and imaging of Wnt-induced repair in human 3D lung tissue cultures. Eur Respir J. 2015;46(4):1150–66. https://doi.org/10.1183/09031936.00183214.
Lee JH, Tammela T, Hofree M, Choi J, Marjanovic ND, Han S, Canner D, Wu K, Paschini M, Bhang DH, Jacks T, Regev A, Kim CF. Anatomically and functionally distinct lung mesenchymal populations marked by Lgr5 and Lgr6. Cell. 2017;170(6):1149–6312. https://doi.org/10.1016/j.cell.2017.07.028.
Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, Morrisey EE. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell. 2017;170(6):1134–4810. https://doi.org/10.1016/j.cell.2017.07.034.
Wang C, de Mochel NSR, Christenson SA, Cassandras M, Moon R, Brumwell AN, Byrnes LE, Li A, Yokosaki Y, Shan P, Sneddon JB, Jablons D, Lee PJ, Matthay MA, Chapman HA, Peng T. Expansion of hedgehog disrupts mesenchymal identity and induces emphysema phenotype. J Clin Invest. 2018;128(10):4343–58. https://doi.org/10.1172/JCI99435.
Xie T, Kulur V, Liu N, Deng N, Wang Y, Rowan SC, Yao C, Huang G, Liu X, Taghavifar F, Liang J, Hogaboam C, Stripp B, Chen P, Jiang D, Noble PW. Mesenchymal growth hormone receptor deficiency leads to failure of alveolar progenitor cell function and severe pulmonary fibrosis. Sci Adv. 2021. https://doi.org/10.1126/sciadv.abg6005.
Hogan B, Tata PR. Cellular organization and biology of the respiratory system. Nat Cell Biol. 2019. https://doi.org/10.1038/s41556-019-0357-7.
Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, Matthay MA, Rock JR, Chapman HA. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517(7536):621–5. https://doi.org/10.1038/nature14112.
Ghosh M, Miller YE, Nakachi I, Kwon JB, Baron AE, Brantley AE, Merrick DT, Franklin WA, Keith RL, Vandivier RW. Exhaustion of airway basal progenitor cells in early and established chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(7):885–96. https://doi.org/10.1164/rccm.201704-0667OC.
Carraro G, Mulay A, Yao C, Mizuno T, Konda B, Petrov M, Lafkas D, Arron JR, Hogaboam CM, Chen P, Jiang D, Noble PW, Randell SH, McQualter JL, Stripp BR. Single-cell reconstruction of human basal cell diversity in normal and idiopathic pulmonary fibrosis lungs. Am J Respir Crit Care Med. 2020;202(11):1540–50. https://doi.org/10.1164/rccm.201904-0792OC.
Greenberg AK, Yee H, Rom WN. Preneoplastic lesions of the lung. Respir Res. 2002;3:20. https://doi.org/10.1186/rr170.
Ray S, Chiba N, Yao C, Guan X, McConnell AM, Brockway B, Que L, McQualter JL, Stripp BR. Rare SOX2(+) airway progenitor cells generate KRT5(+) cells that repopulate damaged alveolar parenchyma following influenza virus infection. Stem Cell Rep. 2016;7(5):817–25. https://doi.org/10.1016/j.stemcr.2016.09.010.
Schupp JC, Ahangari F, McDonough JE, DeIuliis G, Poli S, Rosas IO, Yan X. Kaminski single cell RNA velocity analysis of aberrant basaloid cells in pulmonary fibrosis reveals trajectory towards an alveolar type I like cell state. American Thoracic Society; 2021: Am J Respir Crit Care Med 2021. p. A4350.
Carraro G, Langerman J, Sabri S, Lorenzana Z, Purkayastha A, Zhang G, Konda B, Aros CJ, Calvert BA, Szymaniak A, Wilson E, Mulligan M, Bhatt P, Lu J, Vijayaraj P, Yao C, Shia DW, Lund AJ, Israely E, Rickabaugh TM, Ernst J, Mense M, Randell SH, Vladar EK, Ryan AL, Plath K, Mahoney JE, Stripp BR, Gomperts BN. Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Nat Med. 2021;27(5):806–14. https://doi.org/10.1038/s41591-021-01332-7.
Goodwin K, Ellis SJ, Lostchuck E, Zulueta-Coarasa T, Fernandez-Gonzalez R, Tanentzapf G. Basal cell-extracellular matrix adhesion regulates force transmission during tissue morphogenesis. Dev Cell. 2016;39(5):611–25. https://doi.org/10.1016/j.devcel.2016.11.003.
Bilodeau C, Shojaie S, Goltsis O, Wang J, Luo D, Ackerley C, Rogers I, Cox B, Post M. TP63 basal cells are indispensable during endoderm differentiation into proximal airway cells on acellular lung scaffolds. Npj Regen Med. 2021;6(1):12. https://doi.org/10.1038/s41536-021-00124-4.
Ma Q, Ma Y, Dai X, Ren T, Fu Y, Liu W, Han Y, Wu Y, Cheng Y, Zhang T, Zuo W. Regeneration of functional alveoli by adult human SOX9(+) airway basal cell transplantation. Protein Cell. 2018;9(3):267–82. https://doi.org/10.1007/s13238-018-0506-y.
Rockich BE, Hrycaj SM, Shih HP, Nagy MS, Ferguson MA, Kopp JL, Sander M, Wellik DM, Spence JR. Sox9 plays multiple roles in the lung epithelium during branching morphogenesis. Proc Natl Acad Sci U S A. 2013;110(47):E4456–64. https://doi.org/10.1073/pnas.1311847110.
Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, Virasami A, Sebire NJ, Kinsler V, Valdovinos A, LeSaux CJ, Passos JF, Antoniou A, Rustin MHA, Campisi J, Akbar AN. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat Commun. 2019;10(1):2387. https://doi.org/10.1038/s41467-019-10335-5.
Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6(28):aba1983. https://doi.org/10.1126/sciadv.aba1983.
Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, Brown M, Chang C, Troncoso L, Chen ST, Sastre-Perona A, Park MD, Tabachnikova A, Dhainaut M, Hamon P, Maier B, Sawai CM, Agullo-Pascual E, Schober M, Brown BD, Reizis B, Marron T, Kenigsberg E, Moussion C, Benaroch P, Aguirre-Ghiso JA, Merad M. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595(7868):578–84. https://doi.org/10.1038/s41586-021-03651-8.
Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16(1):51–66. https://doi.org/10.1016/j.stem.2014.11.004.
Cassandras M, Wang C, Kathiriya J, Tsukui T, Matatia P, Matthay M, Wolters P, Molofsky A, Sheppard D, Chapman H, Peng T. Gli1(+) mesenchymal stromal cells form a pathological niche to promote airway progenitor metaplasia in the fibrotic lung. Nat Cell Biol. 2020;22(11):1295–306. https://doi.org/10.1038/s41556-020-00591-9.
Rao W, Wang S, Duleba M, Niroula S, Goller K, Xie J, Mahalingam R, Neupane R, Liew AA, Vincent M, Okuda K, O’Neal WK, Boucher RC, Dickey BF, Wechsler ME, Ibrahim O, Engelhardt JF, Mertens TCJ, Wang W, Jyothula SSK, Crum CP, Karmouty-Quintana H, Parekh KR, Metersky ML, McKeon FD, Xian W. Regenerative metaplastic clones in COPD lung drive inflammation and fibrosis. Cell. 2020;181(4):848-64e18. https://doi.org/10.1016/j.cell.2020.03.047.
McGroder CF, Zhang D, Choudhury MA, Salvatore MM, D’Souza BM, Hoffman EA, Wei Y, Baldwin MR, Garcia CK. Pulmonary fibrosis 4 months after COVID-19 is associated with severity of illness and blood leucocyte telomere length. Thorax. 2021. https://doi.org/10.1136/thoraxjnl-2021-217031.
Ambardar SR, Hightower SL, Huprikar NA, Chung KK, Singhal A, Collen JF. Post-COVID-19 pulmonary fibrosis: novel sequelae of the current pandemic. J Clin Med. 2021. https://doi.org/10.3390/jcm10112452.
Bharat A, Querrey M, Markov NS, Kim S, Kurihara C, Garza-Castillon R, Manerikar A, Shilatifard A, Tomic R, Politanska Y, Abdala-Valencia H, Yeldandi AV, Lomasney JW, Misharin AV, Budinger GRS. Lung transplantation for pulmonary fibrosis secondary to severe COVID-19. medRxiv. 2020. https://doi.org/10.1101/2020.10.26.20218636.
Gao J, Chu W, Duan J, Li J, Ma W, Hu C, Yao M, Xing L, Yang Y. Six-month outcomes of post-ARDS pulmonary fibrosis in patients with H1N1 pneumonia. Front Mol Biosci. 2021;8:640763. https://doi.org/10.3389/fmolb.2021.640763.
Lee SW, Sharma L, Kang YA, Kim SH, Chandrasekharan S, Losier A, Brady V, Bermejo S, Andrews N, Yoon CM, Liu W, Lee JY, Kang MJ, Dela Cruz CS. Impact of cigarette smoke exposure on the lung fibroblastic response after influenza pneumonia. Am J Respir Cell Mol Biol. 2018;59(6):770–81. https://doi.org/10.1165/rcmb.2018-0004OC.
Rane CK, Jackson SR, Pastore CF, Zhao G, Weiner AI, Patel NN, Herbert DR, Cohen NA, Vaughan AE. Development of solitary chemosensory cells in the distal lung after severe influenza injury. Am J Physiol Lung Cell Mol Physiol. 2019;316(6):L1141–9. https://doi.org/10.1152/ajplung.00032.2019.
Popova AP. Mechanisms of bronchopulmonary dysplasia. J Cell Commun Signal. 2013;7(2):119–27. https://doi.org/10.1007/s12079-013-0190-x.
Hurskainen M, Mizikova I, Cook DP, Andersson N, Cyr-Depauw C, Lesage F, Helle E, Renesme L, Jankov RP, Heikinheimo M, Vanderhyden BC, Thebaud B. Single cell transcriptomic analysis of murine lung development on hyperoxia-induced damage. Nat Commun. 2021;12(1):1565. https://doi.org/10.1038/s41467-021-21865-2.
Revenis ME, Kaliner MA. Lactoferrin and lysozyme deficiency in airway secretions: association with the development of bronchopulmonary dysplasia. J Pediatr. 1992;121(2):262–70. https://doi.org/10.1016/s0022-3476(05)81201-6.
Kneidinger N, Yildirim AO, Callegari J, Takenaka S, Stein MM, Dumitrascu R, Bohla A, Bracke KR, Morty RE, Brusselle GG, Schermuly RT, Eickelberg O, Konigshoff M. Activation of the WNT/beta-catenin pathway attenuates experimental emphysema. Am J Respir Crit Care Med. 2011;183(6):723–33. https://doi.org/10.1164/rccm.200910-1560OC.
Conlon TM, John-Schuster G, Heide D, Pfister D, Lehmann M, Hu Y, Ertuz Z, Lopez MA, Ansari M, Strunz M, Mayr C, Angelidis I, Ciminieri C, Costa R, Kohlhepp MS, Guillot A, Gunes G, Jeridi A, Funk MC, Beroshvili G, Prokosch S, Hetzer J, Verleden SE, Alsafadi H, Lindner M, Burgstaller G, Becker L, Irmler M, Dudek M, Janzen J, Goffin E, Gosens R, Knolle P, Pirotte B, Stoeger T, Beckers J, Wagner D, Singh I, Theis FJ, de Angelis MH, O’Connor T, Tacke F, Boutros M, Dejardin E, Eickelberg O, Schiller HB, Konigshoff M, Heikenwalder M, Yildirim AO. Inhibition of LTbetaR signalling activates WNT-induced regeneration in lung. Nature. 2020;588(7836):151–6. https://doi.org/10.1038/s41586-020-2882-8.
Lehmann M, Hu Q, Hu Y, Hafner K, Costa R, van den Berg A, Konigshoff M. Chronic WNT/beta-catenin signaling induces cellular senescence in lung epithelial cells. Cell Signal. 2020;70:109588. https://doi.org/10.1016/j.cellsig.2020.109588.
Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–28. https://doi.org/10.1016/s0092-8674(00)80545-0.
Kathiriya JJ, Wang C, Zhou M, Brumwell A, Cassandras M, Le Saux CJ, Cohen M, Alysandratos KD, Wang B, Wolters P, Matthay M, Kotton DN, Chapman HA, Peng T. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5(+) basal cells. Nat Cell Biol. 2021. https://doi.org/10.1038/s41556-021-00809-4.
Kiyokawa H, Yamaoka A, Matsuoka C, Tokuhara T, Abe T, Morimoto M. Airway basal stem cells reutilize the embryonic proliferation regulator, Tgfbeta-Id2 axis, for tissue regeneration. Dev Cell. 2021;56(13):1917–29. https://doi.org/10.1016/j.devcel.2021.05.016.
Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl AT, Funari VA, Gokey JJ, Stripp BR, Whitsett JA. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1(20):e90558. https://doi.org/10.1172/jci.insight.90558.
Jiang P, GildeRubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, Omary MB, Ridge KM, Zemans RL. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8(hi) transitional state. Am J Respir Crit Care Med. 2020;201(11):1443–7. https://doi.org/10.1164/rccm.201909-1726LE.
Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, Thannickal VJ. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. 2014;6(231):231ra47. https://doi.org/10.1126/scitranslmed.3008182.
Li Y, Liang J, Yang T, Monterrosa Mena J, Huan C, Xie T, Kurkciyan A, Liu N, Jiang D, Noble PW. Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix Biol. 2016;55:35–48. https://doi.org/10.1016/j.matbio.2016.03.004.
Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, Belperio JA, Chen P, Jiang D, Noble PW, Stripp BR. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med. 2021;203(6):707–17. https://doi.org/10.1164/rccm.202004-1274OC.
DePianto DJ, Heiden JAV, Morshead KB, Sun KH, Modrusan Z, Teng G, Wolters PJ, Arron JR. Molecular mapping of interstitial lung disease reveals a phenotypically distinct senescent basal epithelial cell population. JCI Insight. 2021. https://doi.org/10.1172/jci.insight.143626.
Li Y, Zhao H, Huang X, Tang J, Zhang S, Li Y, Liu X, He L, Ju Z, Lui KO, Zhou B. Embryonic senescent cells re-enter cell cycle and contribute to tissues after birth. Cell Res. 2018;28(7):775–8. https://doi.org/10.1038/s41422-018-0050-6.
Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, Kurkciyan A, Mena JM, Stripp BR, Jiang D, Noble PW. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med. 2016;22(11):1285–93. https://doi.org/10.1038/nm.4192.
Beppu A, Zhao J, Yao C, Carraro G, Israely E, Coelho AL, Stripp BR. Epithelial plasticity and innate immune activation promote lung tissue remodeling following respiratory viral infection. biorxiv. 2021. https://doi.org/10.1101/2021.09.22.461381.
Rawlins EL, Perl AK. The a"MAZE"ing world of lung-specific transgenic mice. Am J Respir Cell Mol Biol. 2012;46(3):269–82. https://doi.org/10.1165/rcmb.2011-0372PS.
Peng T, Frank DB, Kadzik RS, Morley MP, Rathi KS, Wang T, Zhou S, Cheng L, Lu MM, Morrisey EE. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature. 2015;526(7574):578–82. https://doi.org/10.1038/nature14984.
Frank DB, Peng T, Zepp JA, Snitow M, Vincent TL, Penkala IJ, Cui Z, Herriges MJ, Morley MP, Zhou S, Lu MM, Morrisey EE. Emergence of a wave of Wnt signaling that regulates lung alveologenesis by controlling epithelial self-renewal and differentiation. Cell Rep. 2016;17(9):2312–25. https://doi.org/10.1016/j.celrep.2016.11.001.
Zhang K, Yao E, Lin C, Chou YT, Wong J, Li J, Wolters PJ, Chuang PT. A mammalian Wnt5a-Ror2-Vangl2 axis controls the cytoskeleton and confers cellular properties required for alveologenesis. Elife. 2020. https://doi.org/10.7554/eLife.53688.
Ikonomou L, Herriges MJ, Lewandowski SL, Marsland R 3rd, Villacorta-Martin C, Caballero IS, Frank DB, Sanghrajka RM, Dame K, Kandula MM, Hicks-Berthet J, Lawton ML, Christodoulou C, Fabian AJ, Kolaczyk E, Varelas X, Morrisey EE, Shannon JM, Mehta P, Kotton DN. The in vivo genetic program of murine primordial lung epithelial progenitors. Nat Commun. 2020;11(1):635. https://doi.org/10.1038/s41467-020-14348-3.
Liu K, Tang M, Liu Q, Han X, Jin H, Zhu H, Li Y, He L, Ji H, Zhou B. Bi-directional differentiation of single bronchioalveolar stem cells during lung repair. Cell Discov. 2020;6:1. https://doi.org/10.1038/s41421-019-0132-8.
Alvarez-Palomo B, Sanchez-Lopez LI, Moodley Y, Edel MJ, Serrano-Mollar A. Induced pluripotent stem cell-derived lung alveolar epithelial type II cells reduce damage in bleomycin-induced lung fibrosis. Stem Cell Res Ther. 2020;11(1):213. https://doi.org/10.1186/s13287-020-01726-3.
Serrano-Mollar A, Nacher M, Gay-Jordi G, Closa D, Xaubet A, Bulbena O. Intratracheal transplantation of alveolar type II cells reverses bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2007;176(12):1261–8. https://doi.org/10.1164/rccm.200610-1491OC.
Zhou Q, Ye X, Sun R, Matsumoto Y, Moriyama M, Asano Y, Ajioka Y, Saijo Y. Differentiation of mouse induced pluripotent stem cells into alveolar epithelial cells in vitro for use in vivo. Stem Cells Transl Med. 2014;3(6):675–85. https://doi.org/10.5966/sctm.2013-0142.
Cores J, Hensley MT, Kinlaw K, Rikard SM, Dinh PU, Paudel D, Tang J, Vandergriff AC, Allen TA, Li Y, Liu J, Niu B, Chi Y, Caranasos T, Lobo LJ, Cheng K. Safety and efficacy of allogeneic lung spheroid cells in a mismatched rat model of pulmonary fibrosis. Stem Cells Transl Med. 2017;6(10):1905–16. https://doi.org/10.1002/sctm.16-0374.
Serrano-Mollar A, Gay-Jordi G, Guillamat-Prats R, Closa D, Hernandez-Gonzalez F, Marin P, Burgos F, Martorell J, Sanchez M, Arguis P, Soy D, Bayas JM, Ramirez J, Tetley TD, Molins L, de la Bellacasa JP, Rodriguez-Villar C, Rovira I, Fibla JJ, Xaubet A, Pneumocyte Study G. Safety and tolerability of alveolar type II cell transplantation in idiopathic pulmonary fibrosis. Chest. 2016;150(3):533–43. https://doi.org/10.1016/j.chest.2016.03.021.
Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, Niklason L, Calle E, Le A, Randell SH, Rock J, Snitow M, Krummel M, Stripp BR, Vu T, White ES, Whitsett JA, Morrisey EE. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15(2):123–38. https://doi.org/10.1016/j.stem.2014.07.012.
Acknowledgements
Not applicable.
Funding
T.X. is funded by the American Heart Association Career Development Award #19CDA34660211, and Cedars-Sinai Medical Center CSRI – Clinical Scholars Award.
Author information
Authors and Affiliations
Contributions
T.X. took part in conceptualization; T.X. and H.L. wrote and prepared the original draft; T.X., H.L., W.P., B.S., P.C., D.J., and P.N. were involved in writing, reviewing, and editing. All authors have read, approved, and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xie, T., Lynn, H., Parks, W.C. et al. Abnormal respiratory progenitors in fibrotic lung injury. Stem Cell Res Ther 13, 64 (2022). https://doi.org/10.1186/s13287-022-02737-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-022-02737-y