
Abstract
Background
Previous reports of chromosomal aberrations in different forms of congenital diaphragmatic hernia have been described as comprising aneuploidies (for example, trisomy 21), microdeletions, and duplications (for example, monosomy 15q24, 22q11.2).
Case presentation
We describe the first association of a de novo partial tetrasomy 4q35.2 in a father with left-sided, isolated renal agenesis and left-sided, isolated congenital diaphragmatic hernia in his son, who inherited the chromosomal aberration from his father.
Conclusions
Given that the aberration occurred de novo in the father and was transmitted to his son, with both presenting with unilateral left-sided developmental field defects, we suggest a gene dosage effect of the tetrasomic region to be involved in the phenotype of our two patients. Furthermore, we suggest performing a genetic workup in multiplex families with congenital malformations.
Similar content being viewed by others


Background
Congenital diaphragmatic hernia (CDH) represents a spectrum of rare developmental defects resulting from an aberrant embryonic morphogenesis of the developing diaphragm. The diaphragmatic defect allows an upward displacement of the abdominal organs into the thorax with a consecutive compression of the intrathoracic compartment, leading to hypoplasia of the lung, secondary pulmonary hypertension, and intestinal malrotation [1]. CDH occurs with a birth prevalence of 1 in 3000–4000 [2]. Long-term complications are gastroesophageal reflux disease, failure to thrive, neurocognitive defects, and hearing loss [3, 4]. In approximately 50–60 % of patients, CDH occurs as isolated malformation [2, 5]; about 40 % present with additional anomalies of other organ systems, including the central nervous system (5–75 %), the cardiovascular system (4–63 %), the genitourinary system (5–27 %), and the gastrointestinal tract (1–20 %) [6]. Among the associated genitourinary anomalies, renal anomalies are common, comprising renal agenesis, dysplasia, hypoplasia, or hydronephrosis [7]. In the past, conventional karyotyping identified chromosomal anomalies in about 6.3 % of cases [2]; in recent studies, researchers used microarray analysis and identified smaller genomic loci, with and without candidate genes [1, 8]. However, the etiology of CDH is still unknown in more than 70 % of patients [2]. In this report, we present the first case of a de novo partial tetrasomy 4q35.2 in a father with isolated unilateral renal agenesis and isolated CDH in his son, respectively.
Case presentation
The index patient is the first child of his nonconsanguineous parents. Due to reduced fertility of the father, assisted reproduction in the form of intracytoplasmic sperm injection had been performed to achieve pregnancy in the couple. Besides his reduced fertility, the father was born with unilateral agenesis of the left kidney and a poorly formed hypoplastic helix of his left ear. He was a university graduate and was working in a management position.
The achieved pregnancy was uneventful until the second trimester, when prenatal ultrasound of the fetus revealed isolated left-sided CDH. The mother gave birth to a boy at 27 + 2 weeks of gestation by cesarean section due to preterm rupture of the membranes and contractions despite intravenous tocolysis. The boy’s birth weight was 950 g (25th percentile), length was 36.6 cm (50th percentile), and head circumference was 25 cm (30th percentile). His Apgar scores were 6 at 1 minute, 8 at 5 minutes, and 8 at 10 minutes. He was intubated immediately after delivery and ventilated. A chest X-ray confirmed left-sided CDH (Fig. 1a). After his circulation and respiration stabilized, the boy underwent surgical correction of the CDH on the 11th day of life (Fig. 1b). He was successfully weaned off mechanical ventilation on the 26th day of life, and oxygen supplementation could be seized on day 61 of life. The neonatal period was complicated by an intraventricular hemorrhage with consecutive dilation of the ventricles, which was eventually treated by implantation of a Rickham reservoir on the 28th day of life. A ventriculoperitoneal shunt was implemented 4 months after birth. By the time of hospital discharge, the boy presented with general developmental delay. However, because he was born prematurely and had periods of severe hypoxia due to oxygenation difficulties due to his CDH condition, and because his neonatal period was further complicated by an intraventricular hemorrhage, it was impossible to distinguish whether his developmental delay was primary or secondary.

Preoperative (a) and postoperative (b) x-rays of the index patient
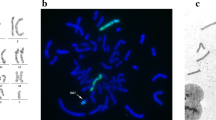
During the pregnancy of the index patient, amniocentesis and conventional karyotyping based on GTG staining at 450 bands per haploid genome level at 16 + 5 weeks of gestation was suggestive of partial tetrasomy 4q35.2 in the male fetus (Fig. 2a). For further characterization, fluorescence in situ hybridization (FISH) analysis was performed, which revealed an increased signal at 4qter only. Multiple ligation-dependent probe amplification (genomic region chr4:185,963,614–189,037,586, kits P036-E1, P070-B1, and P264-A1; MRC-Holland, Amsterdam, the Netherlands) of the specific region confirmed a triplication of chromosomal region 4q35.2 spanning approximately 3.73–4.43 Mb ([46,XY,qdp{4}{q35.2qter}]) in one of the two chromosomes 4 of the fetus. To test whether the chromosomal aberration occurred de novo, and because the father presented with reduced fertility and unilateral renal and helix anomaly, consecutive karyotyping of the parents was performed, which revealed the same aberration in the father (Fig. 2b). Cytogenetic analysis of the mother was unremarkable, as was the FISH analysis of the subtelomeric regions of both her chromosomes 4.

GTG banding of amniotic fluid cells in the fetus (a) and of peripheral blood lymphocytes from the father (b) revealed a triplication of chromosomal region 4q35.2 spanning approximately 3.73–4.43 Mb ([46,XY,qdp{4}{q35.2qter}]) (see arrows)
Genetic counseling of the parents during the hospital course of their son prompted them to ask the paternal grandparents of the index patient to have their karyotypes analyzed to see if the triplication of chromosomal region 4q35.2 had occurred de novo in the father. Conventional karyotyping of the paternal grandparents showed normal results, confirming that the partial tetrasomy of chromosomal region 4q35.2 in the father had occurred de novo.
Discussion
In this report, we present the first case of partial tetrasomy 4q35.2 [(46,XY,qdp{4}{q35.2qter})] and associated CDH, unilateral renal agenesis, and ear anomaly in the carriers. Previous reports of chromosomal aberrations in different forms of CDH described aneuploidies (for example, trisomy 21), microdeletions and duplications (for example, monosomy 15q24, 22q11.2) [6, 8, 9]. Approximately 10 % of patients with CDH have it in the context of monogenic syndromes such as Fryns syndrome [6]. While CDH usually occurs sporadically, reports of familial cases are suggestive of an underlying Mendelian mode of inheritance in some of the families [10, 11]. In accordance with this, patients with CDH and single-gene mutations in FOG2 [12], GATA4 [13], and FREM1 [14] have been reported.
Regarding the aberrant region on 4q35.2 that we describe in this report, we conducted a medical database search using PubMed (www.ncbi.nlm.nih.gov/pubmed) and retrieved several reports of patients with overlapping deletions. Some of these patients were described to have birth defects [15, 16]. However, we retrieved far fewer reports of overlapping duplications. Among these, Lin et al. [17] described a patient with congenital bilateral choanal atresia and several other phenotypic anomalies. Interestingly, we retrieved 94 entries with overlapping duplications of chromosomal region 4q35.2 from the DECIPHER database v9.0 in September 2015 (Database of Chromosomal Imbalance and Phenotype in Humans; https://decipher.sanger.ac.uk/). One of these was a female patient [accession number 4647] presenting with, among other anomalies, unilateral renal agenesis, low-set ears, and ventricular septal defect. Furthermore, a male patient [accession number 278863] presented with, among other anomalies, CDH and posteriorly rotated ears. The phenotypic variation between the father and the son described in the present report might be due to different genetic mutations in the genes downstream of the triplicated genes or that other genes not involved in the triplication may compensate for or inhibit the pathogenesis of the triplication to different degrees [18]. Hence, triplication of the region may have induced the CDH and renal agenesis indirectly by promoting or silencing genes lying downstream. It is also possible that a direct dosage effect of the comprised genes (for example, FAT1) lead to CDH and renal agenesis. FAT1 (NM_005245) encodes a tumor suppressor essential for controlling cell proliferation during Drosophila development. According to the Mouse Genome Informatics database (http://www.informatics.jax.org/), homozygous, targeted null mutations exhibit holoprosencephaly, anophthalmia, kidney defects, and perinatal lethality.
Conclusions
Overall, our observations suggest that duplication or triplication of chromosomal region 4q35.2 carries susceptibility for the development of renal, ear, and diaphragmatic anomalies. Furthermore, we suggest performing a genetic workup of multiplex families with congenital malformations.
Consent
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- CDH:
-
congenital diaphragmatic hernia
- FISH:
-
fluorescence in situ hybridization
References
Slavotinek AM. The genetics of common disorders – congenital diaphragmatic hernia. Eur J Med Genet. 2014;57:418–23.
Pober BR. Genetic aspects of human congenital diaphragmatic hernia. Clin Genet. 2008;74:1–15.
Terui K, Taguchi T, Goishi K, Hayakawa M, Tazuke Y, Yokoi A, et al. Prognostic factors of gastroesophageal reflux disease in congenital diaphragmatic hernia: a multicenter study. Pediatr Surg Int. 2014;30:1129–34.
Peetsold MG, Heij HA, Kneepkens CMF, Nagelkerke AF, Huisman J, Gemke RJBJ. The long-term follow-up of patients with a congenital diaphragmatic hernia: a broad spectrum of morbidity. Pediatr Surg Int. 2009;25:1–17.
Brady PD, Srisupundit K, Devriendt K, Fryns J, Deprest JA, Vermeesch JR. Recent developments in the genetic factors underlying congenital diaphragmatic hernia. Fetal Diagn Ther. 2011;29:25–39.
Enns GM, Cox VA, Goldstein RB, Gibbs DL, Harrison MR, Golabi M. Congenital diaphragmatic defects and associated syndromes, malformations, and chromosome anomalies: a retrospective study of 60 patients and literature review. Am J Med Genet. 1998;79:215–25.
Siebert JR, Benjamin DR, Juul S, Glick PL. Urinary tract anomalies associated with congenital diaphragmatic defects. Am J Med Genet. 1990;37:1–5.
Yu L, Wynn J, Ma L, Guha S, Mychaliska GB, Crombleholme TM, et al. De novo copy number variants are associated with congenital diaphragmatic hernia. J Med Genet. 2012;49:650–9.
Stark Z, Behrsin J, Burgess T, Ritchie A, Yeung A, Tan TY, et al. SNP microarray abnormalities in a cohort of 28 infants with congenital diaphragmatic hernia. Am J Med Genet A. 2015;167A:2319–26.
Mitchell SJ, Cole T, Redford DH. Congenital diaphragmatic hernia with probable autosomal recessive inheritance in an extended consanguineous Pakistani pedigree. J Med Genet. 1997;34:601–3.
Farag TI, Bastaki L, Marafie M, al-Awadi SA, Krsz J. Autosomal recessive congenital diaphragmatic defects in the Arabs. Am J Med Genet. 1994;50:300–1.
Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, et al. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. 2005;1:58–65.
Yu L, Wynn J, Cheung YH, Shen Y, Mychaliska GB, Crombleholme TM, et al. Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic hernia. Hum Genet. 2013;132:285–92.
Beck TF, Shchelochkov OA, Yu Z, Kim BJ, Hernández-García A, Zaveri HP, et al. Novel Frem1-related mouse phenotypes and evidence of genetic interactions with Gata4 and Slit3. PLoS One. 2013;8:e58830.
Pickard BS, Hollox EJ, Malloy MP, Porteous DJ, Blackwood DHR, Armour JAL, et al. 4q35.2 subtelomeric deletion identified in a screen of patients with co-morbid psychiatric illness and mental retardation. BMC Med Genet. 2004;5:21.
Cuturilo G, Menten B, Krstic A, Drakulic D, Jovanovic I, Parezanovic V, et al. 4q34.1-q35.2 deletion in a boy with phenotype resembling 22q11.2 deletion syndrome. Eur J Pediatr. 2011;170:1465–70.
Lin S, Kirk EPE, McKenzie F, Francis C, Shalhoub C, Turner AM. De novo interstitial duplication 4(q28.1q35) associated with choanal atresia. J Paediatr Child Health. 2004;40:401–3.
Wentzel C, Fernström M, Ohrner Y, Annerén G, Thuresson A. Clinical variability of the 22q11.2 duplication syndrome. Eur J Med Genet. 2008;51:501–10.
Acknowledgments
We thank the family for their participation. Furthermore, we thank Mrs. Schlösser of the Department of Radiology, University of Bonn, for her technical assistance with preparing the radiographs.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
UG carried out the amniocentesis and performed the prenatal ultrasound. NK performed the genetic testing. AM, HR, and FK were involved in the medical care of the patient. TB, AM, and HR contributed to the discussion of this case. HR and TB designed the case report of the patient. HR also took care of the patient while being an inpatient. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bogs, T., Kipfmüller, F., Kohlschmidt, N. et al. Familial tetrasomy 4q35.2 associated with congenital diaphragmatic hernia and unilateral renal agenesis: a case report. J Med Case Reports 10, 76 (2016). https://doi.org/10.1186/s13256-016-0855-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-016-0855-1