
Abstract
Background
Over the past decade genome-wide association studies (GWAS) have been applied to aid in the understanding of the biology of traits. The success of this approach is governed by the underlying effect sizes carried by the true risk variants and the corresponding statistical power to observe such effects given the study design and sample size under investigation. Previous ASD GWAS have identified genome-wide significant (GWS) risk loci; however, these studies were of only of low statistical power to identify GWS loci at the lower effect sizes (odds ratio (OR) <1.15).
Methods
We conducted a large-scale coordinated international collaboration to combine independent genotyping data to improve the statistical power and aid in robust discovery of GWS loci. This study uses genome-wide genotyping data from a discovery sample (7387 ASD cases and 8567 controls) followed by meta-analysis of summary statistics from two replication sets (7783 ASD cases and 11359 controls; and 1369 ASD cases and 137308 controls).
Results
We observe a GWS locus at 10q24.32 that overlaps several genes including PITX3, which encodes a transcription factor identified as playing a role in neuronal differentiation and CUEDC2 previously reported to be associated with social skills in an independent population cohort. We also observe overlap with regions previously implicated in schizophrenia which was further supported by a strong genetic correlation between these disorders (Rg = 0.23; P = 9 × 10−6). We further combined these Psychiatric Genomics Consortium (PGC) ASD GWAS data with the recent PGC schizophrenia GWAS to identify additional regions which may be important in a common neurodevelopmental phenotype and identified 12 novel GWS loci. These include loci previously implicated in ASD such as FOXP1 at 3p13, ATP2B2 at 3p25.3, and a ‘neurodevelopmental hub’ on chromosome 8p11.23.
Conclusions
This study is an important step in the ongoing endeavour to identify the loci which underpin the common variant signal in ASD. In addition to novel GWS loci, we have identified a significant genetic correlation with schizophrenia and association of ASD with several neurodevelopmental-related genes such as EXT1, ASTN2, MACROD2, and HDAC4.
Similar content being viewed by others
Background
Autism spectrum disorder (ASD) is diagnosed in roughly 1% of the population [1, 2] and has complex genetic roots. The recurrence risk of developing ASD in siblings of an affected individual is approximately 7–19% [3,4,5], and estimates of heritability are high from both twin (64−91%) [6] and whole genome genotyping studies (31−71%) [7]. Analysis of rare and de novo structural and sequence variation in ASD has had recent success in identifying genes and the biology underpinning ASD, albeit with direct relevance to only a small proportion of cases. The establishment of a number of robust risk genes such as CHD8, GRIN2B, SCN2A, and SYNGAP1 [8], and gene-set analyses from associated structural variation have identified synaptic functioning, chromatin remodelling, Wnt signalling, transcriptional regulation, fragile X mental retardation protein (FMRP) interactors and, more broadly, MAPK signalling, as putative biological processes that are disrupted in ASD [9,10,11,12,13].
Importantly, common genetic variation explains roughly half of this genetic risk in ASD [7], making the genome-wide association study (GWAS) an efficient design for identifying risk variants. Early GWAS [12, 14,15,16,17] were performed using a variety of genotyping arrays, and the independent samples sizes were of low statistical power to robustly identify genome-wide significant (GWS) loci at the lower effect sizes (OR <1.15) [18]. Recently, large-scale coordinated international collaborations have been established to combine independent genotyping data to improve statistical power, a strategy that has been fruitful for both schizophrenia [19] and bipolar disorder [20]. In this study, we report the first meta-analyses of a coordinated international effort in ASD from the ASD Working Group of the Psychiatric Genomics Consortium (PGC). By combining published and unpublished GWAS data, we are now able to provide more robust estimates of the underlying common variant structure.
In addition to identifying risk loci, we have examined whether gene-sets previously implicated in ASD are similarly impacted with associated common genetic risk variants. The converging evidence across the variant spectrum should reinforce and expand our understanding of ASD biology. To uncover new biology, we have also examined enrichment of association across numerous functional and cellular annotations, as well as within canonical gene-sets.
Finally, evidence that common structural variation is shared by individuals with ASD, schizophrenia and intellectual disability (ID) continues to fuel a common biological model of ID-ASD-schizophrenia [21]. For example, FMRP biology has also been implicated in all three diagnoses [11]. The hypothesis of a shared pathophysiology for neurodevelopmental disorders is not novel, with Craddock and Owen [22] suggesting that autism exists along a continuum between mental retardation (intellectual disability (ID)) and schizophrenia. Using results from the PGC Schizophrenia Working Group GWAS [19], we have directly tested the relationship between ASD and schizophrenia and extended the meta-analyses by combining these data to identify neurodevelopment-related variants implicated across disorders.
Methods
Participants
Using meta-analysis, we examined association from 14 independent cohorts contributed by eight academic studies (see Table 1). Each contributing site confirmed all affected individuals had an ASD diagnosis; details of diagnostic processes are provided in the Additional file 1 and where available, study specific details are described elsewhere [7, 12, 16, 17, 23,24,25]. Where data permitted, we excluded individuals assessed at under 36 months of age or if there was any evidence of diagnostic criteria not being met from either the Autism Diagnostic Interview-Revised (ADI-R) [26] or the Autism Diagnostic Observation Schedule (ADOS) [27]. The primary meta-analysis (Worldwide ancestry (WW)) was based on data from 7387 ASD cases and 8567 controls. An additional meta-analysis on a more ancestrally homogenous subset (see Additional file 1) was also performed; this subset included data from 6197 ASD cases and 7377 controls of ‘European ancestry’.
We sought independent replication of our results using summary GWAS findings from two additional sources; the Danish iPSYCH Project (7783 ASD cases and 11359 controls) and a combined deCODE Collection (from Iceland plus a collection of individuals from Ukraine, Georgia and Serbia) and the ‘Study to Explore Early Development’ (SEED) (1369 ASD cases and 137308 controls). A detailed description of each cohort is provided in the Additional file 1.
Statistical analyses
Genotyping quality control
Genotyping quality control and imputation of the 14 independent cohorts were performed by the PGC Statistical Analysis Group. Each dataset was processed separately. Experimental details are described in the Additional file 1.
Association, meta-analyses, and binomial sign test
We tested all 14 cohorts individually for association under an additive logistic regression model using PLINK [28]. For samples derived from parent child trios, we applied a case-pseudocontrol design in which the pseudocontrol was created with the non-transmitted alleles from the parents. Since these pseudocontrols are perfectly matched to each case, no covariates were used in association analysis from these trio cohorts. For the non-trio cohorts, each regression included derived principal components as covariates [29]. To minimise the putative reduction in power observed when non-confounding covariates are included in GWAS for rare disease [30], sex was not included as a covariate in these analyses. Individual PP plots for each cohort are reported in the Additional file 1: Figure S1).
We performed a meta-analysis of the individual GWAS using an inverse-weighted fixed effects model [31] implemented in METAL (http://csg.sph.umich.edu//abecasis/Metal/) [32]. A fixed effects meta-analysis was chosen over a random effects model to maximise power and improved discover of associated SNPs [33]. An additional meta-analysis was performed including 13 of the cohorts, omitting the Swedish PAGES collection, this was named noSWM3. The Swedish PAGES collection include control individuals that overlap with the PGC schizophrenia GWAS, and we wished to preclude any potential for confounding of our results which rely upon comparison of these datasets. We performed a cross-disorder meta-analysis of the noSWM3 ASD GWAS and the PGC schizophrenia GWAS [19] using an inverse-weighted fixed effects model as described above. We applied a GWS threshold of P = 5 × 10−8. This is based on the Bonferroni approach, controlling the observed associations at P = 0.05 given approximately 1000000 independent tests.
To aid interpretation, we report findings as linkage disequilibrium (LD) independent SNPs and the corresponding ranges attributed to the LD. LD pruning was performed using the clump flag in PLINK v1.9 [28, 34]. Clumping was used to link additional associated markers within a 0.5Mb window surrounding the primary association. Markers were linked if they were also associated at P < .05 and had an estimated LD with the index SNP of r2 ≥ 0.2. Associated regions were defined for each index SNP as the location spanning all linked markers. All LD statistics were calculated using the 1000 genomes project phase 1 integrated reference haplotypes.
Binomial sign tests to evaluate the random direction effect between studies were implemented in STATA (version 13, Statacorp, College Station, TX, USA).
Gene-based and gene-set analyses
Combining association signals for multiple loci across genes and gene-sets has the potential to capture a greater proportion of variance leading to an increase in power [35]. We performed gene-based association analyses, using the VEGAS2 method (performed at: https://vegas2.qimrberghofer.edu.au/) [36]. This method calculates a test statistic from the sum of test statistics within a gene region. The LD between markers within a gene region is calculated and adjusted for within the software using the 1000 genomes reference genotypes. Analyses were limited to the top 10%, by P value, of SNPs per gene, an approach which has been shown to give rise to higher sensitivity and lower false positive rates compared to other gene-set methods [37].
To explore the converging biology hypothesis of ASD we examined enrichment within gene-sets derived from previously implicated genes and pathways (candidate gene-set) using the Interval-based Enrichment Analysis Tool for Genome Wide Association Studies (INRICH) method [38]. To identify new biology, we examined gene-set enrichment using established canonical gene-sets including gene ontology and the Kyoto Encyclopedia of Genes and Genomes (KEGG) gene-sets. INRICH is a pathway-based GWAS tool that tests for enrichment of association signals against predefined gene-sets across independent genomic intervals. INRICH analysis was performed in interval mode. The interval mode describes an enrichment statistic E for each gene-set that is the number of intervals that overlap at least one target gene in the gene-set. The significance of E is approximated using permutation and an empirical P value is generated from a null set of intervals which match the interval size, overlapping gene, and SNP number to the original input intervals. A detailed description of the gene-set compositions for the candidate gene-set and canonical gene-sets is reported in the Additional file 1. A summary of the sources of each candidate gene-set are also reported in Additional file 1: Table S1.
LD score-based heritability
SNP-based heritability, genetic correlation estimation, and partitioned heritability analyses were performed using the LD score approach [39] (scripts available at https://github.com/bulik/ldsc). Partitioned heritability was performed to examine enrichment of the heritability estimates within SNPs annotated per functional classes based on gene structure (promoter, intron, coding, UTR) and regulatory elements (Histone and DNASE-I hypersensitivity sites). We also examined cell type-specific histone binding elements to identify whether enrichment was limited to specific cell and tissue types. Finally, we applied the partitioned heritability method to examine whether enrichment existed in genes and gene-pathways previously implicated in ASD (see Additional file 1).
Results
Association analyses
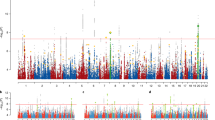
Following quality control, the primary meta-analysis (Worldwide ancestry (WW)) included data on 6695386 common variants across all chromosomes (1−22, X) (minor allele frequency, MAF >0.05; imputation quality score (INFO) >0.60). The secondary GWAS, restricted to individuals defined as being of European (EUR) ancestry, included data on 6632956 common variants that surpassed quality control criteria. Summary Manhattan and PP plots for each analysis are reported in Additional file 1: Figures S2 (WW) and S3 (EUR). We applied a genomewide significance threshold of P ≤ 5 × 10−8. None of the markers investigated exceeded this threshold in the WW meta-analysis. A summary table, containing details of linkage independent associations at P < 10−4 is reported in the Additional file 2 (WW) and Additional file 3 (EUR). Complete summary statistics for these analyses are available at https://www.med.unc.edu/pgc/results-and-downloads.
Replication
Although none of the discovery markers exceeded the GWS threshold, we wanted to further test the existence of true positive signal in our top associated regions and sought replication in independent samples. Previously, when comparing the results of an early iteration of the PGC schizophrenia GWAS (PGC1) [40], which is of similar size to that of reported here for the PGC ASD GWAS, to more recent and larger PGC schizophrenia GWAS (PGC2) [19], we noted that of the association signals 5 × 10−8 ≤ P < 10−4 in the schizophrenia PGC1 study, over 10% (20 of 183 independent loci) were subsequently reported as GWS in the schizophrenia PGC2 analysis. Others have also observed this, revealing in their sample that a substantial minority of associations with borderline genome-wide significance represent replicable and possibly genuine associations [41].
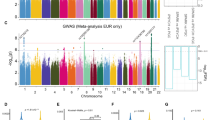
Therefore, we sought replication of the primary PGC ASD GWAS (WW). Summary association data were obtained from the Danish iPSYCH ASD GWAS for all 180 LD-independent markers that were associated at P < 10−4. Of these, 11 (6.1%) met the nominal P < 0.05 threshold in the iPSYCH sample, a non-significant enrichment (Pr(K > = 11/180) = 0.29 where K is the number with P < 0.05). A step-wise binomial sign test was then performed to evaluate the concordance of direction of the effect for each pair of markers below a given rank. This analysis revealed significant enrichment for markers ranked within the top 100 associations (see Fig. 1a). Of the 11 (of 180) markers that were nominally associated in the Danish iPSYCH ASD GWAS, all ranked within the top 100 PGC ASD GWAS association signals (see Additional file 4).

Sign test of concordance of the direction of effect (odds ratio) of the discovery (PGC worldwide) and the replication sample (a replication set 1: iPSYCH; b replication set 2: deCODE/SEED). Blue line is the –log10(P) of the binomial sign test for all associated markers below the rank. The green line describes the concordance, and the grey markers the association in the discovery set
A second replication set from the deCODE/SEED ASD GWAS was also available. Due to platform differences, information from only 159 of the 180 LD-independent markers was available. From these 159 markers, 8 (5%) resulted in association exceeding the nominal P < .05 (Pr(K > = 8/159 = 0.54). The step-wise binomial sign test revealed a smaller concordance effect, with the maximum concordance achieved with approximately the top 70 ranked PGC ASD GWAS association signals, again all nominally associated markers reside within this set (see Fig. 1b).
When the top association signals from the PGC ASD GWAS (P < 10−4) were meta-analysed against the Danish iPSYCH data a single GWS association was observed for rs1409313-T (OR = 1.12 (95% CI 1.08–1.16); P = 1.058 × 10−8). This marker has previously been implicated as a paternally inherited risk marker for ASD within the Simons Simplex Collection (SSC) data [25]. The SSC is not independent as the PGC ASD GWAS data as these individuals are included within our analyses. Examination of the LD between adjacent associated markers and rs1409313 using the clump routine in PLINK, reveals that rs1409313 is correlated with nominally associated markers across a 405 kb region on chromosome 10q24.32 that includes 13 genes (C10orf76, CUEDC2, ELOVL3, FBXL15, GBF1, HPS6, LDB1, MIR146B, NFKB2, NOLC1, PITX3, PPRC1, and PSD). A summary linkage disequilibrium locus plot of these data is shown in Fig. 2.

Association locus plot for the index SNP rs1409313 in the GWAS of all (worldwide) ancestries autism spectrum disorder. The GWS association (pink diamond) refers to the combined PGC-iPSYCH meta-analyses. Additional panels include gene location and location of eQTL markers
Meta-analysis of the PGC ASD—deCODE/SEED data did not result in any GWS findings. However, the top-ranked loci do identify genes previously implicated in ASD such as EXOC4 [42], ANO4 [43], EXT1 [44], and ASTN2 [45]. Similarly, a combined meta-analysis of both PGC plus iPSYCH plus deCODE/SEED datasets did not identify markers exceeding the GWS threshold. The top-ranked locus resulting from this analysis was again rs1409313-T, albeit not achieving GWS (OR = 1.10 (95% CI 1.06–1.14); P = 1.47 × 10−6). In addition to rs1409313-T, the top-ranked associations include markers tagging HDAC4, MACROD2, and EXOC4. A summary of the meta-analysis results is provided in the Additional file 4.
SNP-heritability estimate
We performed LD-score regression to determine the additive heritability attributed to the genome-wide SNPs. The heritability from the WW sample on the observed scale is 0.326 (SE = 0.043, unconstrained, intersect 0.97 (0.007), liability scale h 2 (assuming population prevalence of 1%; 0.18 (SE = .02)). This estimate was based on data from 1095,173 high-confidence SNPs, which show an association inflation, Lambda = 1.06. For the European ancestry samples, the heritability estimate is nearly identical (1,081,358 SNPs; observed scale h 2 = 0.334 (SE = 0.035; unconstrained; intersect 0.99 (0.008); liability scale h 2 (assuming population prevalence of 1%; 0.19 (SE = .02); Lambda = 1.07)). Both estimates are consistent with previous PGC-based estimates of SNP heritability from a subset of these data (liability scale h 2 = 0.17 (SE = 0.025) [46]).
Using the noSWM3 ASD data and the summary GWAS data from the PGC schizophrenia study, we estimate the genetic correlation between ASD and schizophrenia at approximately 23% (genetic correlation (Rg) = 0.23 (SE = 0.05); P = 9 × 10−6; total liability scale genetic covariance = 0.09 (SE 0.02)). This genetic covariance is almost threefold larger than previous reports [47]. As a null comparison, we estimated the genetic correlation against the unrelated Rheumatoid Arthritis GWAS and found no significant correlation with ASD.
Gene-based and gene-set analyses
Gene-based association, performed using the VEGAS2 algorithm, tested the significance of 17,322 genes (Bonferroni threshold P < 2.89 × 10−6) (see Additional file 5). No gene-level associations were significant after Bonferroni correction. The minimum P value achieved, P = 7 × 10−6, was for 3 genes from chromosome 6p21.1 (ENPP4, ENPP5, and CLIC5) and was driven by association of rs7762549, the 4th ranked association interval. To the best of our knowledge, none of these genes have previously been implicated in ASD or ASD-related traits.
Association enrichment was performed using the INRICH method [38]. Using the WW data, we observe enrichment at an empirical P ≤ .05 for the synaptic co-expression network M13 [48], Mendelian disease genes [11], and both human (HARs) and primate-accelerated regions (PARs) [49]. None of these enrichments exceeded the experiment-wise corrected P ≤ .05. Due to overlapping samples between the ASD and PGC GWAS, INRICH analyses against the PGC schizophrenia study [19] was restricted to the noSWM3 ASD GWAS including the worldwide dataset minus the Swedish PAGES sample. This set was analysed in isolation and exceeded the experiment-wise correction (P = .008), with 19 of the 82 blocks included in the analysis overlapping these annotations. Finally, none of the 9708 canonical gene-sets examined in these analyses met the experiment-wise corrected P ≤ .05. A full summary of the INRICH analyses and associated files are given in the Additional files 6, 7, 8, 9, 10, 11, and 12. Of the top-ranked enrichments from canonical pathways, some of the gene-sets overlap those previously highlighted in ASD biology and include processes such as glutamate receptor activity, adheren/cell junctions and the beta-catenin nuclear pathway.
Using the LD Score approach, we also estimated the proportion of heritability that can be attributed to specific partitions of the genome, such as those attributed to gene-sets [39]. Analyses were divided into two sets; WW ASD GWAS against candidate gene-sets, functional annotations and cell type annotation and noSWM3 ASD GWAS against PGC schizophrenia GWS loci. A summary of observed enrichment at an uncorrected P ≤ .05 is given in Table 2, full enrichment data are provided in the Additional file 13. We again observe evidence of an overlap with schizophrenia, with a 2.5-fold enrichment in heritability for those markers within the PGC schizophrenia GWS loci (P = 0.021). The most significant enrichments were observed for annotations tagging gene enhancers, conserved elements and histone marks indexing expression in the mid-frontal lobe. We do not observe evidence for FMRP targets in either the INRICH or LD score analyses.
Overlap of ASD and schizophrenia GWAS
In addition to the significant genetic correlation between ASD and schizophrenia, and enrichment in the heritability, we see further support for an etiological overlap when considering the PGC ASD data as a ‘replication set’ for schizophrenia. When considering the GWS loci reported in the PGC schizophrenia GWAS, 118 of the markers pass QC in the ASD WWW GWAS sample. Eleven of these 118 schizophrenia-associated markers are also associated with ASD at P ≤ .05 (Pr(K > = 11/118) = 0.035). Moreover, when applying the binomial sign test, we observe strong concordance of the direction of these markers (concordance 64.4%; Pr(K > = 76/118) = 0.0011) (see Additional file 1: Figure S14 Panel A). As a comparison, we did not observe any similar enrichment with a Rheumatoid Arthritis dataset (see Additional file 1: Figure S14 Panel B).
Given these observations regarding ASD and schizophrenia, and to further identify novel loci, we meta-analysed the PGC schizophrenia and PGC ASD GWAS data (see Additional file 1: Figure S12 for Manhattan and PP plot). After removing significant loci (+/−1 Mb) previously reported in the PGC schizophrenia analyses, we observed 12 new GWS loci (see Table 3; Additional file 14 and Additional file 15).
Discussion
Genomewide association study of ASD
We present here the results from a large international collaborative GWAS meta-analysis and follow up of 16539 ASD cases and 157234 controls. Despite the considerable increase in the sample size and statistical power of these new analyses to identify associations, we do not observe individual variants that exceed the accepted GWS threshold (P ≤ 5 × 10−8) in the discovery GWAS (n = 7387 ASD cases and 8567 controls). This does not, however, disqualify the loci which fall within the upper-ranked associations from further interrogation, through replication or through supporting biology. There is evidence to support the hypothesis that a substantial proportion of ‘borderline’ association represent genuine associations and deserve an attempt at replication [41]. A PGC schizophrenia GWAS [40], which was of comparable size to this, yielded 5 GWS loci and 183 non-GWS loci at P < 10−4. In a follow-up study [19], 20 of these markers were elevated beyond GWS; these were not limited to the most highly ranked markers, as the newly GWS markers ranged from 7 to 188 in the original study.
We have sought to assess the veracity of these upper-ranked associations through replication and meta-analysis using summary GWAS findings from the Danish iPSYCH Project (n = 7783 ASD cases and 11359 controls) and the combined deCODE and SEED collection (n = 1369 ASD cases and 137308 controls). These analyses show that the concordance of the direction of effect is highly significant for the top 100 and top 70 markers, for the iPSYCH and deCODE/SEED ASD datasets, respectively.
The combined meta-analysis of the PGC ASD GWAS and iPSYCH samples show a single GWS association at rs1409313-T, a marker located on chromosome 10q24.32 within intron 1 of CUEDC2. CUEDC2 encodes the protein ‘CUE domain containing 2’, shown to be involved in the ubiquitination-proteasomal degradation pathway. Others have previously identified associations at this region with the social skills domain of the autism quotient in a population cohort (independent of the PGC ASD GWAS [50]); identifying an association with rs927821 (P = 3.4 × 10−6), a marker in strong linkage disequilibrium with rs1409313 (1000 genomes project all ancestries: r 2 = 0.82; D’ = 0.91; European ancestries: r 2 = 0.68; D’ = 0.85). Other genes in this region include PITX3 which encodes the transcription factor Pitx3 (paired-like homeodomain 3) which plays an important role in the differentiation and maintenance of midbrain dopaminergic neurons during development. It is important to note that the PGC ASD association does not overlap with the 10q24.32 association observed in the recent PGC schizophrenia GWAS. The ASD signal occurs directly upstream of the schizophrenia signal and is separated by a recombination hotspot (see Additional file 1: Figure S13). Other notable genes within the top-ranked associations observed in the PGC-iPSYCH meta-analysis are HDAC4 and MACROD2. GWS associations have previously been reported for MACROD2 by the Autism Genome Project, a subset of the data included in these analyses [12]. HDAC4 encodes the gene histone deacetylase 4 and is involved in the deacetylation of core histones. Dosage of HDAC4 has been implicated in ASD, with observed overexpression in the post-mortem brain tissue of individuals with ASD [51]; conversely, deletions of the HDAC4 loci have been reported in individuals with syndromic autism [52]. Annotation of the associated region implicated in this study (chr2:240183467 to 240327863) does not reveal eQTLs that may support a functional link (see Additional file 2).
The meta-analysis of the PGC–deCODE/SEED data failed to identify GWS SNPs. However, the top-ranked findings did identify genes previously implicated in ASD such as EXOC4 [42], ANO4 [43], EXT1 [44, 53], and ASTN2 [45]. ASTN2 was implicated because of association with the marker rs7026354. Although relatively modest in this meta-analysis, rs7026354 is reported as the 10th ranked association in the PGC-deCODE/SEED ASD analysis (OR = 1.10 (95% CI 1.06–1.14); P = 4.96 × 10−6), this marker is noteworthy because it passes the GWS threshold in the European ancestry only data (rs7026354-A; OR = 1.15 (95% CI 1.09–1.20); P = 4.99 × 10−8 (see Additional file 1: Figure S9 for corresponding locus plot). ASTN2 encodes the protein, astrotactin 2 (ASTN2). Astrotactin 1 (ASTN1) is a membrane protein which forms adhesions between neurons and astroglia [54]. ASTN2 interacts with ASTN1, regulating its expression, thereby playing a role in neuronal-glial adhesion during migration [55]. ASTN2 has previously gained recognition due to the presence of rare CNV losses in ASD [56, 57]. In a recent study of 89,985 individuals, approximately 71% of whom were reported to have a neurodevelopmental disorder (NDD), a total of 46 deletions and 12 duplication mutations were observed in the ASTN2 gene [58]. Exonic deletions were significantly enriched in the NDD cohort (OR = 2.69 (95% CI 1.13–7.77); P = 0.01). The peak of our association signal at 9q33.1 extends over the 3’ end of ASTN2 corresponding to a region of the gene that encodes the C terminus of the protein, a region with cross-species sequence conservation and strong enrichment of exonic deletions in male NDD cases [58].
To garner additional information regarding the biology of ASD, we explored enrichment of association signals across a range of gene-sets and genomic annotations. Of the top canonical results, we observed enrichment in gene-sets related to synaptic structure and function. PDZ domain-binding, beta-catenin nuclear pathways, glutamate receptor activity, and adherens junctions all fit this categorization. PDZ domains (PSD95-disc large-zonula occludens-1) are found in scaffolding proteins, and those at neuronal excitatory synapses are thought to play a key role in synapse organisation and structure. These proteins organise glutamate receptors and associated protein composition at the synapse, subsequently determining the size and strength of the synapse (reviewed in [59]). Moreover, the beta-catenin and adherens gene-sets describe proteins that are involved in establishing synaptic contacts. Genetic insult which disrupts the synapse as a model of ASD is not novel [60], with genes encoding members of the SHANK, neurexin and neuroligin families, all well established as risk factors for ASD. Our data implicating gene-sets that interact with these genes confer additional support for the synaptic hypothesis of ASD.
The candidate gene-set analysis demonstrates a consistent relationship between schizophrenia and ASD. We observed strong genetic correlation between ASD and schizophrenia and a striking concordance in terms of direction of effect (binomial sign test of effect). Moreover, when examining only those genes within the schizophrenia GWS loci, we observed enrichment through gene-based and partitioned heritability analyses. We have excluded confounding due to known sample overlap.
Cross-disorder genome-wide association study of ASD and schizophrenia
We extended our analyses to perform a cross-disorder ASD and schizophrenia meta-analyses to identify putative neurodevelopmental loci; again, we utilised the ASD GWAS excluding known shared controls. We identified 12 GWS loci not previously identified as GWS in the PGC schizophrenia GWAS.
The strongest independent locus, rs57709857-A (OR 0.93, 95% CI 0.91–0.95; P = 4.15 × 10−9) identifies a region previously branded as a neurodevelopmental hub on chromosome 8p (8p11.23). Other GWS associations include the marker rs9879311-T, indexing an association signal within the ATP2B2 gene located at 3p25.3 (OR 1.06, 95% CI 1.04–1.08; P = 6.04 × 10−9) and rs6803008-T, indexing the FOXP1 locus at 3p13 (OR 0.94, 95% CI 0.93–0.96; P = 1.34 × 10−8) (see Additional file 1: Figure S16 for corresponding locus plots). ATP2B2 (ATPase, Ca(2+)-transporting, plasma membrane, 2) plays an important role in intracellular calcium homeostasis and has previously been implicated in ASD through reported genetic associations in North American, Italian, and Chinese samples [61,62,63], as well as through differential expression in ASD brain tissue [64]. FOXP1, a member of the Forkhead Box P family of transcription factors has been implicated in ASD aetiology based on observations of multiple de novo SNVs [65,66,67]. FOXP1 has also been implicated in several related cognitive phenotypes including language impairment and intellectual disability [68,69,70]. Moreover, in a murine Foxp1 KO mouse model, in addition to observable neuronal phenotypes, mice exhibit many behavioural phenotypes associated with ASD [71]. The genetic relationship we observed for common variation shared between schizophrenia and ASD is striking; alongside shared rare structural variation, such as that observed in the 22q11.2 deletion syndrome [72], these data suggest a common risk and a shared biology leading to related but distinct outcomes. We must also consider potential confounding; there is some evidence to support increased assortative mating within and across psychiatric illnesses. Consequently, the evidence from a cross-disorder meta-analysis may not be due to pleiotropy (or not entirely) but may instead be an artefact of a residual genetic background from assortative mating between individuals with these diagnoses [73]. Whether the observed degree of such assortative unions can explain the observed correlations will require further investigation.
To make further progress with the investigation of common variation in ASD, several strategies are being implemented. First, substantial increases in sample size are necessary. This is on the near horizon, with the ongoing activities from groups such as the iPSYCH collaborative likely to bring data from thousands of additional ASD cases to the GWAS effort. Secondly, genetic designs for studies of complex developmental disorders, including ASD and ADHD, have tended to favour a trio-based design and family-based association testing. Although this has provided strength in reducing artefacts resulting from population stratification, recent simulations advise against their use in common and complex polygenic traits, especially where assortative mating may be involved and where the family is known to be multiplex [74]. In such cases, the trio design can underestimate the SNP heritability and the power to observe association.
Conclusions
This study provides an additional step towards understanding the genetic architecture of ASD. We show a robust relationship with the genetic risk identified in schizophrenia GWAS and have highlighted loci such as 10q24.32 and gene-sets that have been previously and independently implicated in ASD and related disorders. Shared heritability findings and our cross-disorder meta-analysis reveals additional GWS loci that may be important in neurodevelopment including a region flagged as a neurodevelopmental hub on chromosome 8p as well as the ATP2B2 gene located at 3p25.3, a gene previously implicated in ASD through both genetic association [61,62,63] and differential expression in the post-mortem brain tissues of individuals with ASD [64].
Like other GWAS of similar size, our ASD-only results are not definitive with the observed associations falling short of accepted statistical significance thresholds. However, we view these data as an important step in the ongoing endeavour to identify the loci which underpin the common variant signal in ASD and we anticipate that some of these loci of ‘borderline’ significance, especially those with additional corroborating evidence such as EXT1, ASTN2, MACROD2, and HDAC4, to eventually garner sufficient evidence to become established robust ASD risk loci.
Abbreviations
- 95% CI:
-
95% Confidence interval boundaries
- ADI-R:
-
Autism Diagnostic Interview-Revised
- ADOS:
-
Autism diagnostic observation schedule
- ASD:
-
Autism spectrum disorder
- EUR:
-
European ancestry
- FMRP:
-
Fragile X mental tetardation protein
- GWAS:
-
Genome-wide association study
- GWS:
-
Genome-wide significant
- H2:
-
Heritability
- ID:
-
Intellectual disability
- INFO:
-
Imputation Quality Score
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- MAPK:
-
Mitogen-activated protein kinase
- noSWM3:
-
ASD GWAS meta-analysis excluding the Swedish PAGES GWAS
- OR:
-
Odds ratio
- PGC:
-
Psychiatrics Genomics Consortium
- Rg:
-
Genetic correlation
- SE:
-
Standard error
- UTR:
-
Untranslated region of a transcript
- WW:
-
Worldwide ancestry
References
Fombonne E. Epidemiology of pervasive developmental disorders. Pediatr Res. 2009;65(6):591–8.
Fernell E, Gillberg C. Autism spectrum disorder diagnoses in Stockholm preschoolers. Res Dev Disabil. 2010;31(3):680–5.
Gronborg TK, Schendel DE, Parner ET. Recurrence of autism spectrum disorders in full- and half-siblings and trends over time: a population-based cohort study. JAMA Pediatr. 2013;167(10):947–53.
Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311(17):1770–7.
Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L, Bryson S, Carver LJ, Constantino JN, Dobkins K, et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics. 2011;128(3):e488–495.
Tick B, Bolton P, Happe F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry. 2016;57(5):585–95.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–5.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–15.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–94.
Krumm N, O'Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37(2):95–105.
Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–21.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–72.
Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu X-Q, Vincent JB, Skaug JL, Thompson AP, Senman L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39(3):319–28.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459(7246):528–33.
Ma D, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Andersen AN, Hoffman JD, Slifer SH, Hedges DJ, Cukier HN, et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann Hum Genet. 2009;73(Pt 3):263–73.
Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461(7265):802–8.
Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet. 2012;21(21):4781–92.
Devlin B, Melhem N, Roeder K. Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Res. 2011;1380:78–84.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–7.
Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, Edenberg HJ, Nurnberger Jr JI, Rietschel M, Blackwood D, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43(10):977–83.
Kirov G. CNVs in neuropsychiatric disorders. Hum Mol Genet. 2015;24(R1):R45–49.
Craddock N, Owen MJ. The Kraepelinian dichotomy - going, going… but still not gone. Br J Psychiatry. 2010;196(2):92–5.
Lajonchere CM, Consortium A. Changing the landscape of autism research: the autism genetic resource exchange. Neuron. 2010;68(2):187–91.
Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, Ducat L, Spence SJ, Committee AS. The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet. 2001;69(2):463–6.
Chaste P, Klei L, Sanders SJ, Hus V, Murtha MT, Lowe JK, Willsey AJ, Moreno-De-Luca D, Yu TW, Fombonne E, et al. A genome-wide association study of autism using the Simons Simplex Collection: does reducing phenotypic heterogeneity in autism increase genetic homogeneity? Biol Psychiatry. 2015;77(9):775–84.
Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24(5):659–85.
Lord C, Rutter M, Goode S, Heemsbergen J, Jordan H, Mawhood L, Schopler E. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J Autism Dev Disord. 1989;19(2):185–212.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Pe'er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32(4):381–5.
Pirinen M, Donnelly P, Spencer CC. Including known covariates can reduce power to detect genetic effects in case-control studies. Nat Genet. 2012;44(8):848–51.
de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17(R2):R122–128.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–1.
Evangelou E, Ioannidis JP. Meta-analysis methods for genome-wide association studies and beyond. Nat Rev Genet. 2013;14(6):379–89.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, Investigators A, Hayward NK, Montgomery GW, Visscher PM, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87(1):139–45.
Mishra A, Macgregor S. VEGAS2: software for more flexible gene-based testing. Twin Res Hum Genet. 2015;18(1):86–91.
Wojcik GL, Kao WH, Duggal P. Relative performance of gene- and pathway-level methods as secondary analyses for genome-wide association studies. BMC Genet. 2015;16:34.
Lee PH, O'Dushlaine C, Thomas B, Purcell SM. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28(13):1797–9.
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics C, Patterson N, Daly MJ, Price AL, Neale BM. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–5.
Schizophrenia Psychiatric Genome-Wide Association Study C. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969–76.
Panagiotou OA, Ioannidis JP, Genome-Wide Significance P. What should the genome-wide significance threshold be? Empirical replication of borderline genetic associations. Int J Epidemiol. 2012;41(1):273–86.
Ben-David E, Granot-Hershkovitz E, Monderer-Rothkoff G, Lerer E, Levi S, Yaari M, Ebstein RP, Yirmiya N, Shifman S. Identification of a functional rare variant in autism using genome-wide screen for monoallelic expression. Hum Mol Genet. 2011;20(18):3632–41.
Wittkowski KM, Sonakya V, Bigio B, Tonn MK, Shic F, Ascano M, Nasca C, Gold-Von Simson G. A novel computational biostatistics approach implies impaired dephosphorylation of growth factor receptors as associated with severity of autism. Transl Psychiatry. 2014;4:e354.
Li H, Yamagata T, Mori M, Momoi MY. Association of autism in two patients with hereditary multiple exostoses caused by novel deletion mutations of EXT1. J Hum Genet. 2002;47(5):262–5.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–73.
Cross-Disorder Group of the Psychiatric Genomics Consortium P, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45(9):984–94.
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, ReproGen C, Psychiatric Genomics C, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control C, Duncan L, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–41.
Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155(5):1008–21.
Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478(7370):476–82.
Jones RM, Cadby G, Melton PE, Abraham LJ, Whitehouse AJ, Moses EK. Genome-wide association study of autistic-like traits in a general population study of young adults. Front Hum Neurosci. 2013;7:658.
Nardone S, Sams DS, Reuveni E, Getselter D, Oron O, Karpuj M, Elliott E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl Psychiatry. 2014;4:e433.
Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87(2):219–28.
Irie F, Badie-Mahdavi H, Yamaguchi Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc Natl Acad Sci U S A. 2012;109(13):5052–6.
Fishell G, Hatten ME. Astrotactin provides a receptor system for CNS neuronal migration. Development. 1991;113(3):755–65.
Wilson PM, Fryer RH, Fang Y, Hatten ME. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J Neurosci. 2010;30(25):8529–40.
Prasad A, Merico D, Thiruvahindrapuram B, Wei J, Lionel AC, Sato D, Rickaby J, Lu C, Szatmari P, Roberts W, et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3. 2012;2(12):1665–85.
Walker S, Scherer SW. Identification of candidate intergenic risk loci in autism spectrum disorder. BMC Genomics. 2013;14:499.
Lionel AC, Tammimies K, Vaags AK, Rosenfeld JA, Ahn JW, Merico D, Noor A, Runke CK, Pillalamarri VK, Carter MT, et al. Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum Mol Genet. 2014;23(10):2752–68.
Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5(10):771–81.
Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16(9):551–63.
Carayol J, Sacco R, Tores F, Rousseau F, Lewin P, Hager J, Persico AM. Converging evidence for an association of ATP2B2 allelic variants with autism in male subjects. Biol Psychiatry. 2011;70(9):880–7.
Yang W, Liu J, Zheng F, Jia M, Zhao L, Lu T, Ruan Y, Zhang J, Yue W, Zhang D, et al. The evidence for association of ATP2B2 polymorphisms with autism in Chinese Han population. PLoS One. 2013;8(4):e61021.
Prandini P, Pasquali A, Malerba G, Marostica A, Zusi C, Xumerle L, Muglia P, Da Ros L, Ratti E, Trabetti E, et al. The association of rs4307059 and rs35678 markers with autism spectrum disorders is replicated in Italian families. Psychiatr Genet. 2012;22(4):177–81.
Vardarajan BN, Eran A, Jung JY, Kunkel LM, Wall DP. Haplotype structure enables prioritization of common markers and candidate genes in autism spectrum disorder. Transl Psychiatry. 2013;3:e262.
Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87(6):1215–33.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–9.
Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, Foomani G, Dobrzeniecka S, Krebs MO, Joober R, et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. 2010;87(5):671–8.
Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149(3):525–37.
Horn D, Kapeller J, Rivera-Brugues N, Moog U, Lorenz-Depiereux B, Eck S, Hempel M, Wagenstaller J, Gawthrope A, Monaco AP, et al. Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat. 2010;31(11):E1851–1860.
Le Fevre AK, Taylor S, Malek NH, Horn D, Carr CW, Abdul-Rahman OA, O'Donnell S, Burgess T, Shaw M, Gecz J, et al. FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am J Med Genet A. 2013;161A(12):3166–75.
Bacon C, Schneider M, Le Magueresse C, Froehlich H, Sticht C, Gluch C, Monyer H, Rappold GA. Brain-specific Foxp1 deletion impairs neuronal development and causes autistic-like behaviour. Mol Psychiatry. 2015;20(5):632–9.
Vorstman JA, Breetvelt EJ, Thode KI, Chow EW, Bassett AS. Expression of autism spectrum and schizophrenia in patients with a 22q11.2 deletion. Schizophr Res. 2013;143(1):55–9.
Nordsletten AE, Larsson H, Crowley JJ, Almqvist C, Lichtenstein P, Mataix-Cols D. Patterns of nonrandom mating within and across 11 major psychiatric disorders. JAMA Psychiat. 2016;73(4):354–61.
Peyrot WJ, Boomsma DI, Penninx BW, Wray NR. Disease and polygenic architecture: avoid trio design and appropriately account for unscreened control subjects for common disease. Am J Hum Genet. 2016;98(2):382–91.
Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Sykes N, Pagnamenta AT, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19(20):4072–82.
Auranen M, Vanhala R, Varilo T, Ayers K, Kempas E, Ylisaukko-Oja T, Sinsheimer JS, Peltonen L, Jarvela I. A genomewide screen for autism-spectrum disorders: evidence for a major susceptibility locus on chromosome 3q25-27. Am J Hum Genet. 2002;71(4):777–90.
Ylisaukko-oja T, Nieminen-von Wendt T, Kempas E, Sarenius S, Varilo T, von Wendt L, Peltonen L, Jarvela I. Genome-wide scan for loci of Asperger syndrome. Mol Psychiatry. 2004;9(2):161–8.
Rehnstrom K, Ylisaukko-oja T, Nieminen-von Wendt T, Sarenius S, Kallman T, Kempas E, von Wendt L, Peltonen L, Jarvela I. Independent replication and initial fine mapping of 3p21-24 in Asperger syndrome. J Med Genet. 2006;43(2):e6.
Kilpinen H, Ylisaukko-oja T, Rehnstrom K, Gaal E, Turunen JA, Kempas E, von Wendt L, Varilo T, Peltonen L. Linkage and linkage disequilibrium scan for autism loci in an extended pedigree from Finland. Hum Mol Genet. 2009;18(15):2912–21.
Bertoglio K, Jill James S, Deprey L, Brule N, Hendren RL. Pilot study of the effect of methyl B12 treatment on behavioral and biomarker measures in children with autism. J Altern Complement Med. 2010;16(5):555–60.
Lit L, Sharp FR, Bertoglio K, Stamova B, Ander BP, Sossong AD, Hendren RL. Gene expression in blood is associated with risperidone response in children with autism spectrum disorders. Pharmacogenomics J. 2012;12(5):368–71.
Gorrindo P, Williams KC, Lee EB, Walker LS, McGrew SG, Levitt P. Gastrointestinal dysfunction in autism: parental report, clinical evaluation, and associated factors. Autism Res. 2012;5(2):101–8.
Hertz-Picciotto I, Croen LA, Hansen R, Jones CR, van de Water J, Pessah IN. The CHARGE study: an epidemiologic investigation of genetic and environmental factors contributing to autism. Environ Health Perspect. 2006;114(7):1119–25.
Schendel DE, Diguiseppi C, Croen LA, Fallin MD, Reed PL, Schieve LA, Wiggins LD, Daniels J, Grether J, Levy SE, et al. The Study to Explore Early Development (SEED): a multisite epidemiologic study of autism by the Centers for Autism and Developmental Disabilities Research and Epidemiology (CADDRE) network. J Autism Dev Disord. 2012;42(10):2121–40.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667–75.
Acknowledgements
We would like to thank the families who participated in and contributed towards the many cohorts used in these studies. Secondly, this extensive international collaboration required the extended effort of many clinical and non-clinical scientists over many years, not all of whom are co-authors of this manuscript. Their collegial effort in supporting these collaborative efforts is essential in the continuing efforts to understanding the genetic architecture of ASD.
The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium
*These authors contributed equally to this work.
†These authors contributed equally to this work.
Richard JL Anney, Email: anneyr@cardiff.ac.uk, Affiliation/s: MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University, Cardiff, CF24 4HQ, UK; Department of Psychiatry, Trinity College Dublin, Dublin, D8, Ireland.
Stephan Ripke, Email: ripke@atgu.mgh.harvard.edu, Affiliation/s: Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research and Program in Medical and Population Genetic, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA; Department of Psychiatry and Psychotherapy, Charité Universitätsmedizin Berlin, CCM, Berlin 10117, Germany.
Verneri Anttila, Affiliation/s: Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research and Program in Medical and Population Genetic, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA.
Jakob Grove, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; iSEQ, Centre for Integrative Sequencing, Aarhus University, Aarhus, DK-8000, Denmark; Department of Biomedicine - Human Genetics, Aarhus University, Aarhus, DK-8000, Denmark; Bioinformatics Research Centre, Aarhus University, Aarhus, Denmark.
Peter Holmans, Affiliation/s: MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University, Cardiff, CF24 4HQ, UK.
Hailiang Huang, Affiliation/s: Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research and Program in Medical and Population Genetic, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA.
Lambertus Klei, Affiliation/s: Department of Psychiatry, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
Phil H Lee, Affiliation/s: Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research and Program in Medical and Population Genetic, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA; Department of Psychiatry, Harvard Medical School, Boston, MA 02115, USA.
Sarah E Medland, Affiliation/s: Queensland Institute of Medical Research, Brisbane, QLD, 4006, Australia.
Benjamin Neale, Affiliation/s: Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research and Program in Medical and Population Genetic, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA.
Elise Robinson, Affiliation/s: Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research and Program in Medical and Population Genetic, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA.
Lauren A Weiss, Affiliation/s: Department of Psychiatry, University of California San Francisco, San Francisco, CA 94143, USA; Inst Human Genetics, University of California San Francisco, San Francisco, CA 94143, USA.
Lonnie Zwaigenbaum, Affiliation/s: Department of Pediatrics, University of Alberta, Edmonton, AB, T6G 1C9, Canada.
Timothy W Yu, Affiliation/s: Division of Genetics, Children’s Hospital Boston, Harvard Medical School, Boston, MA 02115, USA.
Kerstin Wittemeyer, Affiliation/s: School of Education, University of Birmingham, Birmingham, B15 2TT, UK.
A.Jeremy Willsey, Affiliation/s: Department of Psychiatry, University of California San Francisco, San Francisco, CA 94143, USA.
Ellen M Wijsman, Affiliation/s: Department of Medicine, University of Washington, Seattle, WA 98195, USA; Department of Biostatistics, University of Washington, Seattle, WA 98195, USA.
Thomas Werge, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; Institute of Biological Psychiatry, MHC Sct Hans, Mental Health Services Copenhagen, Roskilde, Denmark; Department of Clinical Medicine, University of Copenhagen, Copenhagen, DK-2200, Denmark.
Thomas H Wassink, Affiliation/s: Department of Psychiatry, Carver College of Medicine, Iowa City, IA 52242, USA.
Regina Waltes, Affiliation/s: Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, JW Goethe University Frankfurt, Frankfurt am Main, 60528, Germany.
Christopher A Walsh, Affiliation/s: Division of Genetics, Children’s Hospital Boston, Harvard Medical School, Boston, MA 02115, USA; Program in Genetics and Genomics, Harvard Medical School, Boston, MA 02115, USA; Howard Hughes Medical Institute, Harvard Medical School, Boston, MA 02115, USA; Department of Pediatrics, Harvard Medical School, Boston, MA 02115, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA;
Simon Wallace, Affiliation/s: Department of Psychiatry, University of Oxford and Warneford Hospital, Oxford, OX3 7JX, UK.
Jacob AS Vorstman, Affiliation/s: Department of Psychiatry, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, 3584 CG, The Netherlands.
Veronica J Vieland, Affiliation/s: Battelle Center for Mathematical Medicine, The Research Institute at Nationwide Children’s Hospital, Columbus, OH 43205, USA.
Astrid M Vicente, Affiliation/s: Instituto Nacional de Saúde Dr Ricardo Jorge, Lisboa, 1600, Portugal; Center for Biodiversity, Functional and Integrative Genomics, Campus da FCUL, Lisboa, 1649, Portugal.
Herman vanEngeland, Affiliation/s: Department of Psychiatry, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, 3584 CG, The Netherlands.
Kathryn Tsang, Affiliation/s: Department of Psychiatry, University of California San Francisco, San Francisco, CA 94143, USA; Inst Human Genetics, University of California San Francisco, San Francisco, CA 94143, USA.
Ann P Thompson, Affiliation/s: Department of Psychiatry and Behavioral Neurosciences, McMaster University, Hamilton, ON, L8S 4L8, Canada.
Peter Szatmari, Affiliation/s: Department of Psychiatry, University of Toronto, ON, M5T 1R8, Canada.
Oscar Svantesson, Affiliation/s: Karolinska Institutet, Solna, SE-171 77, Sweden.
Stacy Steinberg, Affiliation/s: deCODE Genetics, Reykjavik, IS-101, Iceland.
Kari Stefansson, Affiliation/s: deCODE Genetics, Reykjavik, IS-101, Iceland.
Hreinn Stefansson, Affiliation/s: deCODE Genetics, Reykjavik, IS-101, Iceland.
Matthew W State, Affiliation/s: Department of Psychiatry, University of California San Francisco, San Francisco, CA 94143, USA.
Latha Soorya, Affiliation/s: Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Rush University Medical Center, Chicago, IL 60612, USA.
Teimuraz Silagadze, Affiliation/s: Department of Psychiatry and Drug Addiction, Tbilisi State Medical University, Tbilisi, 0186, Georgia.
Stephen W Scherer, Affiliation/s: The Centre for Applied Genomics, The Hospital for Sick Children, Toronto, ON, M5G 1L4, Canada; McLaughlin Centre, University of Toronto, Toronto, ON, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, Toronto, ON, M5S 1A8, Canada.
Gerard D Schellenberg, Affiliation/s: Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA 19102, USA.
Sven Sandin, Affiliation/s: Karolinska Institutet, Solna, SE-171 77, Sweden.
Stephan J Sanders, Affiliation/s: Department of Psychiatry, University of California San Francisco, San Francisco, CA 94143, USA.
Evald Saemundsen, Affiliation/s: State Diagnostic and Counseling Centre, Kopavogur, IS-201, Iceland.
Guy A Rouleau, Affiliation/s: Montreal Neurological Institute, Dept of Neurology and Neurosurgery, McGill University, Montreal, QC, H3A 2B4, Canada.
Bernadette Rogé, Affiliation/s: Centre d’Etudes et de Recherches en Psychopathologie, Toulouse University, Toulouse, 31058, France.
Kathryn Roeder, Affiliation/s: Department of Computational Biology, Carnegie Mellon University, Pittsburgh, PA 15213, USA; Department of Statistics, Carnegie Mellon University, Pittsburgh, PA 15213, USA.
Wendy Roberts, Affiliation/s: Autism Research Unit, The Hospital for Sick Children, Toronto, ON, M5G 1L4, Canada.
Jennifer Reichert, Affiliation/s: Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Abraham Reichenberg, Affiliation/s: Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Karola Rehnström, Affiliation/s: Sanger Institute, Hinxton, CB10 1SA, UK.
Regina Regan, Affiliation/s: National Childrens Research Centre, Our Lady’s Hospital Crumlin, Dublin, D12, Ireland; Academic Centre on Rare Diseases, University College Dublin, Dublin, D4, Ireland.
Fritz Poustka, Affiliation/s: Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, JW Goethe University Frankfurt, Frankfurt am Main, 60528, Germany.
Christopher S Poultney, Affiliation/s: Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Joseph Piven, Affiliation/s: University of North Carolina, Chapel Hill, NC 27599, USA.
Dalila Pinto, Affiliation/s: Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; The Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; The Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA
Margaret A Pericak-Vance, Affiliation/s: The John P Hussman Institute for Human Genomics, University of Miami, Miami, FL 33101, USA.
Milica Pejovic-Milovancevic, Affiliation/s: Institute of Mental Health and Medical Faculty, University of Belgrade, Belgrade, 11 000, Serbia.
Marianne Giørtz Pedersen, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; National Centre for Register-based Research, Aarhus University, Aarhus, Denmark; Centre for Integrated Register-based Research, Aarhus University, Aarhus, Denmark.
Carsten Bøcker Pedersen, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; National Centre for Register-based Research, Aarhus University, Aarhus, Denmark; Centre for Integrated Register-based Research, Aarhus University, Aarhus, Denmark.
Andrew D Paterson, Affiliation/s: Department of Molecular Genetics, University of Toronto, Toronto, ON, M5S 1A8, Canada; The Centre for Applied Genomics, The Hospital for Sick Children, Toronto, ON, M5G 1L4, Canada; Dalla Lana School of Public Health, Toronto, ON, M5T 3M7, Canada.
Jeremy R Parr, Affiliation/s: Institute of Neuroscience, Newcastle University, Newcastle Upon Tyne, NE2 4HH, UK; Institue of Health and Science, Newcastle University, Newcastle Upon Tyne, NE2 4AX, UK.
Alistair T Pagnamenta, Affiliation/s: Wellcome Trust Centre for Human Genetics, Oxford University, Oxford, OX3 7BN, UK.
Guiomar Oliveira, Affiliation/s: Unidade de Neurodesenvolvimento e Autismo do Serviço do Centro de Desenvolvimento da Criança and Centro de Investigação e Formação Clinica, Pediatric Hospital, Centro Hospitalar e Universitário de Coimbra, Coimbra, 3041-80, Portugal; University Clinic of Pediatrics and Institute for Biomedical Imaging and Life Science, Faculty of Medicine, University of Coimbra, Coimbra, 3041-80, Portugal.
John I Nurnberger, Affiliation/s: Institute of Psychiatric Research, Department of Psychiatry, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Program in Medical Neuroscience, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Merete Nordentoft, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; Mental Health Services in the Capital Region of Denmark, Mental Health Center Copenhagen, University of Copenhagen, Copenhagen, Denmark.
Michael T Murtha, Affiliation/s: Programs on Neurogenetics, Yale University School of Medicine, New Haven, CT 06520, USA.
Susana Mouga, Affiliation/s: Unidade de Neurodesenvolvimento e Autismo do Serviço do Centro de Desenvolvimento da Criança and Centro de Investigação e Formação Clinica, Pediatric Hospital, Centro Hospitalar e Universitário de Coimbra, Coimbra, 3041-80, Portugal; University Clinic of Pediatrics and Institute for Biomedical Imaging and Life Science, Faculty of Medicine, University of Coimbra, Coimbra, 3041-80, Portugal.
Preben Bo Mortensen, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; iSEQ, Centre for Integrative Sequencing, Aarhus University, Aarhus, DK-8000, Denmark; National Centre for Register-based Research, Aarhus University, Aarhus, Denmark; Centre for Integrated Register-based Research, Aarhus University, Aarhus, Denmark,
Ole Mors, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; Psychosis Research Unit, Aarhus University Hospital, Risskov, Denmark.
Eric M Morrow, Affiliation/s: Department of Psychiatry and Human Behaviour, Brown University, Providence, RI 02912, USA.
Daniel Moreno-De-Luca, Affiliation/s: Department of Psychiatry and Human Behaviour, Brown University, Providence, RI 02912, USA.
Anthony P Monaco, Affiliation/s: Wellcome Trust Centre for Human Genetics, Oxford University, Oxford, OX3 7BN, UK; Tufts University, Boston, MA 02155?, USA.
Nancy Minshew, Affiliation/s: Department of Psychiatry, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
Alison Merikangas, Affiliation/s: Department of Psychiatry, Trinity College Dublin, Dublin, D8, Ireland.
William M McMahon, Affiliation/s: Department of Psychiatry, University of Utah, Salt Lake City, UT 84108, USA.
Susan G McGrew, Affiliation/s: Department of Pediatrics, Vanderbilt University, Nashville, TN 37232, USA.
Manuel Mattheisen, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; iSEQ, Centre for Integrative Sequencing, Aarhus University, Aarhus, DK-8000, Denmark; Department of Biomedicine - Human Genetics, Aarhus University, Aarhus, DK-8000, Denmark.
Igor Martsenkovsky, Affiliation/s: Department of Child, Adolescent Psychiatry and Medical-Social Rehabilitation, Ukrainian Research Institute of Social Forensic Psychiatry and Drug Abuse, Kyiv, 04080, Ukraine.
Donna M Martin, Affiliation/s: Department of Pediatrics and Human Genetics, University of Michigan, Ann Arbor, MI 48109, USA.
Shrikant M Mane, Affiliation/s: Yale Center for Genomic Analysis, Yale University School of Medicine, New Haven, CT 06516, USA.
Pall Magnusson, Affiliation/s: Department of Child and Adolescent Psychiatry, National University Hospital, Reykjavik, IS-101, Iceland.
Tiago Magalhaes, Affiliation/s: National Childrens Research Centre, Our Lady’s Hospital Crumlin, Dublin, D12, Ireland; Academic Centre on Rare Diseases, University College Dublin, Dublin, D4, Ireland.
Elena Maestrini, Affiliation/s: Department of Pharmacy and Biotechnology, University of Bologna, Bologna, 40126, Italy.
Jennifer K Lowe, Affiliation/s: Center for Autism Research and Treatment, Semel Institute, David Geffen School of Medicine at University of California Los Angeles, Los Angeles, CA 90095, USA; Program in Neurogenetics, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Neurobehavioral Genetics, Semel Institute, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Catherine Lord, Affiliation/s: Department of Psychiatry, Weill Cornell Medical College, Cornell University, New York, NY 10065, USA.
Pat Levitt, Affiliation/s: Department of Pediatrics, Keck School of Medicine, University of Southern California, Los Angeles, CA 90027, USA.
Christa LeseMartin, Affiliation/s: Autism and Developmental Medicine Institute, Geisinger Health System, Danville, PA 17837, USA.
David H Ledbetter, Affiliation/s: Chief Scientific Officer, Geisinger Health System, Danville, PA 17837, USA.
Marion Leboyer, Affiliation/s: FondaMental Foundation, Créteil, 94000, France; INSERM U955, Paris, 94010, France; Faculté de Médecine, Université Paris Est, Créteil, 94000, France; Department of Psychiatry, Henri Mondor-Albert Chenevier Hospital, Assistance Publique – Hôpitaux de Paris, Créteil, 94000, France,
Ann S LeCouteur, Affiliation/s: Institute of Neuroscience, Newcastle University, Newcastle Upon Tyne, NE2 4HH, UK; Institue of Health and Science, Newcastle University, Newcastle Upon Tyne, NE2 4AX, UK.
Christine Ladd-Acosta, Affiliation/s: Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD 21205, USA.
Alexander Kolevzon, Affiliation/s: Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; The Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA,
Sabine M Klauck, Affiliation/s: Division of Molecular Genome Analysis and Working Group Cancer Genome Research, Deutsches Krebsforschungszentrum, Heidelberg, D-69120, Germany.
Suma Jacob, Affiliation/s: Institute for Juvenile Research, Department of Psychiatry, University of Illinois at Chicago, Chicago, IL 60612, USA; Institute of Translational Neuroscience and Department of Psychiatry, University of Minnesota, Minneapolis, MN 55454, USA.
Bozenna Iliadou, Affiliation/s: Karolinska Institutet, Solna, SE-171 77, Sweden.
Christina M Hultman, Affiliation/s: Karolinska Institutet, Solna, SE-171 77, Sweden.
David M Hougaard, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; Center for Neonatal Screening, Department for Congenital Disorders, Statens Serum Institut, Copenhagen, DK-2300, Denmark.
Irva Hertz-Picciotto, Affiliation/s: Department of Public Health Sciences, School of Medicine, University of California Davis, Davis, CA 95616, USA; The MIND Institute, School of Medicine, University of California Davis, Davis, CA 95817, USA.
Robert Hendren, Affiliation/s: Department of Psychiatry, University of California San Francisco, San Francisco, CA 94143, USA.
Christine Søholm Hansen, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; Center for Neonatal Screening, Department for Congenital Disorders, Statens Serum Institut, Copenhagen, DK-2300, Denmark.
Jonathan L Haines, Affiliation/s: Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 37232, USA.
Stephen J Guter, Affiliation/s: Institute for Juvenile Research, Department of Psychiatry, University of Illinois at Chicago, Chicago, IL 60612, USA.
Dorothy E Grice, Affiliation/s: Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Jonathan M Green, Affiliation/s: Manchester Academic Health Sciences Centre, Manchester, M13 9NT, UK; Institute of Brain, Behaviour, and Mental Health, University of Manchester, Manchester, M13 9PT, UK.
Andrew Green, Affiliation/s: Academic Centre on Rare Diseases, University College Dublin, Dublin, D4, Ireland; Centre for Medical Genetics, Our Lady’s Hospital Crumlin, Dublin, D12, Ireland.
Arthur P Goldberg, Affiliation/s: Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; The Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Christopher Gillberg, Affiliation/s: Gillberg Neuropsychiatry Centre, University of Gothenburg, Gothenburg, S-405 30, Sweden.
John Gilbert, Affiliation/s: The John P Hussman Institute for Human Genomics, University of Miami, Miami, FL 33101, USA.
Louise Gallagher, Affiliation/s: Department of Psychiatry, Trinity College Dublin, Dublin, D8, Ireland.
Christine M Freitag, Affiliation/s: Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, JW Goethe University Frankfurt, Frankfurt am Main, 60528, Germany.
Eric Fombonne, Affiliation/s: Department of Psychiatry and Institute for Development and Disability, Oregon Health and Science University, Portland, OR 97239, USA.
Susan E Folstein, Affiliation/s: Division of Child and Adolescent Psychiatry, Department of Psychiatry, Miller School of Medicine, University of Miami, Miami, FL 33136, USA.
Bridget Fernandez, Affiliation/s: Memorial University of Newfoundland, St John’s, NL, A1B 3X9, Canada.
M.Daniele Fallin, Affiliation/s: Department of Mental Health, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD 21205, USA.
A.Gulhan Ercan-Sencicek, Affiliation/s: Programs on Neurogenetics, Yale University School of Medicine, New Haven, CT 06520, USA.
Sean Ennis, Affiliation/s: Academic Centre on Rare Diseases, University College Dublin, Dublin, D4, Ireland; Centre for Medical Genetics, Our Lady’s Hospital Crumlin, Dublin, D12, Ireland.
Frederico Duque, Affiliation/s: Unidade de Neurodesenvolvimento e Autismo do Serviço do Centro de Desenvolvimento da Criança and Centro de Investigação e Formação Clinica, Pediatric Hospital, Centro Hospitalar e Universitário de Coimbra, Coimbra, 3041-80, Portugal; University Clinic of Pediatrics and Institute for Biomedical Imaging and Life Science, Faculty of Medicine, University of Coimbra, Coimbra, 3041-80, Portugal.
Eftichia Duketis, Affiliation/s: Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, JW Goethe University Frankfurt, Frankfurt am Main, 60528, Germany.
Richard Delorme, Affiliation/s: FondaMental Foundation, Créteil, 94000, France; Human Genetics and Cognitive Functions Unit, Institut Pasteur, Paris, 75015, France; Centre National de la Recherche Scientifique URA 2182 Institut Pasteur, Paris, 75724, France; Department of Child and Adolescent Psychiatry, Robert Debré Hospital, Assistance Publique – Hôpitaux de Paris, Paris, 75019, France,
Silvia DeRubeis, Affiliation/s: Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Maretha V DeJonge, Affiliation/s: Department of Psychiatry, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, 3584 CG, The Netherlands.
Geraldine Dawson, Affiliation/s: Duke Center for Autism and Brain Developments, Duke University School of Medicine, Durham, NC 27705, USA; Duke Institute for Brain Sciences, Duke University School of Medicine, Durham, NC 27708, USA.
Michael L Cuccaro, Affiliation/s: The John P Hussman Institute for Human Genomics, University of Miami, Miami, FL 33101, USA.
Catarina T Correia, Affiliation/s: Instituto Nacional de Saúde Dr Ricardo Jorge, Lisboa, 1600, Portugal; Center for Biodiversity, Functional and Integrative Genomics, Campus da FCUL, Lisboa, 1649, Portugal.
Judith Conroy, Affiliation/s: Academic Centre on Rare Diseases, University College Dublin, Dublin, D4, Ireland; Temple Street Children’s University Hospital, Dublin, D1, Ireland.
Ines C Conceição, Affiliation/s: Instituto Nacional de Saúde Dr Ricardo Jorge, Lisboa, 1600, Portugal; Center for Biodiversity, Functional and Integrative Genomics, Campus da FCUL, Lisboa, 1649, Portugal.
Andreas G Chiocchetti, Affiliation/s: Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, JW Goethe University Frankfurt, Frankfurt am Main, 60528, Germany.
Patrícia BS Celestino-Soper, Affiliation/s: Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Jillian Casey, Affiliation/s: Temple Street Children’s University Hospital, Dublin, D1, Ireland; Academic Centre on Rare Diseases, University College Dublin, Dublin, D4, Ireland.
Rita M Cantor, Affiliation/s: Department of Psychiatry, David Geffen School of Medicine at University of California Los Angeles, Los Angeles, CA 90095, USA; Department of Human Genetics, David Geffen School of Medicine at University of California Los Angeles, Los Angeles, CA 90095, USA.
Cátia Café, Affiliation/s: Unidade de Neurodesenvolvimento e Autismo do Serviço do Centro de Desenvolvimento da Criança and Centro de Investigação e Formação Clinica, Pediatric Hospital, Centro Hospitalar e Universitário de Coimbra, Coimbra, 3041-80, Portugal.
Jonas Bybjerg-Grauholm, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; Center for Neonatal Screening, Department for Congenital Disorders, Statens Serum Institut, Copenhagen, DK-2300, Denmark.
Sean Brennan, Affiliation/s: Department of Psychiatry, Trinity College Dublin, Dublin, D8, Ireland.
Thomas Bourgeron, Affiliation/s: FondaMental Foundation, Créteil, 94000, France; Human Genetics and Cognitive Functions Unit, Institut Pasteur, Paris, 75015, France; Centre National de la Recherche Scientifique URA 2182 Institut Pasteur, Paris, 75724, France; University Paris Diderot, Sorbonne Paris Cité, Paris, 75013, France,
Patrick F Bolton, Affiliation/s: Institute of Psychiatry, Kings College London, London, SE5 8AF, UK; South London and Maudsley Biomedical Research Centre for Mental Health, London, SE5 8AF, UK.
Sven Bölte, Affiliation/s: Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, JW Goethe University Frankfurt, Frankfurt am Main, 60528, Germany; Department of Women’s and Children’s Health, Center of Neurodevelopmental Disorders, Karolinska Institutet, Stockholm, SE-113 30, Sweden; Child and Adolescent Psychiatry, Center for Psychiatry Research, Stockholm County Council, Stockholm, SE-171 77, Sweden.
Nadia Bolshakova, Affiliation/s: Department of Psychiatry, Trinity College Dublin, Dublin, D8, Ireland.
Catalina Betancur, Affiliation/s: INSERM U1130, Paris, 75005, France; CNRS UMR 8246, Paris, 75005, France; Sorbonne Universités, UPMC Univ Paris 6, Neuroscience Paris Seine, Paris, 75005, France.
Raphael Bernier, Affiliation/s: Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, WA 98195, USA.
Arthur L Beaudet, Affiliation/s: Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Agatino Battaglia, Affiliation/s: Stella Maris Institute for Child and Adolescent Neuropsychiatr, Pisa, 56018, Italy.
Vanessa H Bal, Affiliation/s: Department of Psychiatry, University of California San Francisco, San Francisco, CA 94143, USA.
Gillian Baird, Affiliation/s: Paediatric Neurodisability, King’s Health Partners, Kings College London, London, SE1 7EH, UK.
Anthony J Bailey, Affiliation/s: Department of Psychiatry, University of Oxford and Warneford Hospital, Oxford, OX3 7JX, UK; Mental Health and Addictions Research Unit, University of British Colombia, Vancouver, BC, V5Z 4H4, Canada.
Marie Bækvad-Hansen, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; Center for Neonatal Screening, Department for Congenital Disorders, Statens Serum Institut, Copenhagen, DK-2300, Denmark.
Joel S Bader, Affiliation/s: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD 21218, USA.
Elena Bacchelli, Affiliation/s: Department of Pharmacy and Biotechnology, University of Bologna, Bologna, 40126, Italy.
Evdokia Anagnostou, Affiliation/s: Bloorview Research Institute, University of Toronto, Toronto, ON, M4G 1R8, Canada.
David Amaral, Affiliation/s: The MIND Institute, School of Medicine, University of California Davis, Davis, CA 95817, USA; Department of Psychiatry, School of Medicine, University of California Davis, Davis, CA 95817, USA; Department of Behavioural Sciences, School of Medicine, University of California Davis, Davis, CA 95817, USA.
Joana Almeida, Affiliation/s: Unidade de Neurodesenvolvimento e Autismo do Serviço do Centro de Desenvolvimento da Criança and Centro de Investigação e Formação Clinica, Pediatric Hospital, Centro Hospitalar e Universitário de Coimbra, Coimbra, 3041-80, Portugal.
Anders D Børglum, Affiliation/s: iPSYCH, Lundbeck Foundation Initiative for Integrative Psychiatric Research, Aarhus, Denmark; iSEQ, Centre for Integrative Sequencing, Aarhus University, Aarhus, DK-8000, Denmark; Department of Biomedicine - Human Genetics, Aarhus University, Aarhus, DK-8000, Denmark.
Joseph D Buxbaum, Affiliation/s: Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; The Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA
Aravinda Chakravarti, Affiliation/s: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD 21218, USA.
Edwin H Cook, Affiliation/s: Institute for Juvenile Research, Department of Psychiatry, University of Illinois at Chicago, Chicago, IL 60612, USA.
Hilary Coon, Affiliation/s: Department of Psychiatry, University of Utah, Salt Lake City, UT 84108, USA.
Daniel H Geschwind, Affiliation/s: Center for Autism Research and Treatment, Semel Institute, David Geffen School of Medicine at University of California Los Angeles, Los Angeles, CA 90095, USA; Program in Neurogenetics, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Neurobehavioral Genetics, Semel Institute, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Human Genetics, David Geffen School of Medicine at University of California Los Angeles, Los Angeles, CA 90095, USA,
Michael Gill, Affiliation/s: Department of Psychiatry, Trinity College Dublin, Dublin, D8, Ireland.
Hakon Hakonarson, Affiliation/s: The Center for Applied Genomics and Division of Human Genetics, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, Philadelphia, PA 19104, USA; Dept of Pediatrics, University of Pennsylvania, Philadelphia, PA 19104, USA.
Joachim Hallmayer, Affiliation/s: Department of Psychiatry, Stanford University, Stanford, CA 94305, USA.
Aarno Palotie, Affiliation/s: Sanger Institute, Hinxton, CB10 1SA, UK.
Susan L Santangelo, Affiliation/s: Tufts University School of Medicine, Boston, MA 02111, USA; Maine Medical Center Research Institute, Portland, ME 04102, USA.
James S Sutcliffe, Affiliation/s: Vanderbilt Genetics Institute, Vanderbilt University, Nashville, TN 37232, USA; Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 37232, USA.
†Dan E Arking, Email: arking@jhmi.edu, Affiliation/s: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD 21218, USA.
†Bernie Devlin, Email: devlinbj@upmc.edu, Affiliation/s: Department of Psychiatry, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
†Mark J Daly, Email: mjdaly@atgu.mgh.harvard.edu, Affiliation/s: Analytic and Translational Genetics Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research and Program in Medical and Population Genetic, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA.
Funding
The Autism Working Group of the Psychiatric Genomics Consortium was supported by National Institutes of Mental Health (NIMH, USA) grant MH109539, MH094432 and MH094421 to M.J.D. The ACE Network was supported by MH081754 and MH100027 to D.H.G. The Autism Genetic Resource Exchange (AGRE) is a program of Autism Speaks (USA) and was supported by grant MH081810. The Autism Genome Project (AGP) was supported by grants from Autism Speaks, the Canadian Institutes of Health Research (CIHR), Genome Canada, the Health Research Board (Ireland; AUT/2006/1, AUT/2006/2, PD/2006/48), the Hilibrand Foundation (USA), the Medical Research Council (UK), the National Institutes of Health (USA; the National Institute of Child Health and Human Development and the National Institute of Mental Health), the Ontario Genomics Institute, and the University of Toronto McLaughlin Centre. The Simons Simplex Collection (SSC) was supported by a grant from the Simons Foundation (SFARI 124827 to the investigators of the Simons Simplex Collection Genetic Consortium); approved researchers can obtain the SSC population dataset described in this study (http://sfari.org/resources/sfari-base) by applying at https://base.sfari.org. The Gene Discovery Project of Johns Hopkins was funded by MH060007, MH081754, and the Simons Foundation. The MonBos Collection study was funded in part through a grant from the Autism Consortium of Boston. Support for the Extreme Discordant Sib-Pair (EDSP) family sample (part of the MonBos collection) was provided by the NLM Family foundation. Support for the Massachusetts General Hospital (MGH)–Finnish collaborative sample was provided by NARSAD. The PAGES collection was funded by NIMH grant MH097849. The collection of data and biomaterials that participated in the NIMH Autism Genetics Initiative has been supported by National Institute of Health grants MH52708, MH39437, MH00219, and MH00980; National Health Medical Research Council grant 0034328; and by grants from the Scottish Rite, the Spunk Fund, Inc., the Rebecca and Solomon Baker Fund, the APEX Foundation, the National Alliance for Research in Schizophrenia and Affective Disorders (NARSAD), the endowment fund of the Nancy Pritzker Laboratory (Stanford); and by gifts from the Autism Society of America, the Janet M. Grace Pervasive Developmental Disorders Fund, and families and friends of individuals with autism. The iPSYCH project is funded by The Lundbeck Foundation and the universities and university hospitals of Aarhus and Copenhagen. In addition, the genotyping of iPSYCH samples was supported by grants from the Stanley Foundation, the Simons Foundation (SFARI 311789 to MJD), and NIMH (5U01MH094432-02 to MJD).
The Study to Explore Early Development (SEED) was funded by the Centers for Disease Control and Prevention (CDC) grants U10DD000180, U10DD000181, U10DD000182, U10DD000183, U10DD000184, and U10DD000498.
Statistical analyses were carried out on the Genetic Cluster Computer (http://www.geneticcluster.org) hosted by SURFsara and financially supported by the Netherlands Scientific Organization (NWO 480-05-003), along with a supplement from the Dutch Brain Foundation and the VU University Amsterdam. Additional statistical analyses were performed and supported by the Trinity Centre for High Performance Computing (http://www.tchpc.tcd.ie/) funded through Science Foundation Ireland. Computational support for the PAGES collection was provided in part through the computational resources and staff expertise of the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai (https://hpc.mssm.edu). Data QC and statistical analyses of the iPSYCH samples were performed at the high-performance computing cluster GenomeDK (http://genome.au.dk) at the Center for Integrative Sequencing, iSEQ, Aarhus University. iSEQ provided computed time, data storage, and technical support for the study.
Availability of data and materials
Association summary statistics for these analyses are available at https://www.med.unc.edu/pgc/results-and-downloads.
Authors’ contributions
RA and SR were the lead authors and made substantial contributions to conception, analysis, and interpretation of the data. They were involved in all aspects of the study, including manuscript preparation and critically revision for important intellectual content. DEA, ADB, EHC, MJD, BD, JGr, PH, JSS, and SLS were senior contributors and made a substantial input to the conception, design, and interpretation of the data including contributing to the manuscript preparation and critically revision of the manuscript for important intellectual content. VA, JSB, HC, HHu, LK, PHL, MM, BN, ER, and KRo and made substantial contributions to the central data integration, data analyses and data interpretation. DAm, EA, JSB, AJB, GB, VHB, AB, ALB, CB, PFB, TB, JDB, SBo, RMC, PBSC-S, AC, AGC, HC, MLC, GD, MVDeJ, SDeR, RD, SE, MDF, SEF, EF, CMF, LG, DHG, JGi, MG, CGi, AG, DEG, JLH, HHa, JH, RH, IH-P, HHu, CMH, BI, SMK, LK, AK, CL-A, ASLeC, ML, DHL, CLM, PL, CL, JKL, DMM, WMM, SEM, NM, APM, EMM, OM, PBM, BN, MN, JIN, AP, JRP, ADP, MAP-V, DP, JPi, AR, JR, WR, KRo, BR, GR, SJS, SSand, GDS, SWS, LS, MWS, JSS, OS, PS, KT, AMV, VJV, JASV, SW, CAW, THW, LW, TW, EMW, AJW, TWY, LZ, and HvanE, were the key contributors and made substantial contributions to the study conception and design. JA, EA, RA, DEA, AJB, GB, VHB, AB, RB, CB, PFB, JDB, JB-G, SBo, ADB, CC, PBSC-S, AC, AGC, ICC, EHC, HC, CTC, MLC, MJD, MVDeJ, SDeR, RD, ED, FD, AGE-S, MDF, BF, SEF, EF, CMF, LG, DHG, JGi, CGi, DEG, SG, HHa, JH, RH, IH-P, CMH, BI, SJ, SMK, AK, CL-A, ASLeC, DHL, CLM, CL, JKL, SMM, DMM, SGM, WMM, SEM, NM, DM-D-L, EMM, SM, MTM, JIN, GO, ATP, JRP, FP, AR, JR, WR, BR, GR, SJS, SSand, SLS, SWS, LS, MWS, HS, KS, SSt, JSS, OS, PS, KT, AMV, JASV, SW, RW, THW, LW, AJW, TWY, LZ. and HvanE contributed to acquisition and processing of clinical, biological and/ or genotype data. VA, JSB, NB, RMC, BD, APG, JGr, HHu, LK, PHL, BN, ADP, CSP, ER, VJV, and EMW contributed to processing of phenotype and genotype data. DAm, EB, ALB, TB, SBr, MB-H, JCa, JCo, GD, SE, MG, JMGr, CSH, DMH, ML, PL, EM, TM, PM, IM, AM, APM, OM, PBM, MN, AP, CBP, MGP, MP-M, MAP-V, JPi, RR, KRe, ES, GDS, TS, APT, CAW, TW, KW and contributed to acquisition of clinical, biological, and/ or genotype data. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study is a meta-analysis of mostly previously published data. All samples used in this study arose from investigations approved by the individual and respective Institutional Review Boards. Informed consent was obtained for all adult study participants; for children under age 18, both the consent of the parents or guardians and the assent of the child were obtained.
Ethical approval for the Autism Centre of Excellence (ACE) study was provided by the Institutional Review Boards at AGRE, UCLA, and Washington University. The PAGES study has been reviewed and approved by institutional review boards at the Karolinska Institutet, Icahn School of Medicine at Mount Sinai and Carnegie Mellon University. The Simons Simplex Collection study was overseen by the IRB at both Yale (HIC 0301024156) and UCSF (IRB 14-14726, Ref 146621). The data reported in this paper can be downloaded from SFARI Base (http://sfari.org/resources/sfari-base). The iPSYCH study has been approved by the Danish research ethical committee system. Additional ethical approval was provided in accordance with the ethical standards on human experimentation of the participating sites and has been described elsewhere for the Autism Genome Project [9, 12, 17, 75], the Autism Genetic Resource Exchange, NIMH Repository and Montreal Boston Collections [16], the Finnish Autism Case-Control Study; [76,77,78,79], the Weiss Laboratory Autism Collections [80,81,82,83], the SEED collection [84], and the deCODE collection [85].
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Consortia
Additional files
Additional file 1:
Supplementary Text, Figures and Tables. (DOCX 13.3 MB)
Additional file 2:
Summary GWAS finding for ASD Meta-analysis (World-wide Ancestries); Data reported on P < 10E-4. (XLS 99 KB)
Additional file 3:
Summary GWAS finding for ASD Meta-analysis ("European" Ancestries); Data reported on P < 10E-4. (XLS 119 KB)
Additional file 4:
Targetted Association Replication of Discovery Markers (ASD Meta-analysis (World-wide Ancestries): P <1e-4) in independent ASD cohorts. (XLS 5 MB)
Additional file 5:
Summary Gene-based association findings (VEGAS2) for ASD Meta-analysis (World-wide Ancestries); Data reported on ALL and P < .05. (SETS 0.97 MB)
Additional file 6:
INRICH-format gene-sets for "ASD Candidate Gene-sets". (SETS 39.2 MB)
Additional file 7:
INRICH-format gene-sets for "Canonical Gene-sets". (GENES 601 KB)
Additional file 8:
INRICH-format gene co-ordinates (based on hg19). (SETS 17 KB)
Additional file 9:
INRICH-format gene-set derived from "PGC Schizophrenia GWAS - PMID:25056061". (XLS 490 KB)
Additional file 10:
Full Results of Gene-set Enrichment findings (INRICH) for ASD Meta-analysis (World-wide Ancestries) versus Candidate.sets. (XLS 236 KB)
Additional file 11:
Full Results of Gene-set Enrichment findings (INRICH) for ASD Meta-analysis (World-wide Ancestries; no Swedish PAGES) versus PGCSchizophrenia2014.sets. (XLS 1.48 MB)
Additional file 12:
Full Results of Gene-set Enrichment findings (INRICH) for ASD Meta-analysis (World-wide Ancestries) versus Canonical.sets. (XLS 60 KB)
Additional file 13:
Partitioned Heritability analysis (LDSC) for ASD Meta-analysis (World-wide Ancestries). (XLS 119 KB)
Additional file 14:
Summary finding for ASD (World-wide Ancestries; no Swedish PAGES) and Schizophrenia (PMID: 25056061) Meta-analysis; Data reported on P ˂ 5E-8; all associations. (XLS 154 KB)
Additional file 15:
Summary finding for ASD (World-wide Ancestries; no Swedish PAGES) and Schizophrenia (PMID: 25056061) Meta-analysis; Data reported on P ˂ 5E-8; all associations not previously reported at P ˂ 5E-8 in ASD or SCZ. (XLS 28 KB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Molecular Autism 8, 21 (2017). https://doi.org/10.1186/s13229-017-0137-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-017-0137-9