
Abstract
Background
Jervell and Lange-Nielsen syndrome (Online Mendelian Inheritance in Man 220400) is a rare autosomal recessive cardioauditory ion channel disorder that affects 1/200,000 to 1/1,000,000 children. It is characterized by congenital profound bilateral sensorineural hearing loss, a long QT interval, ventricular tachyarrhythmias, and episodes of torsade de pointes on an electrocardiogram. Cardiac symptoms arise mostly in early childhood and consist of syncopal episodes during periods of stress, exercise, or fright and are associated with a high risk of sudden cardiac death. Jervell and Lange-Nielsen syndrome is caused by homozygous or compound heterozygous mutations in KCNQ1 on 11p15.5 or KCNE1 on 1q22.1-q22.2.
Case presentation
We report the case of a 10-year-old Moroccan boy with congenital hearing loss and severely prolonged QT interval who presented with multiple episodes of syncope. His parents are first-degree cousins. We performed Sanger sequencing and identified a homozygous variant in KCNQ1 (c.1343dupC, p.Glu449Argfs*14).
Conclusions
The identification of the genetic substrate in this patient confirmed the clinical diagnosis of Jervell and Lange-Nielsen syndrome and allowed us to provide him with appropriate management and genetic counseling to his family. In addition, this finding contributes to our understanding of genetic disease in the Moroccan population.
Similar content being viewed by others

Background
Jervell and Lange-Nielsen syndrome (JLNS), Mendelian Inheritance in Man (MIM) 220400, is a rare autosomal recessive cardioauditory ion channel disorder that affects 1/200,000 to 1/1,000,000 children [1, 2]. It is characterized by congenital profound bilateral sensorineural hearing loss (SNHL), a long QT interval usually greater than 500 ms, ventricular tachyarrhythmias, and episodes of torsade de pointes on an electrocardiogram (ECG) [2, 3]. Cardiac symptoms mostly arise in early childhood and consist of syncopal episodes during periods of stress, exercise, or fright with a high risk of sudden cardiac death [2]. Homozygous or compound heterozygous loss-of-function mutations in KCNQ1 on 11p15.5 are responsible for 90% of cases of JLNS [4, 5]. Biallelic mutations in KCNE1 on 1q22.1-q22.2 have been identified as an additional cause of JLNS, establishing the genetic heterogeneity of the disease [6]. The two genes encode respectively the α subunits and β subunits of the voltage-gated potassium channel that in the heart conducts the slow delayed rectifying potassium ion (K+) current during cardiomyocyte repolarization, while in the ear it is involved in potassium-rich endolymph production of inner ear hair cells [5, 7]. Here we report the clinical and molecular analysis of a Moroccan family affected by JLNS.
Case presentation
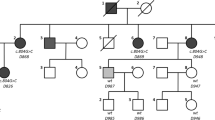
A 10-year-old Moroccan boy was referred by his cardiologist to our medical genetics department (Institut National d’Hygiène, Rabat) for genetic evaluation. He is the firstborn of a healthy consanguineous couple (first-cousins; Fig. 1), both originating from the Northwest of Morocco. There was no family history of sudden death, deafness, syncope, epilepsy, or any other genetic disease. The pregnancy had been medically followed, and no complications were reported. His mother presented with no history of drug ingestion or phytotherapy. His birth weight and length were within normal range and no dysmorphic signs were recorded. At 6 months, he was diagnosed as having severe bilateral SNHL on auditory evoked potential measurement. His first syncopal episode occurred at 24 months of age. His ECG revealed a markedly prolonged QTc interval of 530 ms (corrected by Bazett’s formula) and T-wave alternans on V1 to V4 (Fig. 2). Echocardiography showed a structurally normal heart. Treatment was immediately started with a β-adrenergic blocker. His parents and his two younger brothers, who were 7-years old and 1-year old, were clinically normal. Blood samples from all his family’s members were collected after we were given written informed consent. Deoxyribonucleic acid (DNA) was isolated using standard techniques [8]. Molecular genetic testing of the entire coding region and flanking intronic regions of KCNQ1 and KCNE1 was undertaken by Sanger sequence analysis (details available on request). This led to the identification of a homozygous frameshift mutation c.1343dupC (p.Glu449Argfs*14) in the index patient. Both parents and one sibling (IV-2) were heterozygous for this mutation. The youngest child of the family did not carry the frameshift mutation (IV-3, Fig. 3). This variant was previously reported in a heterozygous state in an individual with long QT syndrome (LQTS) [9].

Pedigree of the studied family. The affected individual is shaded and indicated by an arrow. Family members that were tested for the mutation are marked by an asterisk

Electrocardiogram of the patient

Electropherograms of the identified c.1343dupC; p.Glu449Argfs*14 mutation. The proband IV:1 presented with the homozygous c.1343dupC; p.Glu449Argfs*14 mutation and both parents (III:5 and III:6) and unaffected brother IV:2 are heterozygotes. One healthy brother (IV:3) was homozygous for the wild-type allele. The X indicates the position of detected mutation (Duplication of C base). The arrows indicate the location of the mutated base
Discussion
The primary electric disorders, which among others include LQTS, short-QT syndrome (SQTS), Brugada syndrome (BrS), and catecholaminergic polymorphic ventricular tachycardia (CPVT), are often characterized by specific ECG abnormalities either at baseline or during particular conditions, such as exercise (for example, CPVT and LQTS), fever (for example, BrS), or pharmacological challenge (for example, BrS). The list of familial arrhythmia syndromes has been in recent years expanded by the recognition of two other disorders, namely early repolarization syndrome and idiopathic ventricular fibrillation (VF) [7].
LQTS is a clinically and genetically heterogeneous disorder. Syncopal episodes may occur from infancy through middle age, with risk of sudden death [10]. The autosomal dominant mode of inheritance is typical for all LQTS forms (previously described as the Romano–Ward syndrome). Three genes account for approximately 90% of patients with genotype-positive LQTS. LQT1 is characterized by broad-based T waves and cardiac events during exercise or emotion. It arises from loss-of-function mutations in KCNQ1. In LQT2, T waves are bifid and cardiac events predominantly occur during exercise or emotion. It arises from loss-of-function mutations in KCNH2 (also known as hERG). Gain-of-function mutations in SCN5A are associated to LQT3. These patients show a long ST segment, short T waves, and experience cardiac events predominantly during rest or sleep. Twelve additional genes encoding either ion channel subunits (KCNJ5, KCNE1, KCNE2, and SCN4B) or proteins that regulate ion channel function (AKAP9, CAV3, ANKB, SNT1, CALM1, and CALM2) have been associated with LQTS; however, most of them are only rarely implicated (<1%) [7].
Jervell and Lange-Nielsen syndrome is a rare clinical variant of LQTS that manifests with extracardiac phenotypes and is inherited in an autosomal recessive fashion. Patients with JLNS present with severe prolongation of the QT interval and congenital SNHL. It is one of the most severe forms of LQTS. By the age of 3 years, 50% of patients have had an event and by the age of 18 years, 90% of patients with JLNS have developed symptoms [2]. Therefore, the real incidence of JLNS is probably underestimated because of its high mortality in early infancy, in particular in populations with a high rate of consanguineous marriages like Morocco [11]. In addition, even in the presence of medical therapy the occurrence of sudden cardiac death in JLNS exceeds 25% [2].
To date, approximately 21 distinct KCNQ1 mutations have been characterized in patients with JLNS according to The Human Gene Mutation Database (HGMD) at the Institute of Medical Genetics in Cardiff (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php). Three different mutations in the KCNE1 gene were also reported in patients with JLNS. These mutations have been found homozygously or in a compound heterozygous state (Fig. 4) and, interestingly, one of the families studied in the initial description of KCNE1 in JLNS originated from Morocco [4].

Schematic view of KCNQ1 gene structure and localization of identified KCNQ1 mutations in Jervell and Lange-Nielsen syndrome. The mutations previously reported are indicated with a green frame and the mutation identified in this study is indicated with a red frame
In this study, we reported the molecular characterization of a KCNQ1 homozygous frameshift mutation c.1343dupC (p.Glu449Argfs*14) in a Moroccan patient with JLNS.
This variant was previously identified in a heterozygous state in a 25-month-old girl of Latino origin with severe bilateral SNHL due to a homozygous mutation of connexin 26. She was repeatedly found to have QTc intervals ≥450 ms in a screening program. Sequencing of 12 LQTS genes identified a de novo heterozygous frameshift mutation described in this report (KCNQ1, c.1343dupC; p.Glu449Argfs*14) [9]. To the best of our knowledge, the case of the Moroccan proband reported here is the first case of JLNS carrying this mutation in a homozygous state.
Conclusions
We report here the clinical and molecular description of a Moroccan patient with JLNS. This diagnosis allowed us to provide an appropriate course of management to the patient and to identify and counsel asymptomatic heterozygous carriers.
References
Winbo A, Stattin EL, Diamant UB, Persson J, Jensen SM, Rydberg A. Prevalence, mutation spectrum, and cardiac phenotype of the Jervell and Lange-Nielsen syndrome in Sweden. Europace. 2012;14(12):1799–806.
Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, Shkolnikova M, Berul CI, Bitner-Glindzicz M, Toivonen L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113(6):783–90.
Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54(1):59–68.
Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15(2):186–9.
Tyson J, Tranebjaerg L, McEntagart M, Larsen LA, Christiansen M, Whiteford ML, Bathen J, Aslaksen B, Sorland SJ, Lund O, et al. Mutational spectrum in the cardioauditory syndrome of Jervell and Lange-Nielsen. Hum Genet. 2000;107(5):499–503.
Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med. 1997;336(22):1562–7.
Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res. 2015;116(12):1919–36.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Chang RK, Lan YT, Silka MJ, Morrow H, Kwong A, Smith-Lang J, Wallerstein R, Lin HJ. Genetic variants for long QT syndrome among infants and children from a statewide newborn hearing screening program cohort. J Pediatr. 2014;164(3):590–5. e591–3.
Behere SP, Weindling SN. Inherited arrhythmias: The cardiac channelopathies. Ann Pediatr Cardiol. 2015;8(3):210–20.
Jaouad IC, Elalaoui SC, Sbiti A, Elkerh F, Belmahi L, Sefiani A. Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders. J Biosoc Sci. 2009;41(5):575–81.
Acknowledgements
We thank the patient and his family.
Funding
Not applicable.
Availability of data and materials
Available on request.
Authors’ contributions
NA carried out the molecular genetic studies, and drafted the manuscript. NL participated in the clinical diagnosis and helped to draft the manuscript, RB performed clinical examinations and monitoring of the family, IF participated in the clinical diagnosis, RA participated in the clinical diagnosis, MA participated in the sequence alignment, AS participated in the design of the study, CB participated in the design of the study and helped to draft the manuscript, and IR conceived and coordinated the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian(s) for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal. A consent for publication was obtained from the family. Statements are available on request.
Ethics approval and consent to participate
Ethics approval and consent to participate was provided by all adults and legal guardians of minor individuals involved in this study. Statements of their signed consent are available on request.
This study was approved by the ethics committee of the National Institute of Health in Rabat, Morocco.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Adadi, N., Lahrouchi, N., Bouhouch, R. et al. Clinical and molecular findings in a Moroccan family with Jervell and Lange-Nielsen syndrome: a case report. J Med Case Reports 11, 88 (2017). https://doi.org/10.1186/s13256-017-1243-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-017-1243-1