
Abstract
The prevalence of kidney diseases is emerging as a public health problem. Stem cells (SCs), currently considered as a promising tool for therapeutic application, have aroused considerable interest and expectations. With self-renewal capabilities and great potential for proliferation and differentiation, stem cell therapy opens new avenues for the development of renal function and structural repair in kidney diseases. Mounting evidence suggests that stem cells exert a therapeutic effect mainly by replacing damaged tissues and paracrine pathways. The benefits of various types of SCs in acute kidney disease and chronic kidney disease have been demonstrated in preclinical studies, and preliminary results of clinical trials present its safety and tolerability. This review will focus on the stem cell-based therapy approaches for the treatment of kidney diseases, including various cell sources used, possible mechanisms involved, and outcomes that are generated so far, along with prospects and challenges in clinical application.
Similar content being viewed by others


Introduction
Kidney diseases have become a global public health problem due to their rapidly growing incidence. These diseases affect over 10% of the global population, because of a global rise in the aging population, as well as increase in the frequency of their main etiologies, such as diabetes, cardiovascular diseases, and hypertension [1, 2]. As a routine treatment for kidney diseases, multidrug therapy cannot reverse the process of entering into end-stage renal disease (ESRD) in most of the patients, and those with ESRD require renal replacement therapy, i.e., maintenance dialysis or kidney transplantation [3]. Owing to high medical costs and adverse impacts on the patient’s quality of life, dialysis is not considered as an ideal treatment strategy [4, 5]. Kidney transplantation enables patients to regain their own renal function; however, severe shortage of organ donors and potential organ rejection limited its use. Consequently, exploring novel and better therapeutic options to alleviate, cure, or prevent renal diseases and to improve patients’ survival and quality of life is mandatory.
Stem cells (SCs) are described by their self-renewal abilities and the capability to develop into various functional cells under certain conditions. Based on the advantages of plasticity, infinite amplification, and ease of genetic manipulation, stem cell therapy opens new avenues for almost all human diseases. The application of SC therapy in treating a variety of diseases such as immunological, vascular, cardiac, and renal diseases has been extensively explored [6, 7]. The use of SCs is a promising therapeutic strategy for kidney diseases as well. Increasing results obtained in models of acute kidney injury (AKI) and chronic kidney disease (CKD) document that SCs have therapeutic potential in repair of renal injury, preserving renal function and structure thus prolonging animal survival. The effects were initially attributed to SCs implanting damaged tissue, differentiating, and replacing damaged cells [8]. The subsequent evidence suggests that SCs also act through the secretion of bioactive paracrine factors and/or release of microvesicles with immunomodulatory and proregenerative properties [9, 10]. Additionally, stem cells could be directed to differentiate into kidney lineage cells and generate into kidney organoids, which are expected to be used for disease modeling and drug discovery, and may eventually be applicable for transplantation [11]. In view of the great potential application of SCs in kidney diseases, the need to better understand on how to develop cell-based experimental treatments, and how to implement them in clinical trials, becomes more pressing. In this review, an overview of characteristics of different types of stem cells, the current available evidence from preclinical/clinical studies on cell-based therapy for kidney diseases, and discussion of prospects and challenges of its application in the treatment of kidney diseases were provided.
Potential sources of stem cells for cell-based therapy in kidney diseases
Before we go into further details regarding the current SC-based approaches, a brief summary of the background information regarding potential sources of stem cells has to be put forward. SCs are self-renewable cells that are capable of differentiating into specialized cell types under appropriate conditions, and could be divided into four categories according to their differentiation potential (Fig. 1).

Classification of stem cells based on differentiation potential. TSCs, totipotent stem cells; ESCs, embryonic stem cells; iPSCs, induced pluripotent cells; MSCs, mesenchymal stem cells; HSCs, hematopoietic stem cells; NSCs, neural stem cells; RSPCs, renal stem/progenitor cells
With the greatest differentiation potential, totipotent stem cells (TSCs) possess the omnipotentiality to differentiate into any type of cells, including the extraembryonic lineages and the germline [12, 13]. The zygote and the blastomeres of the early embryo are considered to be totipotent [14, 15]. Providing tools and models for exploring the underlying molecular mechanisms of cell potency, somatic cell nuclear transfer (SCNT) is currently the only way to generate a totipotent cell in vitro, which involved the physical transplantation of a somatic nucleus into an enucleated oocyte. Due to its association with low success rate and requirement of oocytes, there are both scientific and ethical considerations which hold back the broader practical use of this technology in stem cell therapy [16, 17]. But understanding TSCs helps us to understand the molecular mechanisms behind maintenance and manipulation of cell fate and is essential to understand how pluripotent cells form in vivo.
Pluripotency refers to a stem cell with potential to differentiate into any of the three germ lineages: endoderm (interior stomach lining, gastrointestinal tract, the lungs), mesoderm (muscle, bone, blood, urogenital), or ectoderm (epidermal tissues and nervous system) [18]. Pluripotent stem cells (PSCs) are capable of giving rise to all cells of any tissue types except for extraembryonic cells and mainly include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). ESCs are harvested from the inner cell mass of blastocysts and are considered as the initial candidates of cell therapy [18, 19]. However, the clinical use of ESCs generated from surplus embryos has some problems, including bioethical debate, immunological rejections, and the risk of teratoma. One way to circumvent these issues is by generating PSCs directly from the patients’ own cells. Induced pluripotent stem cells are reprogrammed somatic cells that regain pluripotency by inducing a “forced” overexpression of certain transcription factors. iPSCs share many similarities with ESCs in terms of morphology, proliferation, surface markers, gene expression, epigenetic status, etc., and a less risk of immune rejection than ESCs. They are considered to be a potential source for renal generation and promising cell source for patients requiring specific cell therapy.
Adult SCs that exist in the postnatal organisms are either multipotent or unipotent and are restricted to a limited range of cell types if iPSCs were not taken into consideration. Multipotent SCs, including hematopoietic stem cells (HSCs) [20], mesenchymal stem cells (MSCs) [21], neural stem cells (NSCs) [22], and intestinal stem cells (ISCs) [23], can differentiate into several cell types of confined cell lineages. The MSCs, which are also known as the mesenchymal stromal cells (MSCs), can differentiate into cells of mesenchymal cell lineages such as osteocytes, adipocytes, and chondrocytes, and potentially other cell types [21]. In addition, MSCs were isolated virtually from any type of tissues, including the bone marrow stroma, adipose tissue, and umbilical cord blood. It has been hypothesized that MSCs contribute to tissue repair by differentiating into organ-specific cells and replacement of damaged tissues. Recently, a body of studies has confirmed that MSCs act through paracrine/endocrine secretion of bioactive factors and exosomes. MSCs secrete a variety of molecules including cytokines, growth factors, anti-oxidants and pro-angiogenic factors, and those that stimulate cell proliferation and angiogenesis, decrease the response to stress and apoptosis of damaged cells, and regulate the local and systemic inflammatory and immune response, thus contributing to tissue repair [24, 25]. The application of MSCs in treating kidney diseases is widely studied and is superior to the application of other SCs.
Progenitor cells exhibit differentiation potential between SCs and mature cells and were found in many organs, including the bone marrow [26], gastrointestinal mucosa [27], liver, brain, prostate, and skin. These cells play a part in maintaining the wounding process to replace the damaged or dead cells, participating in the normal cell turnover of organs. Whether or not adult kidney SCs exist has been an unsettled controversy to date, but current research has confirmed that the adult mammalian kidney contains multiple cells with self-renewability and differentiation potential, which are known as the renal stem/progenitor cells (RSPCs) [28,29,30,31,32]. With inherent renal differentiation potential and the potential for autologous treatments, RSPCs were considered to be ideal sources of cell therapy for treating kidney diseases.
Embryonic stem cells
ESCs that are characterized by features of self-renewability and multilineage differentiation hold great promise in cell therapy and kidney regeneration. Numerous studies have shown that mouse embryonic stem cells (mESCs) can integrate into kidney compartments, suggesting a possible efficacy for kidney repair. In addition, the exposure of ESCs to factors required for renal specification, such as retinoic acid, activin A, and bone morphogenic proteins (BMPs), induces differentiation of these cells into renal lineage cells in vitro [33,34,35].
Geng et al. have reported that the ESC-loaded gelatin microcryogels on 5/6 nephrectomized rats slowed down the progression of chronic kidney disease and reduced glomerular injury [36]. Similarly, mESC implantation in mice with renal failure induction (RFI) employing cisplatin significantly decreased the mortality, avoiding a greater histological deterioration related to the disease. Glomerulosclerosis or collapsed glomeruli were not observed in RFI mice treated with mESCs; even some regeneration data were found characterized by large nuclei with prominent [37]. Mata-Miranda et al. suggested the beneficial effects of mESCs on AKI were related to the reduction of lipid peroxidation [38].
Patients require renal replacement therapy when their kidney disease progresses to ESRD. Although dialysis offers a temporary solution for patients, it has several limitations. Dialysis does not address the loss of hemostatic and endocrine function of the kidney and its associated complications [5]. Kidney transplantation remains to be the best curative treatment strategy to restore total kidney function, but the demand for organs suitable for transplantation has reached a level that far outstrips the static supply [39]. Due to the precise nature of renal structure, which comprises several kinds of cells and with complex anatomy, the kidney has become the most difficult organs of reconstruction. Many protocols have induced the differentiation of ESCs to generate complex structures resembling kidneys, termed organoids, which contain multiple renal cell types and are capable of self-organization.
Morizane et al. have successfully induced differentiation of ESCs into nephron progenitor cells (NPCs), which were then transformed into self-organized nephron-like structures containing glomeruli, proximal tubules, loop of Henle, and distal tubules in a contiguous, ordered arrangement that are analogous to that of nephrons [40]. Takasato and colleagues have generated kidney organoids that contain not only nephrons and collecting duct but also renal interstitium and an endothelial network. The proximal tubules of these show functional maturity to some extent [41]. The ESC-derived kidney progenitors implanted into immunocompromised mice generated perfused glomeruli containing human capillaries, podocytes with regions of mature basement membrane, and mesangial cells. The vascularized glomeruli showed the ability to produce ultrafiltrate that is processed by adjacent tubules [42]. The latest research is consistent with previous ones. By inducing the directional differentiation of ESCs into ureteric bud progenitor cells and co-culturing with dissociated primary metanephric mesenchyme (pMM), the organoids composed of nephron structures and collecting ducts could be produced, which displayed the presence of endothelial cells forming a vascular network [43].
Although scientists thought they have found a strategy to isolate hESCs from single blastomeres without destroying the embryos, legal and ethical controversies still limit the research and applications of ESCs [44]. Another concern about this technique is that cells and tissues derived from ESCs are at high risk of degenerating into neoplasms, especially teratomas. Yamamoto et al. demonstrated that ESCs can develop teratoma 14 days and 28 days after transplantation into mouse [45]. What is more, ESC-derived differentiated cells are allogenic in nature and inevitably suffer from all the issues related to allografts and immunocompatibility, such as acute and chronic rejection, and graft versus host disease.
Induced pluripotent stem cells
Induced pluripotent stem cells sharing many of the regenerative properties of ESCs are considered effective alternative cell sources for ESCs. Indeed, iPSCs could retain both genetic background and peculiar epigenetic memory of the cells of origin, providing indisputable advantages in cell therapy, kidney regeneration, and other biomedical applications.
According to the earliest report on iPSCs put forward in 2006, scientists obtained PSCs by introducing four transcription factors (OCT4, SOX2, KLF4, and c-MYC) into mouse adult fibroblasts, and this technique has earned Shinya Yamanaka and John Gurdon the Nobel Prize in Physiology or Medicine 2012 [34]. To date, the human iPSCs have been generated from multiple sources, including skin fibroblasts, keratinocytes [46, 47], extraembryonic tissues [48], cord blood [49], peripheral blood cells [50], hepatocytes, stomach cells [51], dental pulp cells [52], and even fully differentiated lymphocytes (such as T and B cells) [53]. Interestingly, the terminally differentiated kidney cells are reprogrammed to pluripotency. Song et al. have reprogrammed normal human mesangial cells to pluripotency by retroviral transduction using defined factors (Oct4, Sox2, Klf4, and c-Myc). The kidney-derived iPSCs resemble that of human embryonic stem cell-like colonies in morphology and gene expression. They were considered as alkaline phosphatase-positive; express OCT3/4, TRA-1 to 60, and TRA-1 to 81 proteins; and form embryoid bodies and express markers of all three germ layers [54]. iPSCs were generated from human renal proximal tubular cells as well. Considering their potential risks as oncogenes Klf4 and c-Myc, Montserrat et al. have generated iPSCs with only two transcription factors, Oct4 and Sox2, by using a single tricistronic vector and without any need of other additional chemical compounds [55]. Moreover, generation of iPSCs from exfoliated renal tubular cells that are present in urine is regarded as a simple, non-invasive method for obtaining iPSCs [56,57,58]. Indeed, iPSCs could retain both genetic background and peculiar epigenetic memory of the cells of origin, providing indisputable advantages in cell therapy, kidney regeneration, and other biomedical applications.
Induced pluripotent stem cells are considered as an effective means to alleviate renal tissue damaged in AKI and CKD, and paracrine pathways may be the major mechanism. The administration of iPSC-derived conditioned medium attenuated AKI by downregulating the oxidative stress response in ischemia-reperfusion rats [59]. The recent result from Collino et al. suggested that iPSC-derived extracellular vesicles contributed to the reduction of macrophage infiltration and protected functional mitochondria and regulated several genes associated with oxidative stress, promoting the renoprotection of AKI rats [60]. In the study conducted by Lee et al., the cell transplantation of iPSCs showed improvement of renal function after AKI and dramatically reduced the mortality associated with AKI, exhibiting anti-oxidative stress, anti-inflammatory, and anti-apoptotic effects. It is worth noting that high doses of iPSC accumulation might lead to renal dysfunction. In the same research, Lee et al. have found that injection with 5 × 107 iPSCs affected the smaller arteries of the kidneys with cellular emboli, leading to the reduction of blood reperfusion after ischemia. When carrying out cell therapy for treatment of AKI using iPSCs, the cell dose for transplantation and the monitoring of renal blood perfusion should be taken care of [61]. For the efficacy of iPSC therapy in chronic kidney disease, Caldas et al. have compared the therapeutic effects of iPSCs and bone marrow-derived mesenchymal stromal cells (BM-MSCs) on the progression of CKD in untreated 5/6 nephrectomized rats. It was shown that both treatments improved function and structure of the kidney. Injection of iPSCs decreased the macrophage infiltration, and TGF-β was reduced in the kidneys in the BMSC group, suggesting an anti-inflammatory response that led to decreased fibrosis. In particularly, proteinuria was reduced only in the iPSC group. However, 5/8 (62.5%) rats treated with iPSCs presented tumors confirmed by histology, the cells are positively stained by PCNA, and Wilms’ tumor protein antibody has the characteristics of Wilms’ tumor. These results indicated the effect of iPSCs on retarding progression of CKD and potential risk of Wilms’ tumor development [62].
The pluripotency of PSCs raises concerns on high risk of maldifferentiation of the cells and even tumor formation, when these cells are administered without pre-differentiation. An attractive alternative consists in the derivation of renal progenitors from iPSCs, which could be achieved by the controlled activation of the correct network of nephric transcription factors. The injected iPSC-derived renal progenitor cells robustly engrafted into damaged tubuli and restored renal function and structure in cisplatin mice with AKI [63]. Similarly, iPSC-derived MSC therapy effectively protected the rat kidney from acute ischemia-reperfusion injury [64]. And iPSC-derived MSCs showed comparable effects in the repair of AKI as compared to adult MSCs [65]. In CKD rat models, the administration of iPSC-derived MSCs selectively mobilized into kidney parenchyma and preserved residual renal function via suppression of the inflammatory reaction, inhibiting apoptosis and regulating cell proliferative/death signaling. All the studies above demonstrate that iPSCs are a valuable source of engraftable cells with regenerative activity for kidney disease and create the basis for future applications in stem cell-based therapy [66].
Induced pluripotent stem cells are regarded as the best source for producing new kidney tissues for transplantation, which are potentially derived from patients and used as a renal replacement therapy without immunosuppression. Although it is difficult to regenerate the entire kidney in vitro, recent advances in the field of SCs have enabled in vitro generation of organoids [67,68,69,70,71,72,73].
The kidney organoid formation involves stimulation of a stepwise differentiation process, in which movement occurs from monolayered iPSCs to primitive streak then intermediate mesoderm to kidney lineage cells [40, 69,70,71, 74]. Several protocols for differentiation of PSCs to kidney cell fates have been established to date. In the early days, several studies have reported successful differentiation of human pluripotent stem cells (hPSCs) into either ureteric epithelium [35, 73, 75] or metanephric mesenchyme [35, 72, 76] in vitro. Takasato et al. have put forward a detailed protocol of inducing human induced pluripotent stem cells (hiPSCs) to generate complex multicellular kidney organoid within which segmented nephrons are connected to the collecting ducts and are surrounded by renal interstitial cells and an endothelial network. The kidney organoid reported is transcriptionally similar to that of fetal human kidneys and showed functional maturity, in which the proximal tubules within the organoids displayed megalin-mediated and cubilin-mediated endocytosis, and responded to a nephrotoxicant to undergo apoptosis [70].
Bioengineering represents one possible approach for generating the complex branching structures of the kidney. Scaffolds are temporary physical support obtained from a variety of biomaterials and help to accommodate cells and support their three-dimensional (3D) growth during tissue developmental stage [77]. Scaffolds have become a hope for clinical translation due to their features of biocompatibility, biochemical and biological cues for cell adhesion, proliferation, migration, differentiation, and continued function [78, 79]. Renal scaffolds were successfully produced from porcine, rat, and human kidneys [80,81,82,83,84]. Considering as a potent cell source for scaffold recellularization, embryonic stem cells and adipose tissue-derived stem cells have been seeded into renal scaffolds, achieving cell adherence, proliferation, differentiation, endothelialization, and vascularization [84,85,86,87]. Du et al. have first attempted to repopulate a kidney scaffold using hiPSC-derived endothelial cells (hiPSC-ECs) [88]. Ciampi et al. have repopulated acellular rat kidney scaffolds by using hiPSC-ECs that properly integrate into kidney structures, forming vessel-like structures [89]. It has been reported that human whole kidney scaffold re-endothelialization can be done using hiPSC-ECs and fully perfused with human whole blood [84]. Microfluidic organ-on-a-chip technology has been used to construct an in vitro model of human kidney glomerulus. Musah et al. have reconstituted kidney glomerular capillary function in vitro through microfluidic organ-on-a-chip device. The hiPSC-derived podocytes were cultured on the top of the laminin-coated membrane and primary human glomerular endothelial cells on the opposite side of the same membrane to recapitulate the podocyte-GBM-endothelial interface. The microfluidic glomerulus chip recapitulates some of the normal molecular filtration properties of the functional human kidney glomerular capillary wall and then replicates pharmacologically induced podocyte injury and albuminuria as seen in patients [90, 91].
Although not yet clinically available for transplantation, hiPSC-derived kidney organoids provide dominant models for studying the pathophysiology of kidney diseases, through which we can better understand gene mutations and disease phenotypes of the disease, mimicking disease progression. Preserving the naturally occurring genetic mutations as well as the genetic background of their parental somatic cells, hiPSCs can be applicable to disease/patient-specific modeling. Genetically characterized hiPSCs have been efficiently derived from patients with autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive PKD (ARPKD), Alport syndrome, systemic lupus erythematosus, and Wilms’ tumor and patients undergoing hemodialysis [58, 92,93,94,95,96,97]. In addition, mouse models commonly used in previous studies could not fully recapitulate human genotypes and phenotypes [98, 99]. For example, KAL-1 encodes a basement membrane protein called anosmin-1 that is present on the surface of ureteric bud (UB). The mouse lacking KAL-1 and humans with KAL-1 mutations have renal agenesis, absence of kidneys and ureters [100]. Another case is that the autosomal dominant polycystic kidney disease in humans often inherits heterozygous loss-of-function mutations in either PKD1 (polycystic kidney disease-1) or PKD2 (polycystic kidney disease-2), while the mice displays only very mild cystic disease in the same situation [101, 102]. Species-specific models developed by hiPSCs that carry naturally occurring human mutations are regarded as important complement to mouse models, showing a great promise in studying kidney development and disease process.
Recently, gene editing technologies such as CRISPR/Cas9 make it possible to treat inherited kidney diseases by correcting the pathogenic mutation in an iPSC line. For example, a family with adult-onset autosomal dominant focal segmental glomerulosclerosis (FSGS) was recently found to carry a new germline missense heterozygous mutation (p.G189R) in the octapeptide domain of the transcription factor PAX2 [103]. Trionfini et al. efficiently corrected this point mutation in patient-derived iPSCs by means of CRISPR-Cas9-based homology-directed repair. Forbes et al. corrected the mutations in IFT140 of iPSCs derived from a patient with nephronophthisis-related ciliopathy and induced the isogenic gene-corrected iPSCs differentiated to kidney organoids. Proband organoid tubules demonstrated shortened, club-shaped primary cilia, whereas gene correction rescued this phenotype [104]. Likewise, genetic correction of the single amino acid mutation of iPSCs generated from a congenital nephrotic syndrome patient due to NPHS1 mutations has restored nephrin localization and phosphorylation and slit diaphragm formation [74].
Mesenchymal stem/stromal cells
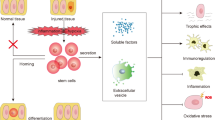
Besides the therapies based on HSCs, MSC-based therapies are currently the most advanced SC-based therapies in terms of clinical testing. Firstly, MSCs can be readily expanded in culture, generating a large number of therapeutic doses. Secondly, MSCs do not express blood group, DR antigens, and costimulatory CD40, CD80, and CD86 proteins to render them hypoimmunogenic, thus facilitating their safe use in allogeneic off-the-shelf protocols [105]. Accumulating pieces of evidence have indicated tremendous treatment potential of MSCs in kidney diseases, including AKI, CKD, diabetic nephropathy (DN), lupus nephritis (LN), polycystic kidney disease (PKD), and renovascular disease (RVD). Although a uniform mechanism governing the MSC-based therapy has not yet been discovered, available data have revealed several working models that promoted their use (Fig. 2).

The diverse mechanism of MSCs in the treatment of kidney diseases
The priority of MSCs to traffic to the sites of ischemia, hypoxia, and inflammatory response following injury plays a crucial role in the success of these as cellular therapy for organ injuries. MSC homing is defined as the arrest of MSCs within the vasculature of a tissue followed by transmigration across the endothelium [106,107,108]. The homing of MSCs to the injured tissues has been driven by a combination of chemokine release from the injured tissue and chemokine receptors that are expressed by MSCs [109]. The stromal derived factor-1 (SDF-1)/chemokine (C-X-C motif) receptor 4 (CXCR4) axis plays a crucial role in the migration of MSCs. Overexpression of CXCR4 gene enhances BM-MSC migration to the kidney area and functional repair after AKI [110]. CD44-hyaluronic acid (CD44-HA) interactions are involved in the process of homing of MSCs to the damaged renal tissue and promote renal function repair following acute tubular injury and chronic renal failure [111]. Due to their vitality in enhancing the kidney-directional migration of transplanted MSCs for increasing the efficiency of tissue repair, some novel preconditioning strategies were explored, including incubation with cytokines or chemical compounds [112,113,114], co-injection [115, 116], hypoxia stimulation [117], and genetic modifications [118, 119]. Also, pulse-focused ultrasound (pFUS) has elicited local molecular responses through mechanotransduction, thus enhancing the renal homing of circulating MSCs [120,121,122].
Treatment with intravenous injection of MSCs has efficiently induced improvement of the repaired kidney both morphologically and functionally. Therapeutic properties of MSCs were originally derived from their engraftment in the injured kidney and subsequent transdifferentiation of MSCs into renal-specific cells to repopulate the kidney. The possible role of MSCs in response to kidney injury was first observed when the female kidney transplants were transferred into male recipients, in which the Y chromosome staining of tubular epithelial cells was found [123, 124]. Several studies have shown that intravenous injection of MSCs homed to the repaired kidney site and localized in the context of tubular epithelial lining and expressed the epithelial markers, indicating engraftment of MSCs [8, 109, 125, 126]. Although MSCs home to the injured sites of kidney via blood circulation after transplantation, the renal MSC engraftment was scarce and transient. Most of injected MSCs remain in other blood-rich organs, such as the lung, liver, and spleen. The numbers of MSC-derived epithelial cells appear to be so low to explain tissue recovery or wound healing (≤ 0.1%), and the hypothesis could not explain the rapid protective effect of injected MSCs which occurred within 24–48 h as well. All these facts suggest that the effects of MSCs may be mediated by another mechanism. The research conducted by Tögel et al. has made a breakthrough in this field. The study reported that differentiation of rat BM-MSCs into a tubular or endothelial cell phenotype was not observed in an ischemic/reperfusion experimental model of AKI. But the expression of proinflammatory cytokines IL-1β, TNF-α, IFN-γ, and inducible nitric oxide synthase showed significant reduction, and that of anti-inflammatory factors IL-10 and bFGF, TGF-α, and Bcl-2 was highly upregulated in the treated kidneys [127]. These studies showed consistent results, in which MSC-conditioned media improved the survival of human proximal tubular cells when exposed to cisplatin in vitro [10, 128, 129]. Now, there is a growing consensus that kidney-protective effects of MSCs are primarily exerted by their paracrine function rather than by direct engraftment.
MSCs secrete a broad repertoire of cytokines, chemokines, and growth factors, including IL-6 and IL-11, vascular endothelial growth factor (VEGF), granulocyte-colony stimulating factor (G-CSF), macrophage-colony stimulating factor (M-CSF), basic fibroblast growth factor (bFGF), monocyte-chemoattractant protein-1 (MCP-1), insulin-like growth factor (IGF), leukemia inhibitory factor (LIF), and hepatocyte growth HGF [10, 130,131,132,133]. These contribute in renal protection by promoting proliferation of epithelial cells and angiogenesis, anti-apoptosis, anti-inflammatory, anti-fibrosis, and other signaling pathways [10, 134,135,136]. Moreover, accumulating evidence also underlined the paracrine actions mediated by extracellular vesicles (EVs), small anuclear membrane-bound particles released from MSCs as a paracrine vehicle to deliver messenger RNA (mRNA), microRNAs, proteins, or bioactive lipids that may reprogram the injured cells or induce secretion of cytoprotective factors [137, 138]. The EVs mainly include exosomes (30–100 nm) that are formed from multivesicular bodies through inward budding of endosome membrane [139] and microvesicles (100–1000 nm) that are released from the cell by outward budding of plasma membrane [140]. Indeed, it has been shown that MSC-derived EVs (MSC-EVs) accelerate renal repair in acute kidney injury (AKI) models. Bruno et al. have demonstrated the protective effects of MSC-EVs in tubular epithelial cells of severe combined immunodeficient (SCID) mice in glycerol-induced AKI. RNase treatment abrogated the protective effects, suggesting an RNA-dependent mechanism and the occurrence of horizontal transfer of human mRNAs in MSC-EVs to target cells [141]. Similar results were obtained by Gatti et al. [142] and Zou et al. [143]. Injections of MSC-EVs reduced the progression of renal injury in experimental models of CKD that was induced by 5/6 nephrectomy [144] and unilateral ureteral obstruction [145, 146]. Eirin and his colleagues have tested the therapeutic effects of EVs in a porcine model with coexistence of RVD and metabolic syndrome. Intrarenal delivery of MSC-EVs attenuated renal inflammation; improved medullary oxygenation and fibrosis; decreased renal vein levels of several pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1-β; and upregulated anti-inflammatory cytokine IL-10 [135]. Moreover, MSC-derived EVs inhibited and reverted fibrosis progression in a diabetic nephropathy mouse model by downregulating fibrosis-related genes [147].
Rather than being a constant mixture of molecular factors, MSCs’ secretome is known to be dependent on the diverse stimuli present in the microenvironment that MSCs encounter. The crosstalk and interplay of MSCs and local environment reversely control and regulate the paracrine activity of MSCs. MSC growth conditions such as oxygen tension, growth factor composition, and mechanical properties may serve to directly influence paracrine activity [148, 149]. Daniëlle et al. cultured human bone marrow and kidney perivascular stromal cells in the TopoWell plate, a custom-fabricated multiwell plate containing 76 unique bioactive surface topographies. Then, they compared the 76 different surface topographies in growth factor and cytokine expression profile; major differences can be observed between secretion levels between topographies of several growth factor and cytokine levels, such as HGF, SDF1α, and trombospondin-I, while others showed a more stable secretion such as VEGF [150]. As the paracrine of MSCs is affected by microenvironment structure, the therapeutic potential of their secretome can be manipulated in an engineered setting. For example, BM-MSCs preconditioned with melatonin express a higher level of catalase and SOD-1 expression and overexpression of basic fibroblast growth factor (bFGF) and HGF. And more surviving grafted MSCs, overstimulation of angiogenesis and proliferation in renal cells, and more rapid renal function recovery were observed in I/R-AKI rats which received the BM-MSCs preconditioned with melatonin [151].
MSCs comprise immunomodulatory and tolerogenic properties. Extensive in vitro and in vivo studies have demonstrated that MSCs are capable of suppressing T cell proliferation, influencing dendritic cell maturation and function, suppressing B cell proliferation and terminal differentiation, and modulating other immune cells such as natural killer (NK) cells and macrophages. Besides, MSCs also induce regulatory T cells (Tregs) and maintain the capability of Tregs to suppress self-reactive T-effector responses [152]. The immunosuppressive capacity of MSCs allows them to act as a potential therapeutic target in renal transplantation as they may inhibit allograft rejection and induce transplant tolerance [153, 154]. On the other hand, immunological properties of MSCs suggest a potential treatment for autoimmune diseases, such as lupus nephritis characterized by autoantibody production and immune complex formation/deposition in glomeruli. A significant reduction in the levels of anti-dsDNA and anti-nuclear autoantibodies, immunoglobulin IgG, IgM, and serum albumin was observed in SLE mice after BM-MSC therapy. And BM-MSC transplantation effectively prevented damage to glomerular morphology/structure and reduced renal complex deposition of both complement component 3 (C3) and IgG [155]. The study conducted by Jang et al. showed that the infusion of BM-MSCs suppressed glomerulonephritis, decreased autoantibody production, reduced proteinuria, and improved overall survival in SLE mice via inhibition of follicular helper cell (Tfh) development and activation of humoral immunity [156]. Dental tissue-derived MSCs are also effective in reducing kidney glomerular lesion and perivascular inflammation infiltration [157]. However, some studies failed to show a positive effect of the treatment with MSCs, and for some aspects, even deleterious results seem to be observed [158].
Despite promising preclinical results obtained in animal models, which indicated effectiveness of MSCs in reducing acute and chronic kidney injuries, clinical trials still remained in the early phases and largely aimed to investigate the safety and efficacy of MSC infusion. To date, more than 40 clinical trials were conducted worldwide, completed or ongoing, involving the use of MSCs in the treatment of kidney diseases as reported by the US National Institute of Health database (ClinicalTrials.gov).
According to the clinical study conducted by Gooch et al., infusion of allogenic MSCs into patients with high risk of AKI after undergoing on-pump cardiac surgery demonstrated no adverse events (AEs) or severe adverse events (SAEs). Moreover, MSC infusion prevented postoperative renal failure (0% versus 20% AKI incidence when compared to case controls) and the length of hospital stay and readmission rates were reduced by 40% [159, 160]. A recent phase 2, randomized, double-blind, placebo-controlled trial conducted by Swaminathan et al. drew inconsistent conclusions to those stated above. No significant difference was observed in the time to recovery of kidney function after treatment of early postoperative AKI with allogeneic MSCs when compared to those treated with placebo [161]. The absence of a significant recovery signal after AKI in patients treated with MSCs might be attributable to several factors. Longer durations of median cardiopulmonary bypass (CPB) time in the MSC group might have masked a beneficial effect. Many patients in this study had impaired kidney function before surgery, and in the setting of already compromised kidney function, beneficial effects of MSCs could be attenuated. What is more, the complexity of postcardiac surgery clinical setting and postoperative course might hinder the ability to observe a modest clinical effect with MSCs or other agents.
Although clinical translation of MSC-based therapeutics for human CKD is still in an early phase, it has demonstrated the potential to modify the progression of CKD. A single-center, randomized, placebo-controlled, phase II/III clinical pilot study investigated the safety and therapeutic efficacy of MSC-derived EVs in CKD patient stage III and IV after following up for a period of 12 months. Although kidney biopsy specimens that are obtained 3 months after the therapy showed no significant histologic changes, EV treatment exhibited a significant improvement in renal function (improved eGFR and decreased serum creatinine, BUN, and albuminuria) [162]. A multicenter, randomized study demonstrated the safety of allogeneic mesenchymal precursor cell (MPC) infusion in diabetic nephropathy. DN patients with eGFR of 20–50 ml/min/1.73 m2 were treated with a single intravenous infusion of two doses of allogenic MPC or placebo. Relative to placebo, a single IV rexlemestrocel-L infusion showed trends of stabilizing or improving eGFR and measured glomerular filtration rate (mGFR) at the prespecified primary endpoint 12 weeks post-infusion, particularly for those with higher baseline eGFR (> 30 ml/min/1.73/m2) and higher serum IL-6 concentration (> 3.5 pg/dl) [163]. In another pilot study, the treatment of autologous adipose tissue-derived MSCs (AT-MSCs) significantly reduced urinary protein excretion of CKD patients, from a median of 0.75 g/day (range, 0.15–9.57) at baseline to 0.54 g/day (range, 0.01v2.66) at month 12. But no improvement in glomerular filtration rate has been observed [164]. Similarly, another 18-month study showed no significant improvements in renal function of CKD patients who received MSC infusion [165]. Saad et al. have reported that intra-arterial infusion of autologous MSCs in patients with RVD showed increased cortical perfusion and renal blood flow (RBF) and reduced renal tissue hypoxia within post-stenotic kidney [166]. The safety and tolerability of a single BM-MSC intravenous infusion in ADPKD patients were observed [167].
The safety and tolerability of MSCs in patients with LN have also been described. Numerous clinical studies have shown that allogeneic MSC transplantation in patients with SLE resulted in the amelioration of disease activity, improvement in serological markers, and stabilization of renal function [168,169,170,171]. A single-center study involving 81 Chinese patients with active and refractory LN reported that 60.5% (49/81) patients have achieved renal remission during 12-month visit by human umbilical cord-derived mesenchymal stem cell (hUC-MSC) transplantation, and 11 of 49 (22.4%) remission patients experienced renal flare at the end of 12 months [172]. Similar findings have been obtained in the research conducted on 3 patients with class IV LN who were treated with allogeneic MSCs. Two patients had early, durable, and substantial complete remissions, while the third patient achieved a partial remission, which permitted reduction of medication doses by 50–90%. Proteinuria levels were improved dramatically during the first month after treatment, and the ameliorations were sustained throughout the 9-month follow-up period [173]. In contrast, a recently published multicenter, randomized, double-blind, placebo-controlled clinical trial in 18 patients with World Health Organization class III or IV LN failed to show any beneficial effect of hUC-MSCs [174].
Given the immune-regulatory properties, MSCs have been administered to kidney transplant recipients, with the aim of controlling the host immune response towards the graft and minimizing immunosuppression, possibly leading to transplant tolerance. A previous study conducted by Perico et al. have reported that 2 recipients of kidney transplant from a living-related donor treated with autologous bone marrow-derived MSC autologous (BM-MSCs) have had transient renal insufficiency (engraftment syndrome) 7–10 days after single infusion [175]. Kidney biopsy revealed infiltration of immune cells with C3 deposits. It is hypothesized that kidney transplantation triggered graft inflammation, causing recruitment of MSCs to the graft and favoring differentiation of MSCs towards a proinflammatory phenotype [176]. Perico et al. have amended the clinical protocol to plan MSC infusion the day before transplantation, and two subsequent patients who received MSCs before transplantation did not experience any engraftment syndrome. Infusion of MSCs has promoted the development of a pro-tolerogenic milieu, which is characterized by an increase in the ratio between Tregs and memory CD8+ T cells in the peripheral blood as well as a profound reduction in ex vivo anti-donor CD8+ T cell cytolytic function. All MSC-treated patients had stable graft function during a 5–7-year follow-up, without increasing the susceptibility to infections or malignancy [177, 178]. Erpicum et al. reported that MSC-treated recipients showed increased frequencies of regulatory T cells on day 30, with no significant change in the B cell frequencies when compared to that of concurrent controls [179]. Clinical researches conducted by Mudrabettu et al. or Reinders et al. were in line with the results of the above research [180, 181]. Notably, administration of MSCs has the potential to reduce the usage of immunosuppressants. Mounting evidence indicates that donor-derived MSCs when combined with low-dose tacrolimus have similar efficacy to that of standard immunosuppression following living-related renal transplantation, allowing 50% reduction of maintenance immunosuppression by calcineurin inhibitors (CNI) [182]. A large randomized clinical trial showed that among patients undergoing renal transplantation, the use of autologous MSCs when compared with anti-IL-2 receptor antibody induction therapy resulted in lowering the incidence of acute rejection, decreasing the risk of opportunistic infection, and better estimated renal function at 1 year follow-up [183] (Table 1).
Renal stem/progenitor cells
It is widely accepted that embryonic renal SCs with nephrogenic potential would deplete soon after (mice) or several weeks before (humans) birth. Interestingly, at least 6000 cells derived from different nephron segments are lost every hour in the urine, suggesting that the progenitor cells are required for continuous replacement of loss of cells during physiological processes [184]. Additional evidence from several studies indicated that adult kidney upon acute or chronic injury has some ability to survive injury and undergo structural remodeling or repair, supporting that it has certain potential to retain regeneration. These have greatly encouraged researchers to search for resident renal stem/progenitor cells (RSPCs).
To our knowledge, there is no single bona fide marker specific for RSPCs at present. Various attempts have been made to identify and characterize RSPCs from differentiated cells in adult kidney, including label-retaining assays, flow cytometry using cell surface markers, and side population assays.
In general, SCs have a slow turnover rate and display minimal physiologic differentiation. When labeled nucleic acids were incorporated, BrdU or 3H-thymidine, SCs retain the label for longer periods of time, while other cells lose [185]. Label-retaining cells (LRCs) are believed to represent the SC compartment. Maeshima and colleagues have first demonstrated the existence of LRCs in renal tubuli of normal rat kidneys. Oliver et al. have identified LRCs in the mouse papilla. These cells form spheres in vitro as well as potentially differentiate towards other lineages [186]. LRCs formed tubule-like structure in 3D culture system and, when LRCs were injected into cultured metanephros, formed nephrons and collecting ducts [187]. The drawback of this technique is that the labels such as BrdU might be released from the dying cells and were taken up from the adjacent dividing cells [188].
By using SC marker expression analyses, the RSPCs were identified in the proximal tubules, in the medullary papilla region, and in the Bowman capsule of human kidneys, which are commonly characterized by the expression of CD133+ and embryonic renal marker PAX2 [30, 189, 190]. Further researches have suggested that CD133+ renal stem cells always co-express CD24 in adult human kidney tissue [191]. The CD24+CD133+ cells had self-renewability and pluripotent differentiation potential, forming a subset of parietal epithelial cells (PECs) in Bowman’s capsule by expressing SC-specific transcription factors Oct-4 and BmI-1 [28]. Numerous studies have demonstrated that CD24+CD133+ cells from adult human kidney represent a multipotent adult resident stem cell population [31, 191,192,193]. It also has been reported that CD24+CD133+cells could be isolated from human urine, which exhibit the same phenotypic and functional features of RSPCs obtained from human kidney tissue and show the potential to differentiate into tubular cells as well as podocytes [194]. Moreover, the RSPCs were further characterized molecularly by expressing the embryonic transcription factor OCT4 and renal developmental genes PAX2, SIX2, and SALL1, and low expression of fully differentiated kidney cell markers [195, 196].
Other selection strategies based on functional assays were utilized to identify and select RSPCs from human renal tissue. With the ability to effectively efflux the Hoechst dye, RSPCs can be detected via flow cytometry and are termed as the side population (SP) cells, which were found in murine kidney interstitial space in several groups [197,198,199,200]. Aldehyde dehydrogenase (ALDH) activity was used to isolate cells with progenitor characteristics from adult human renal cortical tissue. The ALDHhigh cells expressed CD24 and CD133 and displayed typical SC properties such as sphere formation and anchorage-independent growth [29]. Although these cells were identified using a different strategy and were localized in different nephron segments, these all exhibited the capacity to self-renew and the ability to differentiate towards renal tubules or podocytes and different lineages.
Experiments in rat and mouse models of tubular or glomerular damage suggested a therapeutic effect of adult RSPCs of different types and sites of origin. The mechanism of RSPCs involved in kidney repair is associated to integrate into nephrons and is in parallel to a paracrine mechanism based on renoprotective molecules. The activated RSPCs would differentiate along a particular cell lineage and replace the damaged kidney cells. During acute kidney injury, the LRCs in the papilla migrated to the upper papilla and formed a compartment of rapidly proliferating cells, which may play a role in repair after ischemic injury [201]. The eventual descendants of LRCs differentiated into epithelial cells [202]. CD133+ cells are capable of differentiating into epithelial or endothelial cells in in vitro and in vivo [30, 203], and the renal homing and integration of in vitro expanded renal-derived CD133+ cells into some proximal and distal tubules of SCID mice with glycerol-induced acute tubulonecrosis were observed [30, 31, 204]. CD133+CD24+PDX− cells when injected into mice with adriamycin-induced nephropathy glomerular, which are engrafted into the glomerular structures and tubular structures, accounted for 11.08 ± 3.3% of total podocytes and 7.5 ± 1.9% of all proximal tubular cells in mice. The results of intravenous injection of CD133+CD24+PDX− cells reduced proteinuria and improved chronic glomerular damage of SCID mice suffering from nephrotic syndrome [193]. Shen et al. directed the differentiation of hiPSCs into endothelial progenitor cells (iEPCs). As expected, intravenously infused iEPCs were recruited to the injured kidney, replaced injured endothelial cells, and relieve kidney damage in AKI mice. Interestingly, iEPC therapy also ameliorated apoptosis of cardiomyocytes and cardiac dysfunction and no GFP-iEPCs in mouse hearts were observed. It was speculated that the protective effect of iEPC therapy on the heart may have resulted from the reduction of uremic toxin IS and proinflammatory IL-1β in circulation [205]. In addition, the injection of kidney SP cells into the AKI model demonstrated the recovery of renal function [198, 199]. However, some studies did not observe significant renal integration of SP cells and differentiation into renal cells was not observed as expected in some studies [199, 206].
A variety of cytokine/growth factors including IL-15, endothelial growth factor (EGF), and TNF-α that are secreted by RSPCs were detected, supporting a paracrine mechanism, similar to that described for MSCs might also contribute to systemic tissue repair [207, 208]. Bussolati et al. demonstrated that renal CD133+/CD73+ progenitors induce erythropoietin under hypoxia and inhibit prolyl pydroxylase [209]. Aggarwal et al. further provided evidence that the production and release of erythropoietin in in vivo stimulated by CD133+ renal progenitor cells contributed to the protection against fibrosis, limiting the presence of pro-inflammatory in SCID mice with AKI [189]. Recently, it has been reported that RSPCs protect physically injured or chemically damaged renal proximal tubular epithelial cells (RPTECs) by secreting regenerative molecules and microvesicles containing inhibin-A (Inhb-A) and decorin (DCN) [210]. Hishikawa and colleagues have reported that the SP cells isolated from the kidney with acute renal failure expressed high levels of renoprotective factors, such as HGF, VEGF, and leukemia inhibitory factor [198]. Also, the exosomes from conditioned medium of urine-derived stem cells (USCs-Exo) might have the potential to prevent kidney injury from diabetes by inhibiting podocyte apoptosis and promoting vascular regeneration and cell survival [194]. Since these cells could be conveniently obtained through non-invasive methods, urine-derived RSPCs possessed great potential in cell therapy and tissue regeneration. Furthermore, the release of pro-active renal protective factors from distal sites might also be involved, and this is because the intravenously injected RSPCs were localized in extrarenal organs such as the lungs and liver.
Despite the important role played in the repair and regeneration of damaged kidney tissue, a study showed that the dysregulated proliferation of renal progenitor cells generates hyperplastic glomerular lesions, scarring, and nephron loss eventually leading to degenerative disease such as crescentic nephritis or collapsing glomerulopathy [211].
Many researchers believe the presence of renal stem/progenitor cells, but a debate in this field is still going on and some different theories have been put forward. Kusaba et al. have showed that fully differentiated epithelial cells accomplish proximal tubule repair through reversible dedifferentiation and proliferation, without any contribution from the preexisting intratubular SC or progenitor population. In their study, injury to proximal epithelial tubule induced expression of markers of putative epithelial SCs in human kidney (CD24, CD133, and vimentin). What is more, no dilution of fate marker was observed when mice with completely labeled kidneys were subjected to injury and repair, indicating that unlabeled progenitors do not contribute to kidney repair [212]. Berger and colleagues have also suggested that there is no fixed progenitor cell population in the kidney, and tubular cells transiently acquire the phenotype of progenitor cells with reparative characteristics following injury [213].
Challenges in the clinical translation of stem cells
Stem cells have shown great potential in kidney injury repair and disease treatment, but some pressing issues remain to be resolved before the clinical application. ESCs have limited clinical application due to ethical issues regarding their origin, tumorigenicity after transplantation. As the donor cells most likely do not originate from the recipient patient, immunological rejection of the cells differentiated from ESCs upon allogeneic cell transplantation remains a major challenge for cell therapy. Simultaneous administration of immunosuppressive drugs can aid in overcoming these problems but can induce serious side effects [214]. It has been proposed to overcome the immunogenicity of ESC-derived grafts by establishment of ESC donor banks covering various HLAs from selected homozygous HLA-typed volunteers. However, it is a labor-intensive, time-consuming work that is hard to carry out [215]. When it comes to iPSCs, the highly proliferative nature and the use of viral vectors for reprogramming exist with the risk of tumor development following transplantation, although some safer approaches such as the use of small molecules either have less reprogramming efficiency or usually cannot induce pluripotency alone. Reprogramming factors used to induce pluripotency (in particular, the proto-oncogenes c-MYC and KLF4) may become re-activated in the transplanted cells differentiated from iPSCs [216]. Moreover, frustrations due to low replication rate and premature aging were encountered in the administration of iPSCs.
Despite considerable body of evidence in in vitro supports the usefulness of MSCs, controversies about the use of SCs still persist. In clinical trials, the improvement of renal function and structure induced by MSCs in clinical trials is not as obvious as expected. The main reasons for the limited clinical efficacy were the low engraftment, the poor survival rate, and the impaired paracrine capacity of injected cells in vivo. The majority of grafted MSCs might be trapped in the lungs, liver, and spleen. For those cells engrafted into the target tissues, the harsh microenvironment still would induce apoptosis in over 90% of transplanted MSCs in 1 week. To improve the beneficial effects of MSC transplantation, the efficient strategy of administration of MSC needs to be discussed. A recent meta-analysis of MSC administration in kidney disease revealed that cell administration > 1 day after injury yielded greater therapeutic value than within 24 h of injury [217]. Liu et al. proposed that the timing of MSC administration should be immediately after ischemia to 1 h post-I/R. As the lack of inflammation in the early injured kidney was favorable for MSC survival within the tissue, favorable expression of homing adhesion molecules ICAM-1 and VCAM-1 were present, further promoting MSCs to integrate into injured kidney tissue [218]. In the mouse model of graft-versus-host disease, MSCs were most effective when administered at 3, 8, or 20 days after bone marrow transplantation [219]. In contrast, Casiraghi et al. proposed that injection of MSCs before transplantation rather than after it could achieve better immune suppression [176]. Therefore, the optimal timing of MSC administration for different kidney diseases remains under discussion. With more warnings, a long-term examination of intrarenal injection of MSCs in progressive rat model of glomerulonephritis has reported an abnormal and detrimental adipogenic differentiation of MSCs, suggesting the potential harm of administration of MSCs [218]. With regard to RSPCs, which showed the priority in displaying the inherent patient specificity at genomic level and kidney-specific epigenetic changes, the tissues from which autologous RSPCs can be obtained are poorly accessible or insufficient for cell isolation.
The immune rejection of stem cell injection is another challenge that hinders the clinical application of stem cell-based therapy. The allogeneic cells derived from hESCs will be robustly immune rejected by the recipient due to the direct activation of T cells by the foreign MHCs. While the patient-specific iPSCs share the same genetic background, and their derivatives might have a better chance to be immune tolerated, the genomic instability and abnormal epigenetics associated with iPSCs can induce immunogenicity in some cells of their derivatives [220]. MSCs have long been reported to be hypoimmunogenic or “immune privileged.” However, recent studies suggest it is not appropriate to consider MSCs to be immune privileged because they could elicit a humoral and cellular immune response in vivo. For example, whereas culture-expanded MSCs express low levels of MHC class me and are negative for MHC class II, this will likely be activated and/or expressed when MSCs are exposed to IFN-γ or differentiated into mature cell types [221]. Combining MSCs and anti-rejection drugs or a use modified MSC that reduces immunogenicity may be the possible strategies to overcome rejection of all MSCs [222].
Conclusion
Whatever the sources or the approaches utilized such as ESCs, iPSCs, MSCs, or RSPCs, it is widely acceptable that there is an enormous hope underlying the usage of SC therapy as a clinically viable alternative for kidney diseases. Recent advances in cell-based therapy using various cell sources have demonstrated great promise towards restoring normal kidney functions in the AKI, DN, and CKD models, etc. The kidney organoids induced by stem cells provide the authentic and practical models for investigating kidney development and disease and progressing understanding about tissue regeneration, drug screening, and disease modeling. Although the stem cells from different sources show great promise as therapeutic agents for kidney diseases, the amount of research currently available is insufficient to achieve the docking of different kidney diseases with the most suitable transplanted stem cells. But interestingly, because of their immunomodulatory properties, MSCs may be the most promising candidates for stem cell therapy of autoimmune-related kidney diseases. In addition, MSCs obtained from autologous sources in sufficient amounts allow early treatment of patients affected by AKI. There is a need to carry out appropriately designed experimental studies to verify long-term safety of these therapies and to examine the risks of fibrosis, maldifferentiation or malignancy, and AEs of systemic immune suppression. Only the results of larger, well-powered rigorously designed clinical trials will assist in determining the real clinical efficacy of SC therapy in kidney diseases. Additionally, there are more factors that should be taken into consideration in the clinical application of SC therapy, such as appropriate selection of cell types, number of cells required, and appropriate route of administration. These combined efforts favor the translation of experimental results into clinical practice in the near future.
Availability of data and materials
All data generated or analyzed during this study are included in this article.
Abbreviations
- AKI:
-
Acute kidney injury
- AEs:
-
Adverse events
- ADPKD:
-
Autosomal dominant polycystic kidney disease
- AD-MSCs:
-
Adipose-derived mesenchymal stem cells
- ARPKD:
-
Autosomal recessive polycystic kidney disease
- BMPs:
-
Bone morphogenic proteins
- BM-MSCs:
-
Bone marrow-derived mesenchymal stromal cells
- CD44-HA:
-
CD44-hyaluronic acid
- CKD:
-
Chronic kidney disease
- CR:
-
Complete remission
- CXCR4:
-
Chemokine (C-X-C motif) receptor 4
- DN:
-
Diabetic nephropathy
- ESRD:
-
End-stage renal disease
- eGFR:
-
Estimated glomerular filtration rate
- ESCs:
-
Embryonic stem cells
- EVs:
-
Extracellular vesicles
- HGF:
-
Hepatocyte growth factor
- HSCs:
-
Hematopoietic stem cells
- hCB-MSCs:
-
Human cord blood mesenchymal stem cells
- hESCs:
-
Human embryonic stem cells
- hiPSCs:
-
Human induced pluripotent cells
- hPSCs:
-
Human pluripotent stem cells
- hUC-MSCs:
-
Human umbilical cord-derived mesenchymal stem cells
- iPSCs:
-
Induced pluripotent cells
- IM:
-
Intermediate mesoderm
- ISCs:
-
Intestinal stem cells
- LN:
-
Lupus nephritis
- LRCs:
-
Label-retaining cells
- MSCs:
-
Mesenchymal stem cells
- MSC-EVs:
-
Mesenchymal stem cell-derived extracellular vesicles
- MPCs:
-
Mesenchymal precursor cells
- MM:
-
Metanephric mesenchyme
- NSCs:
-
Neural stem cells
- PKD:
-
Polycystic kidney disease
- PSCs:
-
Pluripotent stem cells
- RFI:
-
Renal failure induction
- PR:
-
Partial remission
- RSPCs:
-
Renal stem/progenitor cells
- RVD:
-
Renovascular disease
- SCs:
-
Stem cells
- SAEs:
-
Severe adverse events
- SCID:
-
Severe combined immunodeficient
- TSCs:
-
Totipotent stem cells
- UB:
-
Ureteric bud
- VEGF:
-
Vascular endothelial growth factor
- ZGA or EGA:
-
Activation of zygotic or embryonic genome
References
Jager KJ, Fraser SDS. The ascending rank of chronic kidney disease in the global burden of disease study. Nephrol Dial Transplant. 2017;32:ii121–8.
Levey AS, Coresh J. Chronic kidney disease. Lancet (London, England). 2012;379:165–80.
Couser WG, Remuzzi G, Mendis S, Tonelli M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011;80:1258–70.
Abecassis M, Bartlett ST, Collins AJ, Davis CL, Delmonico FL, Friedewald JJ, Hays R, Howard A, Jones E, Leichtman AB, Merion RM, Metzger RA, Pradel F, Schweitzer EJ, Velez RL, Gaston RS. Kidney transplantation as primary therapy for end-stage renal disease: a National Kidney Foundation/Kidney Disease Outcomes Quality Initiative (NKF/KDOQITM) conference. Clin J Am Soc Nephrol. 2008;3:471–80.
Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, Held PJ, Port FK. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. 1999;341:1725–30.
Ilic D, Ogilvie C. Concise review: human embryonic stem cells-what have we done? What are we doing? where are we going? Stem cells (Dayton, Ohio). 2017;35:17–25.
Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–36.
Herrera MB, Bussolati B, Bruno S, Fonsato V, Romanazzi GM, Camussi G. Mesenchymal stem cells contribute to the renal repair of acute tubular epithelial injury. Int J Mol Med. 2004;14:1035–41.
Ferreira JR, Teixeira GQ, Santos SG, Barbosa MA, Almeida-Porada G, Gonçalves RM. Mesenchymal stromal cell secretome: influencing therapeutic potential by cellular pre-conditioning. Front Immunol. 2018;9:2837.
Imberti B, Morigi M, Tomasoni S, Rota C, Corna D, Longaretti L, Rottoli D, Valsecchi F, Benigni A, Wang J, Abbate M, Zoja C, Remuzzi G. Insulin-like growth factor-1 sustains stem cell mediated renal repair. J Am Soc Nephrol. 2007;18:2921–8.
Kim YK, Nam SA, Yang CW. Applications of kidney organoids derived from human pluripotent stem cells. Korean J Intern Med. 2018;33:649–59.
Ishiuchi T, Torres-Padilla ME. Towards an understanding of the regulatory mechanisms of totipotency. Curr Opin Genet Dev. 2013;23:512–8.
Balakier H, Pedersen RA. Allocation of cells to inner cell mass and trophectoderm lineages in preimplantation mouse embryos. Dev Biol. 1982;90:352–62.
Veiga A, Calderon G, Barri PN, Coroleu B. Pregnancy after the replacement of a frozen-thawed embryo with less than 50% intact blastomeres. Hum Reprod (Oxford, England). 1987;2:321–3.
Van de Velde H, Cauffman G, Tournaye H, Devroey P, Liebaers I. The four blastomeres of a 4-cell stage human embryo are able to develop individually into blastocysts with inner cell mass and trophectoderm. Hum Reprod (Oxford, England). 2008;23:1742–7.
Tachibana M, Amato P, Sparman M, Gutierrez NM, Tippner-Hedges R, Ma H, Kang E, Fulati A, Lee HS, Sritanaudomchai H, Masterson K, Larson J, Eaton D, Sadler-Fredd K, Battaglia D, Lee D, Wu D, Jensen J, Patton P, Gokhale S, Stouffer RL, Wolf D, Mitalipov S. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell. 2013;153:1228–38.
Zhou LQ, Dean J. Reprogramming the genome to totipotency in mouse embryos. Trends Cell Biol. 2015;25:82–91.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science (New York, N.Y.). 1998;282:1145–7.
Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A. 1981;78:7634–8.
Bigas A, Waskow C. Blood stem cells: from beginning to end. Development (Cambridge, England). 2016;143:3429–33.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science (New York, N.Y.). 1999;284:143–7.
Massirer KB, Carromeu C, Griesi-Oliveira K, Muotri AR. Maintenance and differentiation of neural stem cells, Wiley interdisciplinary reviews. Syst Biol Med. 2011;3:107–14.
Stange DE. Intestinal stem cells. Dig Dis (Basel, Switzerland). 2013;31:293–8.
Hu H, Zou C. Mesenchymal stem cells in renal ischemia-reperfusion injury: biological and therapeutic perspectives. Curr Stem Cell Res Ther. 2017;12:183–7.
Oliveira-Sales EB, Boim MA. Mesenchymal stem cells and chronic renal artery stenosis. Am J Physiol Renal Physiol. 2016;310:F6–9.
Jiang Y, Vaessen B, Lenvik T, Blackstad M, Reyes M, Verfaillie CM. Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp Hematol. 2002;30:896–904.
Cui S, Chang PY. Current understanding concerning intestinal stem cells. World J Gastroenterol. 2016;22:7099–110.
Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, Ronconi E, Meini C, Gacci M, Squecco R, Carini M, Gesualdo L, Francini F, Maggi E, Annunziato F, Lasagni L, Serio M, Romagnani S, Romagnani P. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J Am Soc Nephrol. 2006;17:2443–56.
Lindgren D, Bostrom AK, Nilsson K, Hansson J, Sjolund J, Moller C, Jirstrom K, Nilsson E, Landberg G, Axelson H, Johansson ME. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am J Pathol. 2011;178:828–37.
Bussolati B, Bruno S, Grange C, Buttiglieri S, Deregibus MC, Cantino D, Camussi G. Isolation of renal progenitor cells from adult human kidney. Am J Pathol. 2005;166:545–55.
Angelotti ML, Ronconi E, Ballerini L, Peired A, Mazzinghi B, Sagrinati C, Parente E, Gacci M, Carini M, Rotondi M, Fogo AB, Lazzeri E, Lasagni L, Romagnani P. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells (Dayton, Ohio). 2012;30:1714–25.
Gupta S, Verfaillie C, Chmielewski D, Kren S, Eidman K, Connaire J, Heremans Y, Lund T, Blackstad M, Jiang Y, Luttun A, Rosenberg ME. Isolation and characterization of kidney-derived stem cells. J Am Soc Nephrol. 2006;17:3028–40.
Narayanan K, Schumacher KM, Tasnim F, Kandasamy K, Schumacher A, Ni M, Gao S, Gopalan B, Zink D, Ying JY. Human embryonic stem cells differentiate into functional renal proximal tubular-like cells. Kidney Int. 2013;83:593–603.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Takasato M, Er PX, Becroft M, Vanslambrouck JM, Stanley EG, Elefanty AG, Little MH. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat Cell Biol. 2014;16:118–26.
Geng XD, Zheng W, Wu CM, Wang SQ, Hong Q, Cai GY, Chen XM, Wu D. Embryonic stem cells-loaded gelatin microcryogels slow progression of chronic kidney disease. Chin Med J. 2016;129:392–8.
Vazquez-Zapien GJ, Martinez-Cuazitl A, Rangel-Cova LS, Camacho-Ibarra A, Mata-Miranda MM. Biochemical and histological effects of embryonic stem cells in a mouse model of renal failure. Rom J Morphol Embryol. 2019;60:189–94.
Mata-Miranda MM, Bernal-Barquero CE, Martinez-Cuazitl A, Guerrero-Robles CI, Sanchez-Monroy V, Rojas-Lopez M, Vazquez-Zapien GJ. Nephroprotective effect of embryonic stem cells reducing lipid peroxidation in kidney injury induced by cisplatin. Oxidative Med Cell Longev. 2019;2019:5420624.
Garcia GG, Harden P, Chapman J. The global role of kidney transplantation. Curr Opin Nephrol Hypertens. 2012;21:229–34.
Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol. 2015;33:1193–200.
Takasato M, Little MH. A strategy for generating kidney organoids: recapitulating the development in human pluripotent stem cells. Dev Biol. 2016;420:210–20.
Bantounas I, Ranjzad P, Tengku F, Silajdžić E, Forster D, Asselin MC, Lewis P, Lennon R, Plagge A, Wang Q, Woolf AS, Kimber SJ. Generation of functioning nephrons by implanting human pluripotent stem cell-derived kidney progenitors. Stem Cell Rep. 2018;10:766–79.
Tan Z, Rak-Raszewska A, Skovorodkin I, Vainio SJ. Mouse embryonic stem cell-derived ureteric bud progenitors induce nephrogenesis. Cells. 2020;9(2):329.
Klimanskaya I, Chung Y, Becker S, Lu SJ, Lanza R. Human embryonic stem cell lines derived from single blastomeres. Nature. 2006;444:481–5.
Yamamoto M, Cui L, Johkura K, Asanuma K, Okouchi Y, Ogiwara N, Sasaki K. Branching ducts similar to mesonephric ducts or ureteric buds in teratomas originating from mouse embryonic stem cells. Am J Physiol Renal Physiol. 2006;290:F52–60.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
Yang YH, Zhang RZ, Cheng S, Xu B, Tian T, Shi HX, Xiao L, Chen RH. Generation of from human epidermal keratinocytes. Cell Reprogram. 2018;20:356–64.
Spitalieri P, Talarico RV, Botta A, Murdocca M, D'Apice MR, Orlandi A, Giardina E, Santoro M, Brancati F, Novelli G, Sangiuolo F. Generation of human induced pluripotent stem cells from extraembryonic tissues of fetuses affected by monogenic diseases. Cell Reprogram. 2015;17:275–87.
Nishishita N, Takenaka C, Fusaki N, Kawamata S. Generation of human induced pluripotent stem cells from cord blood cells. J Stem Cells. 2011;6:101–8.
Gu H, Huang X, Xu J, Song L, Liu S, Zhang XB, Yuan W, Li Y. Optimizing the method for generation of integration-free induced pluripotent stem cells from human peripheral blood. Stem Cell Res Ther. 2018;9:163.
Aoi T, Yae K, Nakagawa M, Ichisaka T, Okita K, Takahashi K, Chiba T, Yamanaka S. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science (New York, N.Y.). 2008;321:699–702.
Kawano E, Toriumi T, Iguchi S, Suzuki D, Sato S, Honda M. Induction of neural crest cells from human dental pulp-derived induced pluripotent stem cells. Biomed Res (Tokyo, Japan). 2017;38:135–47.
Nagano S, Maeda T, Ichise H, Kashima S, Ohtaka M, Nakanishi M, Kitawaki T, Kadowaki N, Takaori-Kondo A, Masuda K, Kawamoto H. High frequency production of T cell-derived iPSC clones capable of generating potent cytotoxic T cells. Mol Ther Methods Clin Dev. 2020;16:126–35.
Song B, Niclis JC, Alikhan MA, Sakkal S, Sylvain A, Kerr PG, Laslett AL, Bernard CA, Ricardo SD. Generation of induced pluripotent stem cells from human kidney mesangial cells. J Am Soc Nephrol. 2011;22:1213–20.
Montserrat N, Ramirez-Bajo MJ, Xia Y, Sancho-Martinez I, Moya-Rull D, Miquel-Serra L, Yang S, Nivet E, Cortina C, Gonzalez F, Izpisua Belmonte JC, Campistol JM. Generation of induced pluripotent stem cells from human renal proximal tubular cells with only two transcription factors, OCT4 and SOX2. J Biol Chem. 2012;287:24131–8.
Boonkaew B, Thummavichit W, Netsrithong R, Vatanashevanopakorn C, Pattanapanyasat K, Wattanapanitch M. Establishment of an integration-free induced pluripotent stem cell line (MUSIi005-A) from exfoliated renal epithelial cells. Stem Cell Res. 2018;30:34–7.
Zhou T, Benda C, Duzinger S, Huang Y, Li X, Li Y, Guo X, Cao G, Chen S, Hao L, Chan YC, Ng KM, Ho JC, Wieser M, Wu J, Redl H, Tse HF, Grillari J, Grillari-Voglauer R, Pei D, Esteban MA. Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol. 2011;22:1221–8.
Chen W, Huang J, Yu X, Lin X, Dai Y. Generation of induced pluripotent stem cells from renal tubular cells of a patient with Alport syndrome. Int J Nephrol Renov Dis. 2015;8:101–9.
Tarng DC, Tseng WC, Lee PY, Chiou SH, Hsieh SL. Induced pluripotent stem cell-derived conditioned medium attenuates acute kidney injury by downregulating the oxidative stress-related pathway in ischemia-reperfusion rats. Cell Transplant. 2016;25:517–30.
Collino F, Lopes JA, Tapparo M, et al. Extracellular vesicles derived from induced pluripotent stem cells promote Renoprotection in acute kidney injury model. Cells. 2020;9(2):453.
Lee PY, Chien Y, Chiou GY, Lin CH, Chiou CH, Tarng DC. Induced pluripotent stem cells without c-Myc attenuate acute kidney injury via downregulating the signaling of oxidative stress and inflammation in ischemia-reperfusion rats. Cell Transplant. 2012;21:2569–85.
Caldas HC, Lojudice FH, Dias C, Fernandes-Charpiot IMM, Baptista M, Kawasaki-Oyama RS, Sogayar MC, Takiya CM, Abbud-Filho M. Induced pluripotent stem cells reduce progression of experimental chronic kidney disease but develop Wilms' tumors. Stem Cells Int. 2017;2017:7428316.
Imberti B, Tomasoni S, Ciampi O, Pezzotta A, Derosas M, Xinaris C, Rizzo P, Papadimou E, Novelli R, Benigni A, Remuzzi G, Morigi M. Renal progenitors derived from human iPSCs engraft and restore function in a mouse model of acute kidney injury. Sci Rep. 2015;5:8826.
Yuan X, Li D, Chen X, Han C, Xu L, Huang T, Dong Z, Zhang M. Extracellular vesicles from human-induced pluripotent stem cell-derived mesenchymal stromal cells (hiPSC-MSCs) protect against renal ischemia/reperfusion injury via delivering specificity protein (SP1) and transcriptional activating of sphingosine kinase 1 and inhibiting necroptosis. Cell Death Dis. 2017;8:3200.
Huang X, Wang H, Xu Y. Induced pluripotent stem cells (iPSC)-derived mesenchymal stem cells (MSCs) showed comparable effects in repair of acute kidney injury as compared to adult MSCs. Urol J. 2020;17:204–9.
Sheu JJ, Sung PH, Wallace CG, Yang CC, Chen KH, Shao PL, Chu YC, Huang CR, Chen YL, Ko SF, Lee MS, Yip HK. Intravenous administration of iPS-MSC (SPIONs) mobilized into CKD parenchyma and effectively preserved residual renal function in CKD rat. J Cell Mol Med. 2020;24:3593–610.
Becherucci F, Mazzinghi B, Allinovi M, Angelotti ML, Romagnani P. Regenerating the kidney using human pluripotent stem cells and renal progenitors. Expert Opin Biol Ther. 2018;18:795–806.
Taguchi A, Nishinakamura R. Nephron reconstitution from pluripotent stem cells. Kidney Int. 2015;87:894–900.
Takasato M, Er PX, Chiu HS, Little MH. Generation of kidney organoids from human pluripotent stem cells. Nat Protoc. 2016;11:1681–92.
Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, Parton RG, Wolvetang EJ, Roost MS, Lopes SM, Little MH. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2016;536:238.
Takasato M, Little MH. Making a kidney organoid using the directed differentiation of human pluripotent stem cells. Methods Mol Biol (Clifton, N.J.). 2017;1597:195–206.
Lam AQ, Freedman BS, Morizane R, Lerou PH, Valerius MT, Bonventre JV. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J Am Soc Nephrol. 2014;25:1211–25.
Mae SI, Ryosaka M, Toyoda T, Matsuse K, Oshima Y, Tsujimoto H, Okumura S, Shibasaki A, Osafune K. Generation of branching ureteric bud tissues from human pluripotent stem cells. Biochem Biophys Res Commun. 2018;495:954–61.
Tanigawa S, Islam M, Sharmin S, Naganuma H, Yoshimura Y, Haque F, Era T, Nakazato H, Nakanishi K, Sakuma T, Yamamoto T, Kurihara H, Taguchi A, Nishinakamura R. Organoids from nephrotic disease-derived iPSCs identify impaired NEPHRIN localization and slit diaphragm formation in kidney podocytes. Stem Cell Rep. 2018;11:727–40.
Xia Y, Nivet E, Sancho-Martinez I, Gallegos T, Suzuki K, Okamura D, Wu MZ, Dubova I, Esteban CR, Montserrat N, Campistol JM, Izpisua Belmonte JC. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat Cell Biol. 2013;15:1507–15.
Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, Nishinakamura R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell. 2014;14:53–67.
Montserrat N, Garreta E, Izpisua Belmonte JC. Regenerative strategies for kidney engineering. FEBS J. 2016;283:3303–24.
Hussein KH, Park KM, Kang KS, Woo HM. Biocompatibility evaluation of tissue-engineered decellularized scaffolds for biomedical application. Mater Sci Eng. 2016;67:766–78 C, Materials for biological applications.
Rana D, Zreiqat H, Benkirane-Jessel N, Ramakrishna S, Ramalingam M. Development of decellularized scaffolds for stem cell-driven tissue engineering. J Tissue Eng Regen Med. 2017;11:942–65.
Hussein KH, Saleh T, Ahmed E, Kwak HH, Park KM, Yang SR, Kang BJ, Choi KY, Kang KS, Woo HM. Biocompatibility and hemocompatibility of efficiently decellularized whole porcine kidney for tissue engineering. J Biomed Mater Res A. 2018;106:2034–47.
Sullivan DC, Mirmalek-Sani SH, Deegan DB, Baptista PM, Aboushwareb T, Atala A, Yoo JJ. Decellularization methods of porcine kidneys for whole organ engineering using a high-throughput system. Biomaterials. 2012;33:7756–64.
Yu YL, Shao YK, Ding YQ, Lin KZ, Chen B, Zhang HZ, Zhao LN, Wang ZB, Zhang JS, Tang ML, Mei J. Decellularized kidney scaffold-mediated renal regeneration. Biomaterials. 2014;35:6822–8.
Jin M, Yaling Y, Zhibin W, Jianse Z. Decellularization of rat kidneys to produce extracellular matrix scaffolds. Methods Mol Biol (Clifton, N.J.). 2016;1397:53–63.
Leuning DG, Witjas FMR, Maanaoui M, de Graaf AMA, Lievers E, Geuens T, Avramut CM, Wiersma LE, van den Berg CW, Sol W, de Boer H, Wang G, LaPointe VLS, van der Vlag J, van Kooten C, van den Berg BM, Little MH, Engelse MA, Rabelink TJ. Vascular bioengineering of scaffolds derived from human discarded transplant kidneys using human pluripotent stem cell-derived endothelium. Am J Transplant. 2019;19:1328–43.
Xue A, Niu G, Chen Y, Li K, Xiao Z, Luan Y, Sun C, Xie X, Zhang D, Du X, Kong F, Guo Y, Zhang H, Cheng G, Xin Q, Guan Y, Zhao S. Recellularization of well-preserved decellularized kidney scaffold using adipose tissue-derived stem cells. J Biomed Mater Res A. 2018;106:805–14.
Bonandrini B, Figliuzzi M, Papadimou E, Morigi M, Perico N, Casiraghi F, Dipl C, Sangalli F, Conti S, Benigni A, Remuzzi A, Remuzzi G. Recellularization of well-preserved acellular kidney scaffold using embryonic stem cells. Tissue Eng A. 2014;20:1486–98.
Ross EA, Williams MJ, Hamazaki T, Terada N, Clapp WL, Adin C, Ellison GW, Jorgensen M, Batich CD. Embryonic stem cells proliferate and differentiate when seeded into kidney scaffolds. J Am Soc Nephrol. 2009;20:2338–47.
Du C, Narayanan K, Leong MF, Ibrahim MS, Chua YP, Khoo VM, Wan AC. Functional kidney bioengineering with pluripotent stem-cell-derived renal progenitor cells and decellularized kidney scaffolds. Adv Healthc Mater. 2016;5:2080–91.
Ciampi O, Bonandrini B, Derosas M, Conti S, Rizzo P, Benedetti V, Figliuzzi M, Remuzzi A, Benigni A, Remuzzi G, Tomasoni S. Engineering the vasculature of decellularized rat kidney scaffolds using human induced pluripotent stem cell-derived endothelial cells. Sci Rep. 2019;9:8001.
Musah S, Dimitrakakis N, Camacho DM, Church GM, Ingber DE. Directed differentiation of human induced pluripotent stem cells into mature kidney podocytes and establishment of a Glomerulus Chip. Nat Protoc. 2018;13:1662–85.
Musah S, Mammoto A, Ferrante TC, et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary wall function on a chip. Nat Biomed Eng. 2017;1:0069.
Kuebler B, Aran B, Miquel-Serra L, Munoz Y, Ars E, Bullich G, Furlano M, Torra R, Marti M, Veiga A, Raya A. Integration-free induced pluripotent stem cells derived from a patient with autosomal recessive Alport syndrome (ARAS). Stem Cell Res. 2017;25:1–5.
Huang CY, Ho MC, Lee JJ, Hwang DY, Ko HW, Cheng YC, Hsu YH, Lu HE, Chen HC, Hsieh PCH. Generation of induced pluripotent stem cells derived from an autosomal dominant polycystic kidney disease patient with a p.Ser1457fs mutation in PKD1. Stem Cell Res. 2017;24:139–43.
Freedman BS, Lam AQ, Sundsbak JL, Iatrino R, Su X, Koon SJ, Wu M, Daheron L, Harris PC, Zhou J, Bonventre JV. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J Am Soc Nephrol. 2013;24:1571–86.
Son MY, Lee MO, Jeon H, Seol B, Kim JH, Chang JS, Cho YS. Generation and characterization of integration-free induced pluripotent stem cells from patients with autoimmune disease. Exp Mol Med. 2016;48:e232.
Kuebler B, Aran B, Miquel-Serra L, Munoz Y, Ars E, Bullich G, Furlano M, Torra R, Marti M, Veiga A, Raya A. Generation of integration-free induced pluripotent stem cell lines derived from two patients with X-linked Alport syndrome (XLAS). Stem Cell Res. 2017;25:291–5.
Trionfini P, Ciampi O, Romano E, Benigni A, Tomasoni S. Generation of two isogenic knockout PKD2 iPS cell lines, IRFMNi003-A-1 and IRFMNi003-A-2, using CRISPR/Cas9 technology. Stem Cell Res. 2020;42:101667.
Lindstrom NO, Tran T, Guo J, Rutledge E, Parvez RK, Thornton ME, Grubbs B, McMahon JA, McMahon AP. Conserved and divergent molecular and anatomic features of human and mouse nephron patterning. J Am Soc Nephrol. 2018;29:825–40.
Lindstrom NO, Guo J, Kim AD, Tran T, Guo Q, De Sena Brandine G, Ransick A, Parvez RK, Thornton ME, Baskin L, Grubbs B, McMahon JA, Smith AD, McMahon AP. Conserved and divergent features of mesenchymal progenitor cell types within the cortical nephrogenic niche of the human and mouse kidney. J Am Soc Nephrol. 2018;29:806–24.
Duke VM, Winyard PJ, Thorogood P, Soothill P, Bouloux PM, Woolf AS. KAL, a gene mutated in Kallmann’s syndrome, is expressed in the first trimester of human development. Mol Cell Endocrinol. 1995;110:73–9.
Wu G, Tian X, Nishimura S, Markowitz GS, D'Agati V, Park JH, Yao L, Li L, Geng L, Zhao H, Edelmann W, Somlo S. Trans-heterozygous Pkd1 and Pkd2 mutations modify expression of polycystic kidney disease. Hum Mol Genet. 2002;11:1845–54.
Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science (New York, N.Y.). 1996;272:1339–42.
Trionfini P, Ciampi O, Todeschini M, Ascanelli C, Longaretti L, Perico L, Remuzzi G, Benigni A, Tomasoni S. CRISPR-Cas9-mediated correction of the G189R-PAX2 mutation in induced pluripotent stem cells from a patient with focal segmental glomerulosclerosis. CRISPR J. 2019;2:108–20.
Forbes TA, Howden SE, Lawlor K, Phipson B, Maksimovic J, Hale L, Wilson S, Quinlan C, Ho G, Holman K, Bennetts B, Crawford J, Trnka P, Oshlack A, Patel C, Mallett A, Simons C, Little MH. Patient-iPSC-derived kidney organoids show functional validation of a ciliopathic renal phenotype and reveal underlying pathogenetic mechanisms. Am J Hum Genet. 2018;102:816–31.
Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY, Muul L, Hofmann T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci U S A. 2002;99:8932–7.
Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4:206–16.
Sohni A, Verfaillie CM. Mesenchymal stem cells migration homing and tracking. Stem Cells Int. 2013;2013:130763.
De Becker A, Riet IV. Homing and migration of mesenchymal stromal cells: how to improve the efficacy of cell therapy? World J Stem Cells. 2016;8:73–87.
Herrera MB, Bussolati B, Bruno S, Morando L, Mauriello-Romanazzi G, Sanavio F, Stamenkovic I, Biancone L, Camussi G. Exogenous mesenchymal stem cells localize to the kidney by means of CD44 following acute tubular injury. Kidney Int. 2007;72:430–41.
Liu N, Tian J, Cheng J, Zhang J. Migration of CXCR4 gene-modified bone marrow-derived mesenchymal stem cells to the acute injured kidney. J Cell Biochem. 2013;114:2677–89.
Bian XH, Zhou GY, Wang LN, Ma JF, Fan QL, Liu N, Bai Y, Guo W, Wang YQ, Sun GP, He P, Yang X, Su XS, Du F, Zhao GF, Miao JN, Ma L, Zheng LQ, Li DT, Feng JM. The role of CD44-hyaluronic acid interaction in exogenous mesenchymal stem cells homing to rat remnant kidney. Kidney Blood Press Res. 2013;38:11–20.
Masoud MS, Anwar SS, Afzal MZ, Mehmood A, Khan SN, Riazuddin S. Pre-conditioned mesenchymal stem cells ameliorate renal ischemic injury in rats by augmented survival and engraftment. J Transl Med. 2012;10:243.
Saberi K, Pasbakhsh P, Omidi A, Borhani-Haghighi M, Nekoonam S, Omidi N, Ghasemi S, Kashani IR. Melatonin preconditioning of bone marrow-derived mesenchymal stem cells promotes their engraftment and improves renal regeneration in a rat model of chronic kidney disease. J Mol Histol. 2019;50:129–40.
Si X, Liu X, Li J, Wu X. Transforming growth factor-beta1 promotes homing of bone marrow mesenchymal stem cells in renal ischemia-reperfusion injury. Int J Clin Exp Pathol. 2015;8:12368–78.
Liu P, Feng Y, Dong C, Yang D, Li B, Chen X, Zhang Z, Wang Y, Zhou Y, Zhao L. Administration of BMSCs with muscone in rats with gentamicin-induced AKI improves their therapeutic efficacy. PLoS One. 2014;9:e97123.
Liu N, Tian J, Cheng J, Zhang J. Effect of erythropoietin on the migration of bone marrow-derived mesenchymal stem cells to the acute kidney injury microenvironment. Exp Cell Res. 2013;319:2019–27.
Yu X, Lu C, Liu H, Rao S, Cai J, Liu S, Kriegel AJ, Greene AS, Liang M, Ding X. Hypoxic preconditioning with cobalt of bone marrow mesenchymal stem cells improves cell migration and enhances therapy for treatment of ischemic acute kidney injury. PLoS One. 2013;8:e62703.
Liu N, Patzak A, Zhang J. CXCR4-overexpressing bone marrow-derived mesenchymal stem cells improve repair of acute kidney injury. Am J Physiol Renal Physiol. 2013;305:F1064–73.
Wang G, Zhang Q, Zhuo Z, Wu S, Xu Y, Zou L, Gan L, Tan K, Xia H, Liu Z, Gao Y. Enhanced homing of CXCR-4 modified bone marrow-derived mesenchymal stem cells to acute kidney injury tissues by micro-bubble-mediated ultrasound exposure. Ultrasound Med Biol. 2016;42:539–48.
Burks SR, Nagle ME, Bresler MN, Kim SJ, Star RA, Frank JA. Mesenchymal stromal cell potency to treat acute kidney injury increased by ultrasound-activated interferon-gamma/interleukin-10 axis. J Cell Mol Med. 2018;22:6015–25.
Burks SR, Nguyen BA, Tebebi PA, Kim SJ, Bresler MN, Ziadloo A, Street JM, Yuen PS, Star RA, Frank JA. Pulsed focused ultrasound pretreatment improves mesenchymal stromal cell efficacy in preventing and rescuing established acute kidney injury in mice. Stem Cells (Dayton, Ohio). 2015;33:1241–53.
Ziadloo A, Burks SR, Gold EM, Lewis BK, Chaudhry A, Merino MJ, Frenkel V, Frank JA. Enhanced homing permeability and retention of bone marrow stromal cells by noninvasive pulsed focused ultrasound. Stem Cells (Dayton, Ohio). 2012;30:1216–27.
Gupta S, Verfaillie C, Chmielewski D, Kim Y, Rosenberg ME. A role for extrarenal cells in the regeneration following acute renal failure. Kidney Int. 2002;62:1285–90.
Poulsom R, Forbes SJ, Hodivala-Dilke K, Ryan E, Wyles S, Navaratnarasah S, Jeffery R, Hunt T, Alison M, Cook T, Pusey C, Wright NA. Bone marrow contributes to renal parenchymal turnover and regeneration. J Pathol. 2001;195:229–35.
Broekema M, Harmsen MC, Koerts JA, Petersen AH, van Luyn MJ, Navis G, Popa ER. Determinants of tubular bone marrow-derived cell engraftment after renal ischemia/reperfusion in rats. Kidney Int. 2005;68:2572–81.
Kale S, Karihaloo A, Clark PR, Kashgarian M, Krause DS, Cantley LG. Bone marrow stem cells contribute to repair of the ischemically injured renal tubule. J Clin Invest. 2003;112:42–9.
Togel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol. 2005;289:F31–42.
Bi B, Schmitt R, Israilova M, Nishio H, Cantley LG. Stromal cells protect against acute tubular injury via an endocrine effect. J Am Soc Nephrol. 2007;18:2486–96.
Eliopoulos N, Zhao J, Bouchentouf M, Forner K, Birman E, Yuan S, Boivin MN, Martineau D. Human marrow-derived mesenchymal stromal cells decrease cisplatin renotoxicity in vitro and in vivo and enhance survival of mice post-intraperitoneal injection. Am J Physiol Renal Physiol. 2010;299:F1288–98.
Busletta C, Novo E, Valfre Di Bonzo L, Povero D, Paternostro C, Ievolella M, Mareschi K, Ferrero I, Cannito S, Compagnone A, Bandino A, Colombatto S, Fagioli F, Parola M. Dissection of the biphasic nature of hypoxia-induced motogenic action in bone marrow-derived human mesenchymal stem cells. Stem Cells (Dayton, Ohio). 2011;29:952–63.
Togel F, Weiss K, Yang Y, Hu Z, Zhang P, Westenfelder C. Vasculotropic, paracrine actions of infused mesenchymal stem cells are important to the recovery from acute kidney injury. Am J Physiol Renal Physiol. 2007;292:F1626–35.
Liang X, Ding Y, Zhang Y, Tse HF, Lian Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: current status and perspectives. Cell Transplant. 2014;23:1045–59.
Gnecchi M, Danieli P, Malpasso G, Ciuffreda MC. Paracrine mechanisms of mesenchymal stem cells in tissue repair. Methods Mol Biol (Clifton, N.J.). 2016;1416:123–46.
Cetinkaya B, Unek G, Kipmen-Korgun D, Koksoy S, Korgun ET. Effects of Human Placental Amnion Derived Mesenchymal Stem Cells on Proliferation and Apoptosis Mechanisms in Chronic Kidney Disease in the Rat. Int J Stem Cells. 2019;12(1):151-61.
Eirin A, Zhu XY, Puranik AS, Tang H, McGurren KA, van Wijnen AJ, Lerman A, Lerman LO. Mesenchymal stem cell-derived extracellular vesicles attenuate kidney inflammation. Kidney Int. 2017;92:114–24.
Togel F, Zhang P, Hu Z, Westenfelder C. VEGF is a mediator of the renoprotective effects of multipotent marrow stromal cells in acute kidney injury. J Cell Mol Med. 2009;13:2109–14.
Barile L, Vassalli G. Exosomes: therapy delivery tools and biomarkers of diseases. Pharmacol Ther. 2017;174:63–78.
Zhang W, Zhou X, Zhang H, Yao Q, Liu Y, Dong Z. Extracellular vesicles in diagnosis and therapy of kidney diseases. Am J Physiol Renal Physiol. 2016;311:F844–f851.
Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–89.
Burger D, Schock S, Thompson CS, Montezano AC, Hakim AM, Touyz RM. Microparticles: biomarkers and beyond. Clin Sci (London, England : 1979). 2013;124:423–41.
Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, Morando L, Busca A, Falda M, Bussolati B, Tetta C, Camussi G. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20:1053–67.
Gatti S, Bruno S, Deregibus MC, Sordi A, Cantaluppi V, Tetta C, Camussi G. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant. 2011;26:1474–83.
Zou X, Gu D, Xing X, Cheng Z, Gong D, Zhang G, Zhu Y. Human mesenchymal stromal cell-derived extracellular vesicles alleviate renal ischemic reperfusion injury and enhance angiogenesis in rats. Am J Transl Res. 2016;8:4289–99.
He J, Wang Y, Sun S, Yu M, Wang C, Pei X, Zhu B, Wu J, Zhao W. Bone marrow stem cells-derived microvesicles protect against renal injury in the mouse remnant kidney model. Nephrology (Carlton, Vic.). 2012;17:493–500.
He J, Wang Y, Lu X, Zhu B, Pei X, Wu J, Zhao W. Micro-vesicles derived from bone marrow stem cells protect the kidney both in vivo and in vitro by microRNA-dependent repairing. Nephrology (Carlton, Vic.). 2015;20:591–600.
Choi HY, Lee HG, Kim BS, Ahn SH, Jung A, Lee M, Lee JE, Kim HJ, Ha SK, Park HC. Mesenchymal stem cell-derived microparticles ameliorate peritubular capillary rarefaction via inhibition of endothelial-mesenchymal transition and decrease tubulointerstitial fibrosis in unilateral ureteral obstruction. Stem Cell Res Ther. 2015;6:18.
Grange C, Tritta S, Tapparo M, Cedrino M, Tetta C, Camussi G, Brizzi MF. Stem cell-derived extracellular vesicles inhibit and revert fibrosis progression in a mouse model of diabetic nephropathy. Sci Rep. 2019;9:4468.
Kusuma GD, Carthew J, Lim R, Frith JE. Effect of the microenvironment on mesenchymal stem cell paracrine signaling: opportunities to engineer the therapeutic effect. Stem Cells Dev. 2017;26:617–31.
Phinney DG, Pittenger MF. Concise review: msc-derived exosomes for cell-free therapy. Stem Cells (Dayton, Ohio). 2017;35:851–8.
Leuning DG, Beijer NRM, du Fossé NA, Vermeulen S, Lievers E, van Kooten C, Rabelink TJ, Boer J. The cytokine secretion profile of mesenchymal stromal cells is determined by surface structure of the microenvironment. Sci Rep. 2018;8:7716.
Mias C, Trouche E, Seguelas MH, Calcagno F, Dignat-George F, Sabatier F, Piercecchi-Marti MD, Daniel L, Bianchi P, Calise D, Bourin P, Parini A, Cussac D. Ex vivo pretreatment with melatonin improves survival, proangiogenic/mitogenic activity, and efficiency of mesenchymal stem cells injected into ischemic kidney. Stem cells (Dayton, Ohio). 2008;26:1749–57.
Fontaine MJ, Shih H, Schafer R, Pittenger MF. Unraveling the mesenchymal stromal cells’ paracrine immunomodulatory effects. Transfus Med Rev. 2016;30:37–43.
Yu P, Wang Z, Liu Y, Xiao Z, Guo Y, Li M, Zhao M. Marrow mesenchymal stem cells effectively reduce histologic changes in a rat model of chronic renal allograft rejection. Transplant Proc. 2017;49:2194–203.
He Y, Zhou S, Liu H, Shen B, Zhao H, Peng K, Wu X. Indoleamine 2, 3-dioxgenase transfected mesenchymal stem cells induce kidney allograft tolerance by increasing the production and function of regulatory T cells. Transplantation. 2015;99:1829–38.
Sun L, Akiyama K, Zhang H, Yamaza T, Hou Y, Zhao S, Xu T, Le A, Shi S. Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells (Dayton, Ohio). 2009;27:1421–32.
Jang E, Jeong M, Kim S, Jang K, Kang BK, Lee DY, Bae SC, Kim KS, Youn J. Infusion of human bone marrow-derived mesenchymal stem cells alleviates autoimmune nephritis in a lupus model by suppressing follicular helper T-cell development. Cell Transplant. 2016;25:1–15.
Tang X, Li W, Wen X, Zhang Z, Chen W, Yao G, Chen H, Wang D, Shi S, Sun L. Transplantation of dental tissue-derived mesenchymal stem cells ameliorates nephritis in lupus mice. Ann Transl Med. 2019;7:132.
Tani C, Vagnani S, Carli L, Querci F, Kühl AA, Spieckermann S, Cieluch CP, Pacini S, Fazzi R, Mosca M. Treatment with Allogenic Mesenchymal Stromal Cells in a Murine Model of Systemic Lupus Erythematosus. International Journal of Stem Cells. 2017;10(2):160–8.
Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: an update. Cell Transplant. 2016;25:829–48.
Tögel FE, Westenfelder C. Kidney protection and regeneration following acute injury: progress through stem cell therapy. Am J Kidney Dis. 2012;60:1012–22.
Swaminathan M, Stafford-Smith M, Chertow GM, Warnock DG, Paragamian V, Brenner RM, Lellouche F, Fox-Robichaud A, Atta MG, Melby S, Mehta RL, Wald R, Verma S, Mazer CD. Allogeneic mesenchymal stem cells for treatment of AKI after cardiac surgery. J Am Soc Nephrol. 2018;29:260–7.
Nassar W, El-Ansary M, Sabry D, Mostafa MA, Fayad T, Kotb E, Temraz M, Saad AN, Essa W, Adel H. Umbilical cord mesenchymal stem cells derived extracellular vesicles can safely ameliorate the progression of chronic kidney diseases. Biomaterials Res. 2016;20:21.
Packham DK, Fraser IR, Kerr PG, Segal KR. Allogeneic mesenchymal precursor cells (MPC) in diabetic nephropathy: a randomized, placebo-controlled, dose escalation study. EBioMedicine. 2016;12:263–9.
Villanueva S, González F, Lorca E, Tapia A, López VG, Strodthoff R, Fajre F, Carreño JE, Valjalo R, Vergara C, Lecanda M, Bartolucci J, Figueroa FE, Khoury M. Adipose tissue-derived mesenchymal stromal cells for treating chronic kidney disease: a pilot study assessing safety and clinical feasibility. Kidney Res Clin Pract. 2019;38:176–85.
Makhlough A, Shekarchian S, Moghadasali R, Einollahi B, Dastgheib M, Janbabaee G, Hosseini SE, Falah N, Abbasi F, Baharvand H, Aghdami N. Bone marrow-mesenchymal stromal cell infusion in patients with chronic kidney disease: a safety study with 18 months of follow-up. Cytotherapy. 2018;20:660–9.
Saad A, Dietz AB, Herrmann SMS, Hickson LJ, Glockner JF, McKusick MA, Misra S, Bjarnason H, Armstrong AS, Gastineau DA, Lerman LO, Textor SC. Autologous mesenchymal stem cells increase cortical perfusion in renovascular disease. J Am Soc Nephrol. 2017;28:2777–85.
Makhlough A, Shekarchian S, Moghadasali R, Einollahi B, Hosseini SE, Jaroughi N, Bolurieh T, Baharvand H, Aghdami N. Safety and tolerability of autologous bone marrow mesenchymal stromal cells in ADPKD patients. Stem Cell Res Ther. 2017;8:116.
Wang D, Li J, Zhang Y, Zhang M, Chen J, Li X, Hu X, Jiang S, Shi S, Sun L. Umbilical cord mesenchymal stem cell transplantation in active and refractory systemic lupus erythematosus: a multicenter clinical study. Arthritis Res Ther. 2014;16:R79.
Sun L, Wang D, Liang J, Zhang H, Feng X, Wang H, Hua B, Liu B, Ye S, Hu X, Xu W, Zeng X, Hou Y, Gilkeson GS, Silver RM, Lu L, Shi S. Umbilical cord mesenchymal stem cell transplantation in severe and refractory systemic lupus erythematosus. Arthritis Rheum. 2010;62:2467–75.
Wang D, Zhang H, Liang J, Li X, Feng X, Wang H, Hua B, Liu B, Lu L, Gilkeson GS, Silver RM, Chen W, Shi S, Sun L. Allogeneic mesenchymal stem cell transplantation in severe and refractory systemic lupus erythematosus: 4 years of experience. Cell Transplant. 2013;22:2267–77.
Liang J, Zhang H, Hua B, Wang H, Lu L, Shi S, Hou Y, Zeng X, Gilkeson GS, Sun L. Allogenic mesenchymal stem cells transplantation in refractory systemic lupus erythematosus: a pilot clinical study. Ann Rheum Dis. 2010;69:1423–9.
Gu F, Wang D, Zhang H, Feng X, Gilkeson GS, Shi S, Sun L. Allogeneic mesenchymal stem cell transplantation for lupus nephritis patients refractory to conventional therapy. Clin Rheumatol. 2014;33:1611–9.
Barbado J, Tabera S, Sanchez A, Garcia-Sancho J. Therapeutic potential of allogeneic mesenchymal stromal cells transplantation for lupus nephritis. Lupus. 2018;27:2161–5.
Deng D, Zhang P, Guo Y, Lim TO. A randomised double-blind, placebo-controlled trial of allogeneic umbilical cord-derived mesenchymal stem cell for lupus nephritis. Ann Rheum Dis. 2017;76:1436–9.
Perico N, Casiraghi F, Introna M, Gotti E, Todeschini M, Cavinato RA, Capelli C, Rambaldi A, Cassis P, Rizzo P, Cortinovis M, Marasa M, Golay J, Noris M, Remuzzi G. Autologous mesenchymal stromal cells and kidney transplantation: a pilot study of safety and clinical feasibility. Clin J Am Soc Nephrol. 2011;6:412–22.
Casiraghi F, Azzollini N, Todeschini M, Cavinato RA, Cassis P, Solini S, Rota C, Morigi M, Introna M, Maranta R, Perico N, Remuzzi G, Noris M. Localization of mesenchymal stromal cells dictates their immune or proinflammatory effects in kidney transplantation. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg. 2012;12:2373–83.
Perico N, Casiraghi F, Gotti E, Introna M, Todeschini M, Cavinato RA, Capelli C, Rambaldi A, Cassis P, Rizzo P, Cortinovis M, Noris M, Remuzzi G. Mesenchymal stromal cells and kidney transplantation: pretransplant infusion protects from graft dysfunction while fostering immunoregulation. Transplant Int. 2013;26:867–78.
Perico N, Casiraghi F, Todeschini M, Cortinovis M, Gotti E, Portalupi V, Mister M, Gaspari F, Villa A, Fiori S, Introna M, Longhi E, Remuzzi G. Long-term clinical and immunological profile of kidney transplant patients given mesenchymal stromal cell immunotherapy. Front Immunol. 2018;9:1359.
Erpicum P, Weekers L, Detry O, Bonvoisin C, Delbouille MH, Gregoire C, Baudoux E, Briquet A, Lechanteur C, Maggipinto G, Somja J, Pottel H, Baron F, Jouret F, Beguin Y. Infusion of third-party mesenchymal stromal cells after kidney transplantation: a phase I-II, open-label, clinical study. Kidney Int. 2019;95:693–707.
Mudrabettu C, Kumar V, Rakha A, Yadav AK, Ramachandran R, Kanwar DB, Nada R, Minz M, Sakhuja V, Marwaha N, Jha V. Safety and efficacy of autologous mesenchymal stromal cells transplantation in patients undergoing living donor kidney transplantation: a pilot study. Nephrology (Carlton, Vic.). 2015;20:25–33.
Reinders ME, de Fijter JW, Roelofs H, Bajema IM, de Vries DK, Schaapherder AF, Claas FH, van Miert PP, Roelen DL, van Kooten C, Fibbe WE, Rabelink TJ. Autologous bone marrow-derived mesenchymal stromal cells for the treatment of allograft rejection after renal transplantation: results of a phase I study. Stem Cells Transl Med. 2013;2:107–11.
Pan GH, Chen Z, Xu L, Zhu JH, Xiang P, Ma JJ, Peng YW, Li GH, Chen XY, Fang JL, Guo YH, Zhang L, Liu LS. Low-dose tacrolimus combined with donor-derived mesenchymal stem cells after renal transplantation: a prospective, non-randomized study. Oncotarget. 2016;7:12089–101.
Tan J, Wu W, Xu X, Liao L, Zheng F, Messinger S, Sun X, Chen J, Yang S, Cai J, Gao X, Pileggi A, Ricordi C. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA. 2012;307:1169–77.
McMahon AP. Development of the mammalian kidney. Curr Top Dev Biol. 2016;117:31–64.
Rangarajan S, Sunil B, Fan C, Wang PX, Cutter G, Sanders PW, Curtis LM. Distinct populations of label-retaining cells in the adult kidney are defined temporally and exhibit divergent regional distributions. Am J Physiol Renal Physiol. 2014;307:F1274–82.
Oliver JA, Maarouf O, Cheema FH, Martens TP, Al-Awqati Q. The renal papilla is a niche for adult kidney stem cells. J Clin Invest. 2004;114:795–804.
Maeshima A, Sakurai H, Nigam SK. Adult kidney tubular cell population showing phenotypic plasticity, tubulogenic capacity, and integration capability into developing kidney. J Am Soc Nephrol. 2006;17:188–98.
Humphreys BD, Bonventre JV. The contribution of adult stem cells to renal repair. Nephrol Ther. 2007;3:3–10.
Aggarwal S, Grange C, Iampietro C, Camussi G, Bussolati B. Human CD133(+) renal progenitor cells induce erythropoietin production and limit fibrosis after acute tubular injury. Sci Rep. 2016;6:37270.
Grange C, Moggio A, Tapparo M, Porta S, Camussi G, Bussolati B. Protective effect and localization by optical imaging of human renal CD133+ progenitor cells in an acute kidney injury model. Physiol Rep. 2014;2:e12009.
Romagnani P, Remuzzi G. CD133+ renal stem cells always co-express CD24 in adult human kidney tissue. Stem Cell Res. 2014;12:828–9.
Shrestha S, Somji S, Sens DA, Slusser-Nore A, Patel DH, Savage E, Garrett SH. Human renal tubular cells contain CD24/CD133 progenitor cell populations: implications for tubular regeneration after toxicant induced damage using cadmium as a model. Toxicol Appl Pharmacol. 2017;331:116–29.
Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, Lasagni L, Romagnani S, Romagnani P. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol. 2009;20:322–32.
Jiang ZZ, Liu YM, Niu X, Yin JY, Hu B, Guo SC, Fan Y, Wang Y, Wang NS. Exosomes secreted by human urine-derived stem cells could prevent kidney complications from type I diabetes in rats. Stem Cell Res Ther. 2016;7:24.
Becherucci F, Lazzeri E, Lasagni L, Romagnani P. Renal progenitors and childhood: from development to disorders. Pediatr Nephrol (Berlin, Germany). 2014;29:711–9.
Chambers BE, Wingert RA. Renal progenitors: roles in kidney disease and regeneration. World J Stem Cells. 2016;8:367–75.
Hishikawa K, Marumo T, Miura S, Nakanishi A, Matsuzaki Y, Shibata K, Kohike H, Komori T, Hayashi M, Nakaki T, Nakauchi H, Okano H, Fujita T. Leukemia inhibitory factor induces multi-lineage differentiation of adult stem-like cells in kidney via kidney-specific cadherin 16. Biochem Biophys Res Commun. 2005;328:288–91.
Hishikawa K, Marumo T, Miura S, Nakanishi A, Matsuzaki Y, Shibata K, Ichiyanagi T, Kohike H, Komori T, Takahashi I, Takase O, Imai N, Yoshikawa M, Inowa T, Hayashi M, Nakaki T, Nakauchi H, Okano H, Fujita T. Musculin/MyoR is expressed in kidney side population cells and can regulate their function. J Cell Biol. 2005;169:921–8.
Challen GA, Bertoncello I, Deane JA, Ricardo SD, Little MH. Kidney side population reveals multilineage potential and renal functional capacity but also cellular heterogeneity. J Am Soc Nephrol. 2006;17:1896–912.
Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–806.
Oliver JA, Klinakis A, Cheema FH, Friedlander J, Sampogna RV, Martens TP, Liu C, Efstratiadis A, Al-Awqati Q. Proliferation and migration of label-retaining cells of the kidney papilla. J Am Soc Nephrol. 2009;20:2315–27.
Maeshima A, Yamashita S, Nojima Y. Identification of renal progenitor-like tubular cells that participate in the regeneration processes of the kidney. J Am Soc Nephrol. 2003;14:3138–46.
Bruno S, Bussolati B, Grange C, Collino F, di Cantogno LV, Herrera MB, Biancone L, Tetta C, Segoloni G, Camussi G. Isolation and characterization of resident mesenchymal stem cells in human glomeruli. Stem Cells Dev. 2009;18:867–80.
Bussolati B, Collino F, Camussi G. CD133+ cells as a therapeutic target for kidney diseases. Expert Opin Ther Targets. 2012;16:157–65.
Shen WC, Chou YH, Huang HP, Sheen JF, Hung SC, Chen HF. Induced pluripotent stem cell-derived endothelial progenitor cells attenuate ischemic acute kidney injury and cardiac dysfunction. Stem Cell Res Ther. 2018;9:344.
Iwatani H, Ito T, Imai E, Matsuzaki Y, Suzuki A, Yamato M, Okabe M, Hori M. Hematopoietic and nonhematopoietic potentials of Hoechst (low)/side population cells isolated from adult rat kidney. Kidney Int. 2004;65:1604–14.
Ranghino A, Bruno S, Bussolati B, Moggio A, Dimuccio V, Tapparo M, Biancone L, Gontero P, Frea B, Camussi G. The effects of glomerular and tubular renal progenitors and derived extracellular vesicles on recovery from acute kidney injury. Stem Cell Res Ther. 2017;8:24.
Rabb H. Paracrine and differentiation mechanisms underlying stem cell therapy for the damaged kidney. Am J Physiol Renal Physiol. 2005;289:F29–30.
Bussolati B, Lauritano C, Moggio A, Collino F, Mazzone M, Camussi G. Renal CD133(+)/CD73(+) progenitors produce erythropoietin under hypoxia and prolyl hydroxylase inhibition. J Am Soc Nephrol. 2013;24:1234–41.
Sallustio F, Curci C, Aloisi A, Toma CC, Marulli E, Serino G, Cox SN, De Palma G, Stasi A, Divella C, Rinaldi R, Schena FP. Inhibin-A and decorin secreted by human adult renal stem/progenitor cells through the TLR2 engagement induce renal tubular cell regeneration. Sci Rep. 2017;7:8225.
Smeets B, Angelotti ML, Rizzo P, Dijkman H, Lazzeri E, Mooren F, Ballerini L, Parente E, Sagrinati C, Mazzinghi B, Ronconi E, Becherucci F, Benigni A, Steenbergen E, Lasagni L, Remuzzi G, Wetzels J, Romagnani P. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol. 2009;20:2593–603.
Kusaba T, Lalli M, Kramann R, Kobayashi A, Humphreys BD. Differentiated kidney epithelial cells repair injured proximal tubule. Proc Natl Acad Sci U S A. 2014;111:1527–32.
Berger K, Bangen JM, Hammerich L, Liedtke C, Floege J, Smeets B, Moeller MJ. Origin of regenerating tubular cells after acute kidney injury. Proc Natl Acad Sci U S A. 2014;111:1533–8.
Abad M, Mosteiro L, Pantoja C, Cañamero M, Rayon T, Ors I, Graña O, Megías D, Domínguez O, Martínez D, Manzanares M, Ortega S, Serrano M. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502:340–5.
Taylor CJ, Peacock S, Chaudhry AN, Bradley JA, Bolton EM. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell. 2012;11:147–52.
Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, Zhao T, Ye J, Yang W, Liu K, Ge J, Xu J, Zhang Q, Zhao Y, Deng H. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science (New York, N.Y.). 2013;341:651–4.
Wang Y, He J, Pei X, Zhao W. Systematic review and meta-analysis of mesenchymal stem/stromal cells therapy for impaired renal function in small animal models. Nephrology (Carlton, Vic.). 2013;18:201–8.
Liu X, Cai J, Jiao X, Yu X, Ding X. Therapeutic potential of mesenchymal stem cells in acute kidney injury is affected by administration timing. Acta Biochim Biophys Sin. 2017;49:338–48.
Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392–402.
Jin X, Lin T, Xu Y. Stem cell therapy and immunological rejection in animal models. Curr Mol Pharmacol. 2016;9:284–8.
Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringdén O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890–6.
Ferreri AJ, Illerhaus G, Zucca E, Cavalli F. Flows and flaws in primary central nervous system lymphoma. Nat Rev Clin Oncol. 2010;7 https://doi.org/10.1038/nrclinonc.2010.1039-c1031; author reply https://doi.org/1010:1038/nrclinonc.2010.1039-c1032.
Acknowledgements
We thank the Research Institute of Nephrology personnel for their guidance and help.
Funding
This study is supported by grants from the Science and Technology Huimin Project of Henan Province (no. 162207310001), Pathogenesis & Precision diagnosis and treatment of Chronic Kidney Disease (no. 182101510002), and National Science and Technology Major Project of China (No. 2020ZX09201-009).
Author information
Authors and Affiliations
Contributions
LSZ and LDW conceived the idea. CF and PSL prepared the figures and tables. LDW and CF drafted the manuscript. LSZ and LDW edited and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, D., Cheng, F., Pan, S. et al. Stem cells: a potential treatment option for kidney diseases. Stem Cell Res Ther 11, 249 (2020). https://doi.org/10.1186/s13287-020-01751-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-020-01751-2