
Abstract
Background
Protein tyrosine phosphatase 1B (PTP1B) is one of the major negative regulators of leptin and insulin signaling, and has been strongly implicated in insulin resistance development in the course of obesity and metabolic syndrome conditions; however, its exact role in controlling adipose tissue biogenesis is still poorly understood.
Objectives
This investigation aimed to elucidate whether selective inhibition of PTP1B using MSI-1436 compound may improve and restore the defective adipogenicity of ASCs isolated from EMS-affected horses.
Methods
Equine ASC EMS cells were cultured under adipogenic conditions in the presence of PTP1B inhibitor and were subsequently tested for expression of the main adipogenic-related genes using RT-qPCR, changes in free fatty acid profiles by means of GC-MS technique, and for mitochondrial dynamics improvement through the analysis of mitochondrial transmembrane potential and oxidative stress.
Results
Selective inhibition of PTP1B in equine ASC EMS cells improved substantially adipogenic differentiation by promoting cellular proliferation and normalizing expression of C/EBPalpha, PPARγ, and Adipoq markers that are critical for proper adipogenesis. Levels of secreted adiponectin and PPARγ were also shown to be increased in MSI-1436-conditioned cells, while total leptin levels markedly dropped under the same conditions. Moreover, MSI-1436 treatment enabled the regulation of metabolic-related transcripts that are crosslink to adipogenesis, namely Akt1, Akt2, and SHBG. The obtained results demonstrated also an obvious reduction in intracellular accumulated ROS and NO, as well as mitigated ER stress through the downregulation of Chop, Perk, Atf6, Ire1, and Xbp1 transcripts upon PTP1B inhibition. Furthermore, general fluctuations in FFA composition of all differentiated groups have been highlighted, where palmitic acid, palmitoleic acid, stearic acid, and linolelaidic acid that are known to be associated with the development of metabolic disorders were found to be normalized upon PTP1B inhibition during adipogenic differentiation.
Conclusion
The presented data provides the evidence that the use of PTP1B inhibitor may be successful in controlling and enhancing adipogenic differentiation of impaired equine ASCs affected by metabolic syndrome, and thus offers new insights for the management of obesity through the regulation of adipose tissue dynamics.
Graphical abstract

Similar content being viewed by others


Background
Metabolic syndrome in both human as well as in horses (defined as equine metabolic syndrome—EMS) has become more and more frequently diagnosed endocrine disorder [1, 2]. Its development is mainly caused by sedentary lifestyle, overfeeding with carbohydrates combined with lack of physical activity. As a consequence, affected individuals develop insulin resistance and related complications. In both species, metabolic syndrome culminates in the development of fatal cardiovascular diseases (in humans diabetic foot while in horses laminitis) [3]. Horses affected with EMS are characterized by deterioration of lipid metabolism and clinically specific lipid accumulation on the neck or on the base of tail and finally lipotoxicity, which is a key player in the insulin resistance development. Although obesity is not a critical component of EMS, since it was proved that lean horses also suffer for insulin resistance, involvement of adipose tissue deterioration in EMS and local inflammation seem to be indisputable [4,5,6].
Adipose tissue (AT) and adipose stem progenitor cells (ASCs) have been shown to be key factors in the development of obesity as well as insulin resistance [7,8,9,10,11,12]. AT is considered as an essential, active metabolic and hormonal organ that orchestrates many pathways involved in insulin resistance development. AT is responsible for the synthesis and secretion of several hormones including leptin, adiponectin, visfatin, or angiotenstin, which modulates insulin resistance and inflammatory axis [13]. Adipose tissue of EMS horses secretes a large amount of pro-inflammatory mediators including tumor necrosis factor α (TNF-α), interleukin-1 (IL-1), and interleukin-6 [4, 14].
Recently, sex hormone binding globulin (SHBG) and fetuin-A (AHSG) have been shown to play an important regulatory role in insulin resistance, and can thus represent promising targets for future therapeutics. Increased fetuin and deceased SHBG systemic levels are strongly associated with metabolic syndrome and type 2 diabetes development [15]. Growing evidence indicates that fetuin-A can modulate insulin-stimulated insulin receptor (INSR) and insulin receptor substrate 1 (IRS1) phosphorylation through the direct interaction with the insulin receptor β-subunit; moreover, fetuin-A is believed to strongly promote lipid-induced insulin resistance over the enhancement of free fatty acids binding to toll-like receptor 4 (TLR4). Furthermore, fetuin-A has been implicated in the pathophysiological mechanisms that lead to obesity onset; indeed, this protein has been claimed to mediate the reduction of adiponectin synthesis through the modulation of the Wnt3a/PPARγ pathway, and has been proposed as a chemoattractant for macrophage recruitment and migration into adipose tissue, promoting their subsequent polarization to M1 pro-inflammatory subtype, that contributes mainly to the AT inflammation [16]. To another extent, decreased levels of SHBG have been attributed to high levels of circulating insulin and glucose, as well as to monosaccharide-induced lipogenesis in the course of obesity and hyperinsulinemia, that may contribute to the repression of the gene that controls the production of SHBG [17]. Secretion of particular hormones or cytokines in AT in the course of EMS development partially results from the molecular deterioration of adipose stem progenitor cells that reside within. Recent data clearly indicates that ASCs as progenitor cells that give a rise to mature adipocytes play a critical role in the impairment of AT during metabolic syndrome development in both human and horses [18, 19]. Our and other groups’ early findings showed that accumulation of reactive oxygen species (ROS) combined with elevated inflammation is key components that impair adipogenic differentiation potential of ASCs and limit their multipotency [20, 21]. Deterioration of mitochondrial biogenesis and dynamics, and production and secretion of the so-called mitoproteins that are involved in adipocyte maturation are the major mechanisms which underlay that phenomenon. The ASC-derived mitochondria are characterized by membrane raptures, disarrayed cristae, and vacuole formation, which in turn contributes to the development of senescent phenotype and decreased proliferative potential. Furthermore, EMS condition seriously limits ASC viability as well as induces apoptosis of those cells and modulates hypertrophy of mature adipocytes [22,23,24]. Adipose stem progenitor cells under metabolic syndrome condition might be also partially involved in adipose tissue fibrosis, which is an important component of metabolic disorders. An excessive accumulation of oxidative stress factors, lipotoxicity, and inflammation strongly contribute to ASCs’ molecular impairment and in turn to the insulin resistance development [25]. Accumulation of free fatty acids (FFAs) in the blood of patients suffering from metabolic syndrome indicates on the correlation between obesity-related insulin resistance and inflammation; however, as mentioned above, not every horse which suffers from EMS is obese [5, 26]. Therefore, probably some other yet not discovered mechanisms may be involved in the development of adipose tissue insulin resistance. Several researches have underlined the importance of endoplasmic reticulum (ER) stress in the course of insulin resistance and inflammation development, since ER is a major site for the proteins, lipids, and sterol synthesis, which modulate adipocyte metabolism [27, 28]. The deterioration of ER functionality leads to increased accumulation of misfolded or unfolded peptides triggering the state of ER stress. Among proteins involved in the ER stress, inositol-requiring enzyme (IRE)-1, PKR-like ER-regulating kinase (PERK), and activating transcription factor (ATF)-6 play the key role [29, 30]. These proteins trigger the activation of pathways that mitigate ER stress, through the unfolded protein response (UPR). As a consequence, the production of protein chaperones needed for proper protein folding or by slowing protein synthesis is triggered; however, if unsuccessful, the unfolded proteins are removed from cells.
ER stress is also involved in the activation of several serine/threonine kinases, including c-jun NH2-terminal kinase (JNK) and IκB-α kinase (IKK). Moreover, it was shown that formation of the IRE-1α-TRAF 2 complex results in the activation and nuclear translocation of nuclear factor κB, which is a key promoter of inflammation. In turn, IRE-1α-TRAF 2 complex also activates JNK, which modulates the expression of pro-inflammatory cytokines and induces insulin resistance through serine phosphorylation of insulin receptor substrates 1 and 2 [27]. Little is known about the involvement of ER stress in the protein tyrosine phosphates’ (PTPs) activation, which are known to inactivate insulin receptor substrate through its dephosphorylation. Recently, MSI-1436 has been shown to inhibit PTP1B—a key enzyme regulating insulin and leptin signaling, via a non-competitive allosteric mechanism, which improves insulin sensitivity and reduces obesity. MSI-1436 is a specific, reversible, and non-competitive inhibitor of PTP1B, which targets preferentially the long form of PTP1B (1–405), containing the extended C-terminal segment [31]. It was shown that MSI-1436 is a potential anti-diabetic agent that can improve glucose tolerance, improve insulin sensitivity, and increase weight loss in insulin-resistant mice [31]. Therefore, the major goal of the presented investigation was to characterize the effects of MSI-1436 and thus PTP1B inhibition on adipogenic differentiation potential of ASCs isolated from EMS horses, with the special attention to ER stress, SHBG, and fetuin axis in order to define its potent involvement in the modulation of adipose tissue insulin resistance.
Materials and methods
All chemicals and reagents were obtained from Sigma-Aldrich (Poznań, Poland), unless otherwise stated. Cell culture reagents were purchased from BioWest (VWR International, Gdańsk, Poland).
Equine ASC isolation and cell culture
Animals from whose adipose tissue samples were collected were diagnosed in accordance with previously established protocol [20]. Clinical and biochemical parameters of these animals were described previously [20]. Subcutaneous adipose tissue biopsies were sampled from the tail base area of adult healthy and EMS horses, under local anesthesia induced by 2% lidocaine (Polfa S.A., Warsaw, Poland). Samples were immediately rinsed using Hanks’ Balanced Salt Solution (HBSS) supplemented with 1% antibiotics for the minimization of possible microbial contamination. Tissue samples were afterwards finely diced using sterile scalpel-blade, subjected to collagenase type I digestion (0.1 mg/mL) during 40 min at 37 °C and 5% CO2, and centrifuged at 1200×g for 10 min. The obtained cell pellet was resuspended in Dulbecco’s modified Eagle’s medium (DMEM) containing 1000 mg/L glucose, and supplemented with 5% of fetal bovine serum (FBS) and 1% of a penicillin and streptomycin (PS) solution in culture flasks. Cultures were maintained in a humidified CO2 incubator (37 °C, 5% CO2, 95% air atmosphere), passaged every 3 days (80–90% of confluence) using an Accutase® solution, and cells were used at third passage for experiments [32].
Cellular population purity was evaluated by means of fluorescence-activated cell sorting technique (BD FACSCalibur, Becton Dickinson, Franklin Lakes, NJ, USA). ASC phenotyping was assessed by flow cytometry analysis using fluorochrome-conjugated monoclonal antibodies (anti-CD105, Acris, Herford, Germany, SM1177PT; anti-CD45, Novus Biologicals, Littleton, CO, USA, NB1006590APC; anti-CD44, R&D Systems, Minneapolis, MN, USA, MAB5449; anti-CD90, ab225, Abcam, Cambridge, UK). Multipotency of isolated ASCs was evaluated through osteogenic, chondrogenic, and adipogenic differentiation of cells cultured in StemXVivo kits (R&D System). All abovementioned techniques were thoroughly described previously [9, 21, 33].
Isolated ASCs expressed significant amounts of positive cell surface markers, namely CD90 and CD105, and were negative for CD45 and CD34, which excluded their hematopoietic origin. Moreover, multipotency of ASCs was confirmed by efficient differentiation into osteoblast, chondrocytes, or adipocytes in vitro [9, 20, 21].
Adipogenic differentiation of equine ASCs
To investigate the potential effect of PTP1B inhibition using MSI-1436 on adipogenic differentiation efficiency, ASCs derived from either healthy (HE) or EMS horses were firstly seeded onto culture plates in regular DMEM culture medium and cultured until they reached confluence. Adipogenic differentiation was then initiated using the StemPro™ Adipogenesis Differentiation Kit (Gibco™, Thermo Fisher Scientific, Poland), following the manufacturer’s instruction. All groups of cells were cultured in adipogenic medium for 14 days in the presence or absence of MSI-1436 inhibitor at a concentration of 1 μM, and culture media were changed every 3 days.
In parallel, HE and EMS cells incubated in the basal medium including low glucose DMEM and 5% FBS and 1% P/S were used as negative controls for both phenotypes.
At the 15th day of differentiation process, all cultures were stopped and subjected to further analysis described below.
Bromodeoxyuridine incorporation assay
DNA synthesis and cell proliferation extend were assessed using the 5-bromo-2-deoxyuridine (BrdU) Cell Proliferation ELISA Kit (Abcam, Cambridge, UK) according to the manufacturer’s recommendations. Briefly, adipogenic and undifferentiated ASCs were incubated with BrdU reagent 24 h prior to differentiation arrest, and left overnight at 37 °C. Incorporation of BrdU into cellular DNA was determined by labeling fixed cells with anti-BrdU monoclonal antibody, and goat anti-mouse IgG conjugated with horseradish peroxidase (HRP) as a secondary antibody. HRP substrate degradation degree was measured with a spectrophotometer plate reader (Spectrostar Nano; BMG Labtech, Ortenberg, Germany) at a wavelength of 450 nm [34].
Oil red O staining
The intracellular accumulation of neutral lipids within the differentiated and undifferentiated ASCs was evaluated by Oil Red O staining according to supplier’s instructions. Briefly, the cells were fixed in 4% paraformaldehyde for 40 min at room temperature, and additionally for 5 min with 60% isopropanol. The fixed cells were subsequently stained with 0.5 g/mL Oil Red prepared in 60% aqueous isopropanol for 15 min at room temperature, and then washed with 60% aqueous isopropanol and PBS. The nuclei were counterstained with hematoxylin solution for 1 min. All stained cells were monitored under an inverted microscope (Axio Observer A1, Zeiss), and micrographs were acquired using a Canon PowerShot digital camera.
Detection of intracellular lipid droplets
The accumulation of neutral lipid droplets in adipogenic ASCs was visualized using the Oil Red O dye as described previously [35]. Briefly, ASCs were fixed with 4% paraformaldehyde for 40 min at room temperature at the 15th day of differentiation induction, followed by incubation with 60% isopropanol for 5 min. Slides were then incubated with Oil Red O for 15 min. The cells were also counterstained with hematoxylin for 1 min. Excessive dye was washed away with PBS. Photos were acquired using Axio Observer A1 inverted microscope (Zeiss, Oberkochen, Germany), while the photographic documentation was made using Canon PowerShot digital camera (Ota, Tokio, Japan).
Analysis of viability and cell apoptosis using flow cytometry
Adipogenic EMS and healthy ASC apoptosis, dead cells, and viability were assessed using the multifunctional Muse Annexin V & Dead Cell Assay kit™ (Cat. No. MCH100105, Merck Millipore, Darmstadt, Germany), according to the manufacturer’s protocol. All differentiated-treated and untreated cells were collected, washed with HBSS, and labeled with the Annexin V & Dead Cell kit for 20 min at room temperature. The distribution of cells across the four populations (i.e., (i) non-apoptotic cells, not undergoing detectable apoptosis, Annexin V (−) and 7-AAD (−); (ii) early apoptotic cells, Annexin V (+) and 7-AAD (−); (iii) late apoptotic cells, Annexin V (+) and 7-AAD (+); and (iv) cells that have died through non-apoptotic pathway, Annexin V (−) and 7-AAD (+)) was determined by the use of Muse Cell Analyzer (Merck Millipore, Darmstadt, Germany).
Multicaspase activity detection
The activity of caspase 1, 3, 4, 5, 6, 7, 8, and 9 was monitored using a Muse MultiCaspase assay kit (Cat. No. MCH100109, Merck Millipore, Darmstadt, Germany) following the user’s guide. All experimental groups of cells were mixed with 5 μL of Muse™ multicaspase reagent working solution and incubated for 30 min in a 37 °C incubator. Thereafter, 150 μL of Muse™ caspase 7-aminoactinomycin D (7-AAD) working solution was added to each sample and incubated in the dark for 5 min at room temperature. Multicaspase activity was subsequently assessed using a Muse Cell Analyzer (Merck Millipore, Darmstadt, Germany).
Mitochondrial membrane potential detection assay
Changes in the mitochondrial membrane potential (MMP) were analyzed by measuring the incorporated MitoPotential lipophilic cationic dye, using the Muse™ MitoPotential Assay kit (Cat. No. MCH100110, Merck Millipore, Darmstadt, Germany). Following adipogenic differentiation in the presence or absence of MSI-1436 inhibitor, EMS and healthy cells were washed with HBSS and stained with the provided fluorescent dyes for 30 min at 37 °C, and the percentage of total depolarized cells (depolarized live + depolarized dead) was evaluated by the mean of a Muse Cell Analyzer (Merck Millipore, Darmstadt, Germany).
Intracellular reactive oxygen and nitrogen species determination
Quantitative measurements of intracellular ROS and RNS, namely superoxide and nitric oxide radicals, were performed using the flow cytometry-based Muse® Oxidative Stress kit and Muse® Nitric Oxide kit (Cat. No. MCH100111/MCH100112, Merck Millipore, Darmstadt, Germany) respectively according to the users’ guide instructions. Cells from each treatment were collected at the 15th day of differentiation, washed with HBSS, and then resuspended in Muse Oxidative Stress and/or Muse Nitric Oxide Reagent working solutions for 30 min at 37 °C in the dark. Determination of ROS+/NO+ versus ROS−/NO− populations was achieved using a Muse Cell Analyzer (Merck Millipore, Darmstadt, Germany).
Measurement of adiponectin, leptin, and PPARγ levels in cultured media
Secretion of adiponectin, leptin, and PPARγ into the medium of the differentiated and undifferentiated ASCs was evaluated using commercial ELISA kits, as recommended by the manufacturer. In brief, cell-free supernatants were collected following 14 days of adipogenesis in the presence or not of PTP1B inhibitor, as well as from undifferentiated control HE and EMS cells. All conditioned culture media were then placed on ready-to-use microwell plates coated with antibodies against adiponectin, leptin, and PPARγ, respectively (Cat. No. MBS081354/MBS936352/MBS016871, MyBioSource, San Diego, USA), incubated with a biotin-labeled secondary antibody and a streptavidin horseradish-peroxidase (HRP) conjugate. The colorimetric reaction was initiated by addition of an HRP substrate (tetramethylbenzidine/peroxide), and measured at 450 nm in a spectrophotometer plate reader (Spectrostar Nano; BMG Labtech, Ortenberg, Germany). Adiponectin, leptin, and PPARγ concentrations were calculated based on the respective dose-response calibration curves that were built using horse adiponectin, leptin, and PPARγ standards provided in the ELISA kits.
RNA preparation and quantitative RT-PCR for gene expression analysis
Total RNA of MSI-1436-treated adipogenic EMS cells as well as adipogenic and undifferentiated EMS and healthy untreated cells was collected using the TRIzol method according to the manufacturer’s protocol. RNA purity and concentration were established using a nanospectrophotometer (WPA, Biowave II, Germany). Genomic DNA (gDNA) digestion and cDNA synthesis were performed using a PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa, Gdańsk, Poland) by the mean of a T100 Thermal Cycler (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions.
Expression levels of targeted genes (Table 1) were analyzed by real-time reverse transcription polymerase chain reaction (RT-PCR), using a SensiFAST SYBR Green Kit (Bioline, London, UK) in a CFX Connect™ Real-Time PCR Detection System (Bio-Rad). Reactions performed in a 10-μL volume containing 150 ng cDNA were subjected to the following cycling conditions: 95 °C for 2 min, followed by 40 cycles at 95 °C for 15 s, annealing for 15 s, and elongation at 72 °C for 15 s. All results were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. The relative expression level was calculated by comparison of the tested groups with control group using the 2-ΔΔCQ method [36].
Lipid extraction and GC-MS-based lipidomics analysis
Cellular metabolite isolation
Total metabolites were extracted from equine healthy and EMS ASCs cultured under either basal or adipogenic conditions according to the procedure described previously [29]. Briefly, cells from all tested groups were collected following 14 days of differentiation, washed three times with cold HBSS, and suspended in an extracting mixture comprising chloroform/methanol/water mixture in a ratio of 20:50:20. Thereafter, suspended cells were subjected to ultra-sonication at room temperature in a water bath sonicator for 2 h and vortexed for 2 min. All samples were transferred to 1.5 mL centrifugation tubes and centrifuged at 4 °C, 18,000×g for 20 min. Afterwards, the obtained supernatants were collected for each sample and dried completely overnight and subjected to gas chromatography-mass spectrometry (GC-MS) analysis.
Gas chromatography-mass spectrometry fatty acid characterization
The global analysis of adipogenic cells’ free fatty acid profile and quantity was performed according to Bourebaba et al.’s [29] protocol. Briefly, crude lipid extract of adipogenic cells, with 30 μg of heptadecanoic acid methyl ester (Me. C17:0) (Sigma-Aldrich, Steinheim, Germany) as internal standard, was subjected to alkaline hydrolysis with 1 M potassium hydroxide solution in methanol. Thereafter, the solution was extracted with hexane and dried over anhydrous magnesium(II) sulfate. Then, after solvent evaporation, methylation of extracted free fatty acids with 14% solution of boron trifluoride in methanol was carried out. Obtained fatty acid methyl esters were extracted with hexane, washed with saturated sodium chloride solution, and dried over anhydrous magnesium(II) sulfate. Finally, the solution of fatty acid methyl esters was densified under nitrogen (up to 200 μL), placed into vial with glass insert, and subjected to GC/MS analysis.
Shimadzu GCMS-QP2020 (Kyoto, Japan) equipped with a ZB-FAME (Phenomenex, Torrance, CA, USA) column (60 m × 0.25 mm i.d. × 0.25 μm film thickness) was used for GC/MS analysis. GC operational conditions are as follows: injection temperature 280 °C; 80 °C kept for 2 min, then to 180 °C at 3.0 °C/min, and then raised to 240 °C at 8.0 °C/min and kept for 4 min; carrier gas—helium with linear velocity 35.0 cm/s; and split ratio 1:10. MS operational conditions are as follows: ion source temperature 220 °C, interface temperature 250 °C, electron impact (EI) ionization at 70 eV, and scanning mode from 40 to 400 m/z.
Identification of fatty acid methyl esters was performed in comparison with analytical standard Supelco 37 component FAME Mix (Bellefonte, PA, USA) analysis carried out under the same conditions. The identification was confirmed by comparison of the experimentally obtained mass spectra with those available in Lipids GC/MS Shimadzu Library for GCMS-QP2020 single quadrupole (Chromaleont srl, Messina, Italy), and only compounds with similarity score ≥ 90% were considered as correct hits. Quantification was based on the peak area normalization.
Statistics
Results were expressed as mean ± SD (n = 3). All statistical analyses and graphical representations were performed using GraphPad Prism (San Diego, CA, USA). Statistical differences were established with a one-way analysis of variance (ANOVA) followed by Bonferroni post hoc multiple comparison test, as indicated. All p values lower than 0.05 (p < 0.05) are summarized with one asterisk (*), those at p < 0.01 use two asterisks (**), and those at p < 0.001 have three asterisks (***).
Results
Evaluation of morphology, proliferation rate, and adipogenic differentiation
Morphology of cells was investigated with brightfield microscope (Fig. 1a). In comparison to cells cultured in standard medium (undifferentiated), morphology of cells which underwent adipogenic differentiation changed from fibroblastic to spherical. Moreover, accumulation of lipid droplets in the cytoplasm was visible in differentiated cells. Analysis of BrdU incorporation revealed significant decrease in proliferation of ASC EMS ND in comparison to their healthy counterparts (Fig. 1b). Similar phenomenon was observed in the cells cultured in adipogenic differentiation medium. Interestingly, cells treated with PTP1B inhibitor, MSI-1436, displayed increased proliferation rate in comparison to untreated cells.

Evaluation of morphology, proliferation rate, and adipogenic differentiation of investigated cells. Visualization of cells cultured in standard growth medium (undifferentiated—ND) and cells which underwent adipogenic differentiation (AD) (a). BrdU assay for cell proliferation (b). Accumulation of lipid droplets was visualized with Oil Red O staining and cells were pictured with confocal microscope (c). Transcript levels of C/EBPalpha (d), PPARγ (e), Lep (f), and Adipoq (g) were determined with RT-qPCR. ELISAs revealed the extracellular levels of PPARγ (h), leptin (i), and adiponectin (j). Relative changes in the expression of genes related to adipogenesis and metabolic regulation: Akt1 (k), Akt2 (l), AHSG (m), and SHBG (n) established by RT-PCR. Results expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. Magnification × 100; scale bar, 250 μm
In order to visualize accumulation of lipid droplets, cells were stained with Oil Red O (Fig. 1c). Interestingly, ASC CTRL AD accumulated more lipid droplets in comparison to EMS counterparts while treating EMS cells with MSI-1436 inhibitor increased the accumulation of fatty acids. To support the staining results, RT-qPCRs for adipogenic factors were performed as well. We observed decreased expression of C/EBPalpha in both EMS groups (ND and AD) in comparison to healthy cells (ND and AD) (Fig. 1d). Interestingly, treating of EMS cells during adipogenesis with the PTP1B inhibitor significantly increased the expression of C/EBPalpha gene. The same phenomenon was observed for PPARγ expression (Fig. 1e). Interestingly, expression of Lep was elevated in EMS cells (ND and AD) and addition of the inhibitor did not influence its expression in cells (Fig. 1f). Expression of Adipoq was elevated in ASC EMS ND in comparison to their healthy counterparts (Fig. 1g). During adipogenic differentiation, mRNA levels of that gene were increased in CTRL cells in comparison to EMS while addition of the inhibitor reduced its expression. We have also investigated extracellular, secreted levels of proteins involved in adipogenesis progression using ELISAs. There were no differences in PPARγ amount in ND groups (Fig. 1h). Its levels were significantly decreased in ASC EMS AD in comparison to healthy cells while treating cells with MSI-1436 significantly increased its levels. In case of Lep, there were no differences between ND groups. Leptin amount was increased in EMS cells during adipogenic differentiation, and treating those cells with the inhibitor significantly reduced its levels (Fig. 1i). Adiponectin levels were decreased in EMS ND and AD cells in comparison to both CTRL ND and CTRL AD, respectively (Fig. 1j). Treating cells with the inhibitor did not influence adiponectin amount. Gene expression was evaluated with RT-qPCR. Akt1 expression was increased in EMS groups in comparison to healthy cells (Fig. 1k). Treatment of EMS cells with MSI-1436 significantly reduced Akt1 expression. The same phenomenon was noted for Akt2 expression (Fig. 1l). Its mRNA levels were upregulated in EMS cells; however, application of MSI-1436 reduced Akt2 expression. Expression of fetuin was increased in EMS cells in comparison to their healthy counterparts in both control and adipogenic conditions (Fig. 1m). Addition of the inhibitor did not affect AHSG expression. SHBG mRNA levels were diminished in EMS cells when comparing to control, and addition of PTP1B inhibitor did not influence significantly its expression levels (Fig. 1n).
Evaluation of apoptosis
To determine the apoptotic profile of cells, we performed the Muse® Annexin V & Dead Cell assay (Fig. 2a, b). The number of live cells was decreased in EMS undifferentiated cells in comparison to their healthy counterparts, while no significant differences in viable cells were noted between the remaining experimental groups. We also performed RT-qPCR analysis of pro-apoptotic Bax gene expression (Fig. 2c). Interestingly, its expression was increased in EMS ND and decreased in EMS AD in comparison to both healthy cells (ND and AD). Treating cells with the inhibitor increased Bax expression. Anti-apoptotic Bcl-2 mRNA levels were decreased in EMS ND in comparison to CTRL ND (Fig. 2d). During adipogenic differentiation, its expression was significantly reduced in EMS cells; however, addition of MSI-1436 inhibitor elevated its expression. The same phenomenon was noted for p21 (Fig. 2e) and p53 (Fig. 2f) expression. Their mRNA levels were reduced in EMS cells in comparison to the CTRL group; however, addition of the inhibitor increased their amount.

Evaluation of apoptosis. The Muse® Annexin V & Dead Cell assay (a, b) results showing increased apoptosis in EMS ND cells. Data was supported by the analysis of genes involved in the regulation of apoptotic pathway: Bax (c), Bcl-2 (d), p21 (e), and p53 (f). The Muse® MultiCaspases assay results (g, h) indicate increased apoptosis in EMS cells. Furthermore, using RT-qPCR, expression of Casp-3 (i) and Casp-9 (j) was investigated. Results expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001
In order to estimate the caspase activation, we performed the Muse® MultiCaspases assay which allows for the quantitative measurement of caspase 1, 3, 4, 5, 6, 7, 8, and 9 simultaneously (Fig. 2g, h). We found that the number of caspase positive cells was increased in EMS ND in comparison to CTRL ND. Interestingly, no differences were noted between CTRL and EMS cells during adipogenic differentiation; however, addition of MSI-1436 reduced the number of dead cells in culture. Data from Muse® was further supported with RT-qPCR analysis of Casp-3 and Casp-9 expression. Interestingly, mRNA levels of Casp-3 were increased in EMS ND and decreased in EMS AD in comparison to the respective control groups (Fig. 2i). Furthermore, addition of the inhibitor significantly enhanced its expression. Interestingly, the same phenomenon was observed for Casp-9 expression (Fig. 2j).
Evaluation of ER stress
To determine the intensity of ER stress in cultures, we analyzed the mRNA levels of the UPR-related genes. Obtained results revealed significantly increased expression of Chop in EMS AD (Fig. 3a). However, PTP1B inhibition resulted in significantly diminished Chop mRNA levels. The expression of Perk was increased in EMS cells in both ND and AD conditions in comparison to CTRL cells (Fig. 3b). Addition of MSI-1436 ameliorated the expression of Perk in ASC EMS. The same phenomenon was observed for the expression of Atf6 (Fig. 3c) and Ire1 (Fig. 3d) which indicates that PTP1B inhibition diminished ER stress in treated cells. ASC EMS ND were characterized by increased expression of Xbp1 in comparison to control cells (Fig. 3e). Interestingly, there were no significant differences of Xbp1 in adipogenic condition.

Expression of UPR-linked markers during adipogenesis and PTP1B inhibition. Transcript levels of Chop (a), Perk (b), Atf6 (c), Ire1 (d), and Xbp1 (e). Results expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001
Assessment of oxidative stress
To assess the oxidative stress, cells were subjected to flow cytometry-based system analysis. Condition of mitochondria in cells cultured in standard condition and subjected to adipogenic differentiation was established with Muse® MitoPotential kit (Fig. 4a, b). Obtained data showed that the number of depolarized cells was increased in EMS ND in comparison to CTRL ND, while no differences between CTRL and EMS cells during adipogenic differentiation were noted. Interestingly, MSI-1436 inhibitor decreased the number of depolarized cells. Muse® Oxidative Stress assay (Fig. 4c, d) revealed increased accumulation of ROS in EMS ND in comparison to CTRL ND. Furthermore, in case of adipogenesis, treatment of EMS cells with PTP1B inhibitor significantly reduced ROS levels. Using the same machine, we investigated the accumulation of nitric oxide in cells with Muse® Nitric Oxide kit (Fig. 4e, f). Nitric oxide amount was increased in both EMS groups (ND and AD) in comparison to respective controls, while treatment of cells with MSI-1436 significantly diminished its levels. To support Muse findings, we performed RT-qPCR analysis to investigate the expression of antioxidative enzymes. No differences in Cat expression were observed between cells cultured in standard medium while decreased expression was found in EMS cells which underwent adipogenic differentiation in comparison to control (Fig. 4g). Interestingly, PTP1B inhibition significantly enhanced Cat expression in EMS cells during AD. Furthermore, we found that expression of Sod1 was upregulated in EMS cells (ND and AD) in comparison to controls (Fig. 4h). Similar phenomenon was noted for Sod2 expression; however, in that case, PTP1B inhibition resulted in significantly decreased Sod2 mRNA levels (Fig. 4i).

Assessment of oxidative stress in control and adipogenic conditions. Cells were subjected to flow cytometry-based system analysis and analyzed with Muse® MitoPotential kit (a, b), Muse® Oxidative Stress assay (c, d), and Muse® Nitric Oxide kit (e, f). Expression of antioxidative enzymes Cat (g), Sod1 (h), and Sod2 (i) was investigated with RT-qPCR. Results expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001
Evaluation of FFA metabolism
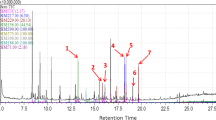
Lipid remodeling changes during adipogenesis of EqASCs were monitored using a GC-MS-based lipidomic profiling technique upon treatment with MSI-1436 inhibitor (Table 2). Overall, significant fluctuations in the levels of all detected fatty acids were observed in all differentiated groups as compared to undifferentiated cells. Among these fatty acids, myristic acid (C14:0), methyl pentadecanoate (C15:0), methyl elaidate (C18:1n, 9E), methyl octadecadienoate/linoleic acid (C18:2n, 9Z,11Z), and oleic acid (C18:1n) were significantly increased following adipogenic differentiation of healthy cells when compared to undifferentiated healthy cells (p < 0.01; p < 0.001). Moreover, decreasing levels of the same myristic acid (C14:0), methyl pentadecanoate (C15:0), methyl elaidate (C18:1n, 9E), and methyl octadecadienoate/linoleic acid (C18:2n, 9Z,11Z) were observed in EMS adipogenic cells in opposition to healthy ASCs differentiated cells (Table 2). By contrast, only myristic acid (C14:0) and methyl octadecadienoate/linoleic acid (C18:2n, 9Z,11Z) levels were maintained in EMS adipogenic MSI-1436-treated cells as compared to both healthy and EMS untreated adipogenic cells. To another extent, GC-MS profiling revealed a consequent decrease in both palmitic acid (C16:0), palmitoleic acid (C16:1), stearic acid (C18:0), and linolelaidic acid (C18:3N, 6Z, 9Z, 12Z) levels in healthy differentiated cells (p < 0.001). EMS differentiated cells exhibited lower amounts of palmitic acid (C16:0) and stearic acid (C18:0), and higher abundance of palmitoleic acid (C16:1) and linolelaic acid (C18:3N, 6Z, 9Z, 12Z) as compared to healthy group of cells (p < 0.001). Inhibition of PTP1B using MSI-1436 compound during adipogenesis induction enabled the normalization of the abovementioned fatty acids’ (palmitic acid, palmitoleic acid, stearic acid, and linolelaidic acid) concentrations in regard to EMS untreated cells (p < 0.001).
Discussion
In the presented study, for the first time, we have investigated the effects of MSI-1436 (trodusquemine) on the adipogenic differentiation of healthy and metabolically impaired mesenchymal stem cells. MSI is a selective, PTP1B inhibitor which protects rodents from diet-induced obesity (DIO) and for that reason is now being tested in clinical trials as an anti-diabetic agent for humans (in accordance with ClinicalTrials.gov, 4 studies have been performed). However, all of the molecular mechanisms of action and outcomes of MSI-1436 in different types of cells remain elusive. We have found that application of MSI-1436 restores adipogenic differentiation potential in EMS derived ASC, ameliorates ER stress, and modulates fatty acid composition of cells and mitochondrial bioenergetics. Our study provides novel evidence for the role of PTP1B in adipocyte dysfunction during obesity and underlines the role of its inhibitor in disease prevention and treatment.
In our previous studies, we have shown that adipogenic differentiation of ASC is altered in EMS animals, which correlates with adipose tissue hypertrophy and inflammation [20, 33]. Dysfunctional adipose tissue remodeling contributes to insulin resistance, accelerates fibrosis and inflammation, and secretes wide range of adipokines, which worsens metabolic state of the organisms [4]. MSI-1436 action has been mostly investigated with HepG2 cells in vitro and in DIO models in vivo [29]. It was shown to improve insulin-stimulated tyrosine phosphorylation of insulin receptor (IR) β and enhance insulin sensitivity, suppress appetite, reduce body weight, and decrease leptin and insulin plasma levels [31, 37]. Yet, little attention has been paid toward molecular mechanisms in adipocytes and changes in their metabolism after treatment. For that reason, to increase the knowledge regarding potential effects of MSI-1436 in the clinic, we decided to investigate its effects on adipogenic differentiation of ASC and their cytobiological properties.
Data from the present and previous studies indicates that deterioration of adipose tissue metabolism and plasticity contributes to the development of metabolic disorders [25]. As MSI-1436 was shown to protect against diabetes and enhance insulin sensitivity, we decided to unravel for the first time its effects on adipose tissue progenitor stem cells during control (standard culture medium, ND) and adipogenic (adipogenic differentiation medium, AD) conditions. We have found that EMS cells displayed decreased proliferation rate in both ND and AD conditions which stands with a good agreement with our and other studies, which showed that metabolically impaired cells suffer from decreased proliferation potential [18, 20, 21]. However, MSI-1436 treatment significantly increased growth kinetics of cells as we observed increased BrdU incorporation. Similarly, Smith et al. [37] revealed enhanced proliferation of zebra fish cardiomyocytes and mouse skeletal muscle satellite cells after MSI-1436 application. Obtained results clearly indicate that MSI-1436 promotes proliferation of metabolically impaired cells and restores progression of cell cycle. It is especially important since it was shown that hyperinsulinemia leads to cell cycle-induced senescence [38]. Adipogenic differentiation is a complex, multistep process in which preadipocyte differentiation is controlled and mediated by the set of signaling proteins and transcription factors. C/EBPalpha and PPARγ play a crucial role in the early stages of the process [39]. In the presented study, we have found that both of these genes’ mRNA levels were downregulated in EMS cells during ND and AD conditions. Similar phenomenon was observed in the study performed by Matulewicz et al. [40], who found decreased expression of C/EBPalpha and PPARγ in the subcutaneous adipose tissue samples of obese patients. Defects in PPARγ expression were shown to deteriorate adipose tissue function, plasticity, and lipotoxicity [41] which correlates with metabolic profile of EMS derived ASC [21]. On the other hand, activation of PPARγ signaling triggers crucial metabolic pathways in adipocytes and maintains their homeostasis. In obesity, PPARγ activation diminishes ectopic lipid accumulation, reduces inflammation, and improves insulin sensitivity and lipid metabolism. For that reason, targeting PPARγ activity in patients with type 2 diabetes and pre-diabetic insulin resistance is considered as a potential therapeutic strategy. Interestingly, in the presented study, we have found that PTP1B inhibition significantly increased PPARγ expression and protein levels which resulted in augmented adipogenic differentiation. It was shown that insulin resistance and inflammation reduce the ability of ASC to differentiate into mature adipocytes by suppressing PPARγ and insulin signaling pathway [42], leading to adipocyte hypertrophy in order to meet the demand for increased triglyceride storage. Furthermore, Bakker et al. have shown that preadipocyte number is significantly reduced in obese subjects [43]. Impaired adipose tissue turnover and disbalance between adipocytes’ death and maturation exaggerate disease progression. For that reason, restoration of adipose tissue plasticity by triggering progenitor stem cell differentiation is considered as an anti-diabetic strategy. Interestingly, in the presented study, we have found that PTP1B inhibition in progenitor cells increased PPARγ activity and enhanced adipogenic differentiation indicating a great therapeutic role of MSI-1436 in the restoration of adipose tissue plasticity. Our findings appear to be consistent with previous investigations performed by Song et al. [44] who showed that PTP1B negatively regulates and impairs adipocyte differentiation in obesity through TNFα. What is more, PTP1B was shown to negatively regulate adipogenesis of brown adipocytes [45]. Another evidence of PTP1B role in adipogenesis comes from the study performed by Owen et al. [46], who revealed that adipocyte-specific PTP1B depletion enhances lipogenesis and regulates glucose homeostasis. Adipose tissue modulates the functions of multiple other tissues in the body through the secretion of wide range of cytokines, the so-called adipokines. Among them, leptin and adiponectin are recognized as key players in the development of insulin resistance, obesity, and diabetes. It was shown that low levels of adiponectin, increased concentration of leptin, and leptin resistance correlate with high risk of developing metabolic and cardiovascular disorders [47]. It stands with a good agreement with our results as we observed increased levels of leptin and decreased amount of adiponectin in adipocytes from EMS individuals. Previous studies demonstrated that PTP1B inhibition enhanced insulin sensitivity and adiponectin serum levels in monkeys [48]. Increased adiponectin concentration and expression following MSI-1436 may be explained by improved insulin sensitivity in adipocytes. Furthermore, activation of PPARγ activity positively regulates adiponectin secretion [49]. Therefore, it is likely that PTP1B inhibition results in the activation of PPARγ activity, which further triggers the cascade of events leading to increased adiponectin secretion and enhanced insulin sensitivity. It is supported by the recent findings by Kumar et al. [50], who revealed that Src homology region 2 domain-containing phosphatase-1 (Shp1)—the member of protein-tyrosine phosphates family—interacts with PPARγ by tyrosine phosphorylation. In case of leptin, it was shown that PTP1B deletion increases leptin sensitivity [51]. Here, we showed that MSI-1436 significantly reduces secretion of leptin by EMS derived adipocytes, which may protect against development of insulin resistance and restore physiological adipocyte secretome. It stands with a good agreement with Lund et al. [52] who revealed that negative regulatory role of PTP1B on leptin signaling is mediated through dephosphorylation of JAK2 and STAT3. Our findings revealed that PTP1B inhibition promotes adipogenesis which is in line with the research performed by Owen et al. [46] who showed enhanced lipogenesis in adipocyte-specific-PTP1B knockout mice. To summarize that part of research, it should be stated that PTP1B acts as a critical regulator of adipogenesis promoting progenitor stem cell differentiation. Yet, the results of further experiments clearly highlight additional signaling pathways that are under PTP1B influence in these cells.
Dimerization of insulin receptor followed by the insulin receptor substrate phosphorylation triggers two major signaling pathways—Ras to mitogen-activated kinases (MAPK) and phosphatidylinositol 3 kinase (PI3K) pathway, which elicits AKT/PKB kinase phosphorylation [53]. AKT is phosphorylated and activated by PDK1/2, and further phosphorylates AS160. As a consequence, GLUT4 translocation to membrane and glucose uptake is triggered. Interestingly, in the presented study, we observed increased expression of both isoforms Akt1 and Akt2 in EMS-derived cell/s in both ND and AD conditions. However, the strong limitation is lack of their protein levels, which may not correlate with mRNA levels. Inhibition of PTP1B resulted in the decreased expression of Akt1 and Akt2. On the contrary, it was shown that PTP1B deficiency enhances AKT activation however in macrophages [54]. Due to relatively low data regarding PTP1B influence on Akt expression, that phenomenon requires further investigation. In the presented study, we also investigated the expression of two novel biomarkers of metabolic disorders—fetuin-A and SHBG. It was shown that fetuin-A levels are increased in adipose tissue of obese subjects [55], which correlates with our results as elevated expression of that gene was observed in EMS-derived cell/s. Contrary phenomenon was observed for SHBG, in which low serum levels correlate with increased susceptibility of type 2 diabetes development [15]. Yet, PTP1B inhibition did not alter expression of these genes, which indicates that PTP1B is not directly involved in the modulation of their action and works through distinct mechanisms.
Our previous, extensive research clearly reported on the increased senescence and apoptosis of EMS derived ASC [20, 22, 33]. In this study, we have shown that EMS cells in ND condition are characterized by increased expression of pro-apoptotic genes, including p53, Bax, and p21. It correlates with the studies performed by our and other research groups which showed that aging, lifestyle, and health condition strongly affect cytophysiological properties of mesenchymal stem cells [18, 21, 56,57,58,59]. Interestingly, we observed increased apoptosis in healthy cells in AD and EMS cells treated with MSI-1436 during differentiation. It can be explained by recent research performed by Hou et al. [60], who revealed that apoptosis is essential for early adipogenesis and that loss of caspase 3 impaired the differentiation capacity of preadipocytes. That phenomenon is explained by the hypothesis in which apoptotic cells stand as a source of factors and nutrients which facilitate adipogenic differentiation. It correlates with our findings which showed reduced caspase 3 in EMS derived ASC suffering from adipogenesis impairment. On the other hand, restoration of caspase 3 activity after MSI-1436 application enhanced adipogenic differentiation of EMS cells. Interestingly, it also enhanced the activity of caspase 9, which indicates that it can also be implicated in the differentiation process. Obtained results reveal a unique role of PTP1B as a modulator of apoptosis in progenitor stem cells triggered by adipogenic differentiation stimuli. This novel mechanism may stand as a potential therapeutic target in the development of strategies for adipose tissue metabolism modulation during insulin resistance.
In the next step of the experiment, we decide to investigate the expression of ER stress-related genes as EMS cells are characterized by UPR activation [22]. Furthermore, it was also shown that progressive ER stress inhibits adipocyte differentiation [61]. It correlates with our results as increased expression of Chop, Perk, Atf6, Ire1, and Xbp1 was observed in EMS cells. However, inhibition of PTP1B significantly reduced the expression of the majority of these genes. Han et al. [61] demonstrated that IF2α-CHOP axis suppressed adipogenesis and limits expansion of fat mass in mice. On the other hand, Panzhinskiy et al. [62] reported that ER stress upregulates expression of PTP1B and impairs glucose uptake in cultured myotubes, while Agouni and colleagues [63] reported that PTP1B inhibition reduces ER stress-induced apoptosis in endothelial cells. For that reason, we can conclude that PTP1B inhibition results in increased adipogenic differentiation through the amelioration of ER stress.
Oxidative stress has been implicated in the development of multiple disorders including insulin resistance and type 2 diabetes [64]. EMS-derived cell/s were characterized by increased ROS and nitric oxide accumulation while MSI-1436 reversed that phenomenon. That fact explains observed here increased adipogenic differentiation after PTP1B silencing as it was shown that ER stress inhibits adipogenesis by the modulation of mitochondrial dynamics [65]. Interestingly, EMS cells cultured in ND condition displayed increased expression of Sod1 and Sod2 while their expression was diminished after MSCI 1436 application. We hypothesize that it can be a compensatory mechanism triggered by cells in order to deal with excessive accumulation of ROS. Yet, a strong limitation is a lack of Sod1 and Sod2 protein levels which may differ from their mRNA amount cells. Given the implication of oxidative stress in the onset of diabetes, increased antioxidative defense of cells treated with PTP1B inhibitor indicates on its potential utility against development of insulin resistance in obese subjects.
Finally, using GC-MS, we analyzed fatty acid composition in investigated cells. Free fatty acids (FFAs) are abundant in obesity leading to impairment of cellular metabolism through mitochondrial damage [66]. Interestingly, EMS differentiated cells exhibited lower amounts of palmitic acid (C16:0) and stearic acid (C18:0), and higher abundance of palmitoleic acid (C16:1) and linoleic acid (C18:3N, 6Z, 9Z, 12Z) as compared to healthy cells. Interestingly, high levels of palmitoleate are associated with the development of metabolic disorders [67]. Similar phenomenon was observed for plasma concentration of linoleic acid [68,69,70]. It was shown that oleic acid regulates adipogenesis through DNA methylation and may predispose to obesity and obesity-related disorders [71]. On the other hand, palmitate was shown to trigger adipogenic differentiation in bone marrow mesenchymal stem cells, which would explain that its insufficient amount in EMS cells may contribute to adipogenesis impairment as well [72]. To summarize, FFA composition strongly affects the adipocyte phenotype and may induce insulin resistance as well as deteriorate adipokine secretion profile. Here, we have shown for the first time that PTP1B inhibition affects composition of FFA in differentiated adipocytes which indicates on the link between PTP1B and lipid metabolism.
Conclusions
Involvement of adipose tissue in the development of insulin resistance, obesity, and type 2 diabetes has been intensively investigated during the recent years. Searching for novel pharmaceuticals targeting that tissue brings hope to affected individuals. Selective chemicals, targeting molecules directly involved with the deterioration of adipocyte metabolism, may restore proper functionality of these cells and restore adipose tissue homeostasis. In the presented research, we tested MSI-1436—a selective PTP1B inhibitor on metabolically impaired progenitor cells—and obtained data revealed that it can restore adipogenic potential of these cells. Here, we have shown that ASC derived from EMS individuals suffers from adipogenesis impairment; however, PTP1B inhibition restores their plasticity and differentiation potential. Obtained results clearly indicate that inhibition of PTP1B modulates adipogenesis and lipid accumulation and highlights the therapeutic potential of its inhibitors against metabolic disorders. Here, we demonstrated that PTP1B acts as a negative regulator of progenitor stem cells plasticity which impairs adipogenic differentiation during insulin-resistant state. Our study provides an evidence of PTP1B role in metabolic disorders and shows that its inhibition during that state remodels adipose tissue plasticity and fatty acid composition restoring efficient differentiation of progenitor stem cells.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 7-AAD:
-
7-Aminoactinomycin D
- AD:
-
Cells which underwent adipogenic differentiation
- ASCs:
-
Adipose stem progenitor cells
- AT:
-
Adipose tissue
- ATF-6:
-
Activating transcription factor
- BrdU:
-
Bromodeoxyuridine
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- DIO:
-
Diet-induced obesity
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- EI:
-
Electron impact
- EMS:
-
Equine metabolic syndrome
- ER:
-
Endoplasmic reticulum
- FFA:
-
Free fatty acid
- GC-MS:
-
Gas chromatography-mass spectrometry
- gDNA:
-
Genomic DNA
- HBSS:
-
Hanks’ Balanced Salt Solution
- HRP:
-
Streptavidin horseradish-peroxidase
- IKK:
-
IκB-α kinase
- IL-1:
-
Interleukin-1
- IRE-1:
-
Inositol-requiring enzyme
- JNK:
-
c-Jun NH2-terminal kinase
- ND:
-
Undifferentiated cells
- PERK:
-
PKR-like ER-regulating kinase
- PS:
-
Penicillin and streptomycin
- PTP1B:
-
Protein tyrosine phosphate 1B
- PTPs:
-
Protein tyrosine phosphates
- ROS:
-
Reactive oxygen species
- RT-PCR:
-
Real-time reverse transcription polymerase chain reaction
- SHBG:
-
Sex hormone binding globulin
- TNF-α:
-
Tumor necrosis factor α
- UPR:
-
Unfolded protein response
References
Frank N. Equine metabolic syndrome. J Equine Vet Sci. 2009;29:259–.
Marycz K, Michalak I, Kornicka K. Advanced nutritional and stem cells approaches to prevent equine metabolic syndrome. Res Vet Sci. 2018;118:115–25.
Ertelt A, Barton A-K, Schmitz RR, Gehlen H. Metabolic syndrome: is equine disease comparable to what we know in humans? Endocr Connect. 2014;3:R81–93.
Basinska K, Marycz K, Śmieszek A, Nicpoń J. The production and distribution of IL-6 and TNF-α in subcutaneous adipose tissue and their correlation with serum concentrations in Welsh ponies with equine metabolic syndrome. J Vet Sci. 2015;16:113–20.
Durham AE, Frank N, McGowan CM, Menzies-Gow NJ, Roelfsema E, Vervuert I, et al. ECEIM consensus statement on equine metabolic syndrome. J Vet Intern Med. 2019;33:335–49.
Johnson PJ, Wiedmeyer CE, LaCarrubba A, Seshu Ganjam VK, Messer NT IV. Laminitis and the equine metabolic syndrome. Vet Clin North Am Equine Pract. 2010;26:239–55.
Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov. 2016;15:639–60.
Kohlgruber A, Lynch L. Adipose tissue inflammation in the pathogenesis of type 2 diabetes. Curr Diab Rep. 2015;15:92.
Nawrocka D, Kornicka K, Śmieszek A, Marycz K. Spirulina platensis improves mitochondrial function impaired by elevated oxidative stress in adipose-derived mesenchymal stromal cells (ASCs) and intestinal epithelial cells (IECs), and enhances insulin sensitivity in Equine Metabolic Syndrome (EMS) horse. Marine Drugs. 2017;15:1–28.
Marycz K, Grzesiak J, Wrzeszcz K, Golonka P. Adipose stem cell combined with plasma-based implant bone tissue differentiation in vitro and in a horse with a phalanx digitalis distalis fracture: a case report. Veterinarni Medicina (Czech Republic). 2012; [cited 2016 Jul 3]; Available from: http://agris.fao.org/agris-search/search.do?recordID=CZ2013000199.
Grzesiak J, Marycz K, Czogala J, Wrzeszcz K, Nicpon J. Comparison of behavior, morphology and morphometry of equine and canine adipose derived mesenchymal stem cells in culture. Int J Morphol. 2011;29:1012–7.
Cislo-Pakuluk A, Marycz K. A promising tool in retina regeneration: current perspectives and challenges when using mesenchymal progenitor stem cells in veterinary and human ophthalmological applications. Stem Cell Rev. 2017;13:598–602.
Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97.
Marycz K, Kornicka K, Grzesiak J, Mieszek A, et al. Macroautophagy and selective mitophagy ameliorate chondrogenic differentiation potential in adipose stem cells of equine metabolic syndrome: new findings in the field of progenitor cells differentiation [Internet]. Oxidative Medicine and Cellular Longevity. 2016 [cited 2017 Jul 27]. Available from: https://www.hindawi.com/journals/omcl/2016/3718468/.
Ding EL, Song Y, Manson JE, Hunter DJ, Lee CC, Rifai N, et al. Sex hormone–binding globulin and risk of type 2 diabetes in women and men. N Engl J Med. 2009;361:1152–63.
Bourebaba L, Marycz K. Pathophysiological implication of fetuin-A glycoprotein in the development of metabolic disorders: a concise review. J Clin Med. 2033;2019:8.
Daka B, Rosen T, Jansson PA, Råstam L, Larsson CA, Lindblad U. Inverse association between serum insulin and sex hormone-binding globulin in a population survey in Sweden. Endocr Connect. 2012;2:18–22.
Kornicka K, Marycz K, Tomaszewski KA, Marędziak M, Śmieszek A. The effect of age on osteogenic and adipogenic differentiation potential of human adipose derived stromal stem cells (hASCs) and the impact of stress factors in the course of the differentiation process. Oxid Med Cell Longev. 2015;309169:2015.
Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci. 2019;20 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6539070/. [cited 2019 Nov 17].
Marycz K, Kornicka K, Basinska K, Czyrek A. Equine metabolic syndrome affects viability, senescence, and stress factors of equine adipose-derived mesenchymal stromal stem cells: new insight into EqASCs isolated from EMS Horses in the context of their aging. Oxidative Med Cell Longevity. 2016;2016:1–17.
Marycz K, Weiss C, Śmieszek A, Kornicka K. Evaluation of oxidative stress and mitophagy during adipogenic differentiation of adipose-derived stem cells isolated from equine metabolic syndrome (EMS) horses. Stem Cells Int. 2018;2018:1–18 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6011082/. [cited 2018 Sep 28].
Kornicka K, Szłapka-Kosarzewska J, Śmieszek A, Marycz K. 5-Azacytydine and resveratrol reverse senescence and ageing of adipose stem cells via modulation of mitochondrial dynamics and autophagy. Journal of Cellular and Molecular Medicine [Internet]. 2018 [cited 2018 Dec 7];0. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/jcmm.13914.
Marycz K, Kornicka K, Irwin-Houston JM, Weiss C. Combination of resveratrol and 5-azacytydine improves osteogenesis of metabolic syndrome mesenchymal stem cells. J Cell Mol Med. 2018;22:4771–93.
Marycz K, Houston JMI, Weiss C, Röcken M, Kornicka K. 5-Azacytidine and resveratrol enhance chondrogenic differentiation of metabolic syndrome-derived mesenchymal stem cells by modulating autophagy [Internet]. Oxidative Medicine and Cellular Longevity. 2019 [cited 2019 Jul 17]. Available from: https://www.hindawi.com/journals/omcl/2019/1523140/.
Kornicka K, Houston J, Marycz K. Dysfunction of mesenchymal stem cells isolated from metabolic syndrome and type 2 diabetic patients as result of oxidative stress and autophagy may limit their potential therapeutic use. Stem Cell Rev and Rep. 2018:14(3):337–45.
Ormseth MJ, Swift LL, Fazio S, Linton MF, Raggi P, Solus JF, et al. Free fatty acids are associated with metabolic syndrome and insulin resistance, but not inflammation in SLE patients. Lupus. 2013;22:26–33.
Ariyasu D, Yoshida H, Hasegawa Y. Endoplasmic reticulum (ER) stress and endocrine disorders. Int J Mol Sci. 2017;18:382.
Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–93.
Bourebaba L, Łyczko J, Alicka M, Bourebaba N, Szumny A, Fal AM, et al. Inhibition of protein-tyrosine phosphatase PTP1B and LMPTP promotes palmitate/oleate-challenged HepG2 cell survival by reducing lipoapoptosis, improving mitochondrial dynamics and mitigating oxidative and endoplasmic reticulum stress. J Clin Med. 2020;9:1294.
Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18:59–68.
Lantz KA, Hart SGE, Planey SL, Roitman MF, Ruiz-White IA, Wolfe HR, et al. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity. 2010;18(8):1516–23.
Bourebaba L, Michalak I, Röcken M, Marycz K. Cladophora glomerata methanolic extract decreases oxidative stress and improves viability and mitochondrial potential in equine adipose derived mesenchymal stem cells (ASCs). Biomed Pharmacother. 2019;111:6–18.
Marycz K, Kornicka K, Marędziak M, Golonka P, Nicpoń J. Equine metabolic syndrome impairs adipose stem cells osteogenic differentiation by predominance of autophagy over selective mitophagy. J Cell Mol Med. 2016;20:2384–404.
Bourebaba L, Michalak I, Baouche M, Kucharczyk K, Marycz K. Cladophora glomerata methanolic extract promotes chondrogenic gene expression and cartilage phenotype differentiation in equine adipose-derived mesenchymal stromal stem cells affected by metabolic syndrome. Stem Cell Res Ther. 2019;10:392.
Marędziak M, Śmieszek A, Chrząstek K, Basinska K, Marycz K. Physical activity increases the total number of bone-marrow-derived mesenchymal stem cells, enhances their osteogenic potential, and inhibits their adipogenic properties [Internet]. Stem Cells International. 2015 [cited 2018 Apr 19]. Available from: https://www.hindawi.com/journals/sci/2015/379093/.
Suszynska M, Poniewierska-Baran A, Gunjal P, Ratajczak J, Marycz K, Kakar SS, et al. Expression of the erythropoietin receptor by germline-derived cells - further support for a potential developmental link between the germline and hematopoiesis. J Ovarian Res. 2014;7:66.
Smith AM, Maguire-Nguyen KK, Rando TA, Zasloff MA, Strange KB, Yin VP. The protein tyrosine phosphatase 1B inhibitor MSI-1436 stimulates regeneration of heart and multiple other tissues. NPJ Regen Med. 2017;2:4.
Chow H-M, Shi M, Cheng A, Gao Y, Chen G, Song X, et al. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat Neurosci. 2019;22:1806–19.
Rosen ED, Hsu C-H, Wang X, Sakai S, Freeman MW, Gonzalez FJ, et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002;16:22–6.
Matulewicz N, Stefanowicz M, Nikołajuk A, Karczewska-Kupczewska M. Markers of adipogenesis, but not inflammation, in adipose tissue are independently related to insulin sensitivity. J Clin Endocrinol Metab. Oxford Academic. 2017;102:3040–9.
Corrales P, Vidal-Puig A, Medina-Gómez G. PPARs and metabolic disorders associated with challenged adipose tissue plasticity. Int J Mol Sci [Internet]. 2018 [cited 2020 Aug 19];19. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6073677/.
Ye J, Gimble JM. Regulation of stem cell differentiation in adipose tissue by chronic inflammation. Clin Exp Pharmacol Physiol. 2011;38:872–8.
Bakker AHF, Nijhuis J, Buurman WA, van Dielen FMH, Greve JWM. Low number of omental preadipocytes with high leptin and low adiponectin secretion is associated with high fasting plasma glucose levels in obese subjects. Diabetes Obes Metab. 2006;8:585–8.
Song D-D, Chen Y, Li Z-Y, Guan Y-F, Zou D-J, Miao C-Y. Protein tyrosine phosphatase 1B inhibits adipocyte differentiation and mediates TNFα action in obesity. Biochim Biophys Acta. 2013;1831:1368–76.
Matsuo K, Bettaieb A, Nagata N, Matsuo I, Keilhack H, Haj FG. Regulation of brown fat adipogenesis by protein tyrosine phosphatase 1B. PLoS One. 2011;6 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3031545/. [cited 2020 Aug 19].
Owen C, Czopek A, Agouni A, Grant L, Judson R, Lees EK, et al. Adipocyte-specific protein tyrosine phosphatase 1B deletion increases lipogenesis, adipocyte cell size and is a minor regulator of glucose homeostasis. PLOS ONE. 2012;e32700:7.
Diwan AG, Kuvalekar AA, Dharamsi S, Vora AM, Nikam VA, Ghadge AA. Correlation of serum adiponectin and leptin levels in obesity and type 2 diabetes mellitus. Indian J Endocrinol Metab. 2018;22:93–9.
Swarbrick MM, Havel PJ, Levin AA, Bremer AA, Stanhope KL, Butler M, et al. Inhibition of protein tyrosine phosphatase-1B with antisense oligonucleotides improves insulin sensitivity and increases adiponectin concentrations in monkeys. Endocrinology. 2009;150:1670–9.
Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–9.
Kumar A, Lavallee-Bourget M-H, Forest M-P, Schwab M, Bellmann K, Houde V, et al. 1880-P: the protein-tyrosine phosphatase Shp1 interacts with PPAR gamma and regulates its transcriptional activity through dephosphorylation of selected tyrosine sites. Diabetes. 2019;68 Available from: https://diabetes.diabetesjournals.org/content/68/Supplement_1/1880-P. [cited 2020 Aug 19].
Tsou RC, Rak KS, Zimmer DJ, Bence KK. Improved metabolic phenotype of hypothalamic PTP1B-deficiency is dependent upon the leptin receptor. Mol Metab. 2014;3:301–12.
Lund IK, Hansen JA, Andersen HS, Møller NPH, Billestrup N. Mechanism of protein tyrosine phosphatase 1B-mediated inhibition of leptin signalling. J Mol Endocrinol. 2005;34:339–51.
Świderska E, Strycharz J, Wróblewski A, Szemraj J, Drzewoski J, Śliwińska A. Role of PI3K/AKT pathway in insulin-mediated glucose uptake. Blood Glucose Levels [Internet]. IntechOpen; 2018 [cited 2020 Aug 19]; Available from: https://www.intechopen.com/books/blood-glucose-levels/role-of-pi3k-akt-pathway-in-insulin-mediated-glucose-uptake.
Través PG, Pardo V, Pimentel-Santillana M, González-Rodríguez Á, Mojena M, Rico D, et al. Pivotal role of protein tyrosine phosphatase 1B (PTP1B) in the macrophage response to pro-inflammatory and anti-inflammatory challenge. Cell Death Dis. 2014;5:e1125.
Khadir A, Kavalakatt S, Madhu D, Hammad M, Devarajan S, Tuomilehto J, et al. Fetuin-A levels are increased in the adipose tissue of diabetic obese humans but not in circulation. Lipids Health Dis [Internet]. 2018 [cited 2020 Aug 19];17. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6303986/.
Marędziak M, Marycz K, Tomaszewski KA, Kornicka K, Henry BM. The influence of aging on the regenerative potential of human adipose derived mesenchymal stem cells [Internet]. Stem Cells International. 2016 [cited 2018 Feb 2]. Available from: https://www.hindawi.com/journals/sci/2016/2152435/.
Nawrocka D, Kornicka K, Szydlarska J, Marycz K. Basic fibroblast growth factor inhibits apoptosis and promotes proliferation of adipose-derived mesenchymal stromal cells isolated from patients with type 2 diabetes by reducing cellular oxidative stress [Internet]. Oxidative Medicine and Cellular Longevity. 2017 [cited 2017 Jul 27]. Available from: https://www.hindawi.com/journals/omcl/2017/3027109/ref/.
Alicka M, Major P, Wysocki M, Marycz K. Adipose-derived mesenchymal stem cells isolated from patients with type 2 diabetes show reduced “stemness” through an altered secretome profile, impaired anti-oxidative protection, and mitochondrial dynamics deterioration. J Clin Med. 2019;8(6):765.
Alicka M, Kornicka-Garbowska K, Kucharczyk K, Kępska M, Rӧcken M, Marycz K. Age-dependent impairment of adipose-derived stem cells isolated from horses. Stem Cell Res Ther. 2020;11:4.
Hou W, Chen Q, Wang H, Qiu P, Lyu X, Chen W, et al. The metabolic footprint during adipocyte commitment highlights ceramide modulation as an adequate approach for obesity treatment. EBioMedicine. 2020;102605:51.
Han J, Murthy R, Wood B, Song B, Wang S, Sun B, et al. ER stress signalling through eIF2α and CHOP, but not IRE1α, attenuates adipogenesis in mice. Diabetologia. 2013;56:911–24.
Panzhinskiy E, Hua Y, Culver B, Ren J, Nair S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia. 2013;56:598–607.
Agouni A, Pasha M, Abdelsalam S, Gamal HE. Protein tyrosine phosphatase (PTP) 1B inhibition improves endoplasmic reticulum stress-induced apoptosis in endothelial cells. FASEB Journal. 2019;33:677 1–677.1.
Hurrle S, Hsu WH. The etiology of oxidative stress in insulin resistance. Biomed J. 2017;40:257–62.
Fernando R, Wardelmann K, Deubel S, Kehm R, Jung T, Mariotti M, et al. Low steady-state oxidative stress inhibits adipogenesis by altering mitochondrial dynamics and decreasing cellular respiration. Redox Biol [Internet]. 2020 [cited 2020 Aug 19];32. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7097524/.
Boden G. 45Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18:139–43.
Mozaffarian D, Cao H, King IB, Lemaitre RN, Song X, Siscovick DS, et al. Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am J Clin Nutr. 2010;92:1350–8.
Yi L, He J, Liang Y, Yuan D, Gao H, Zhou H. Simultaneously quantitative measurement of comprehensive profiles of esterified and non-esterified fatty acid in plasma of type 2 diabetic patients. Chem Phys Lipids. 2007;150:204–16.
Liu L, Li Y, Guan C, Li K, Wang C, Feng R, et al. Free fatty acid metabolic profile and biomarkers of isolated post-challenge diabetes and type 2 diabetes mellitus based on GC-MS and multivariate statistical analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2817–25.
Grapov D, Adams SH, Pedersen TL, Garvey WT, Newman JW. Type 2 diabetes associated changes in the plasma non-esterified fatty acids, oxylipins and endocannabinoids. PLoS ONE. 2012;e48852:7.
Malodobra-Mazur M, Cierzniak A, Dobosz T. Oleic acid influences the adipogenesis of 3T3-L1 cells via DNA methylation and may predispose to obesity and obesity-related disorders. Lipids Health Dis [Internet]. 2019 [cited 2020 Oct 29];18. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6935146/.
Park EY, Yeum CE, Seo G, Lee J-Y, Lee S-B, Chae G-T. The adipogenic effect of palmitate in mouse bone marrow-derived mesenchymal stem cells. Tissue Eng Regen Med. 2013;10:77–85.
Acknowledgements
None.
Funding
The work was supported by grant from the National Science Centre in Poland over the course of the realization of the projects: “Inhibition of tyrosine phosphatase as a strategy to enhance insulin sensitivity through activation of chaperone mediated autophagy and amelioration of inflammation and cellular stress in the liver of equine metabolic syndrome (EMS) horses” (2018/29/B/NZ7/02662) and “Mitochondria transfer as an innovative method of restoring stemness of adipose-derived stem cells isolated from horses suffering from metabolic syndrome (EMS)” (2017/27/N/NZ7/02343).
Author information
Authors and Affiliations
Contributions
L. Bourebaba participated in the writing of manuscript, experiment planning, cell culture, and data analysis, K. Kornicka-Garbowska participated in the writing of manuscript and experiment planning. M. Al Naem and M. Rocken participated in the interpretation of obtained result, validation, methodology, and data curation. Łyczko J performed and described the GC-MS experiments. K. Marycz designed and coordinated the study, wrote the manuscript, and provided the funding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All of the work described was performed with the approval of the local ethics committee for animal experiments at Ludwik Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Weigla Str. 12, 53-114, Wrocław, Poland (the protocol number was 84/2018). All animal experiments were performed according to the local regulations. The horses in this study were examined with the consent of their owners.
Consent for publication
Not applicable.
Competing interests
The authors confirm that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bourebaba, L., Kornicka-Garbowska, K., Al Naem, M. et al. MSI-1436 improves EMS adipose derived progenitor stem cells in the course of adipogenic differentiation through modulation of ER stress, apoptosis, and oxidative stress. Stem Cell Res Ther 12, 97 (2021). https://doi.org/10.1186/s13287-020-02102-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-020-02102-x