
Abstract
Background
To evaluate the effects of fluticasone furoate on the hypothalamic–pituitary–adrenocortical axis, and the safety and tolerability of fluticasone furoate treatment in children with asthma.
Methods
This was a randomized, double-blind, placebo-controlled, multicenter, stratified, parallel-group, non-inferiority study of fluticasone furoate 50 µg inhalation powder administered once daily. The study enrolled children (aged 5–11 years inclusive) with a documented diagnosis of asthma for ≥ 6 months and a Childhood Asthma Control Test score of > 19. After a 7–14-day run-in period, eligible subjects were stratified by age and randomized to fluticasone furoate 50 µg once daily or placebo once daily via ELLIPTA for 6 weeks. The primary endpoint was the change from baseline (expressed as a ratio) in 0–24-h weighted mean serum cortisol at the end of the treatment period.
Results
Fifty-six randomized subjects received fluticasone furoate 50 µg once daily and 55 received placebo. The primary analysis was performed in the serum cortisol population (n = 104) and demonstrated that fluticasone furoate 50 µg once daily was non-inferior to placebo (ratio = 0.93; 95% confidence interval 0.8096, 1.0620), as the lower limit of the 95% confidence interval for the geometric mean treatment ratio of fluticasone furoate 50 µg once daily versus placebo was greater than 0.80. Findings from the intent-to-treat population (n = 111) were similar.
Conclusions
Six weeks of treatment with inhaled fluticasone furoate 50 µg once daily had no clinically relevant effect on the hypothalamic–pituitary–adrenocortical axis function of children, as measured by 24-h serum cortisol profiles. The primary analysis showed that fluticasone furoate 50 µg once daily was non-inferior to placebo. Fluticasone furoate 50 µg once daily was well tolerated and no new safety concerns emerged during the study.
Trial registration
This study is registered in ClinicalTrials.gov (NCT02483975). Date of submission: 25 June 2015.
Similar content being viewed by others


Background
Inhaled corticosteroids (ICS) are a mainstay of treatment in adults and children with persistent asthma of varying severity [1]. ICS are recommended by the Global Initiative for Asthma (GINA) guidelines in children aged 6–11 years with persistent asthma that is not sufficiently controlled by a short-acting β2-agonist (SABA) [1].
Although ICS are effective anti-inflammatory treatments of persistent asthma [1], their excessive or long-term use at high dosages is associated with hypothalamic–pituitary–adrenocortical (HPA) axis suppression in adults and children, potentially leading to adrenal insufficiency and in severe cases, adrenal crisis and death [2,3,4].
Fluticasone furoate (FF), delivered via the ELLIPTA inhaler (GlaxoSmithKline, Middlesex, UK), is an ICS that has been approved as a once-daily (QD) maintenance treatment for asthma prophylaxis in patients ≥ 5 years of age in the USA [5].
The safety of inhaled FF in combination with the long-acting β2-agonist vilanterol (VI) on the HPA axis in adult and adolescent (12–17 years of age) patients with asthma has been established [6]. A phase 2b, dose–response, efficacy and safety study showed that FF was effective and well tolerated in children aged 5–11 years with uncontrolled asthma [7]. Two randomized, double-blind, crossover studies of FF 100 µg or FF/VI 100/25 µg found no evidence of serum cortisol (SC) suppression in children (aged 5–11 years) with persistent asthma, suggesting this FF dose does not affect the HPA axis in this patient population [8, 9].
Nevertheless, data on the effects of FF on HPA axis suppression in children are limited. The primary objective of this study was to evaluate the effect of 6 weeks of treatment with QD inhaled FF 50 µg (FF 50 QD) on the HPA axis in children aged 5–11 years compared with placebo. Secondary objectives included safety and tolerability assessments and to determine the FF plasma concentration at 30 min post-dose after 6 weeks of treatment with FF 50 QD.
Methods
Study design
This was a randomized, double-blind, placebo-controlled, stratified, parallel-group, non-inferiority study (Fig. 1), conducted at 13 clinics in the USA and three clinics in South Africa.

Study design. Subjects received open-label montelukast during the run-in and treatment periods, and albuterol/salbutamol as needed to treat acute asthma symptoms. *Days 5–7 after the end of treatment. FF 50 QD fluticasone furoate 50 µg once daily
After a 7–14-day run-in period, eligible subjects were stratified by age (5–< 8 and 8–< 12 years) and randomized to FF 50 QD or placebo QD via ELLIPTA for 42 days (6 weeks; treatment period). The first dose of study drug was administered in the clinic under supervision at Visit 2 (randomization) after 24-h serum sampling and urine collection. Throughout the run-in and treatment periods, subjects received open-label montelukast. Albuterol/salbutamol was provided to use on an as-needed basis to treat acute asthma symptoms. Serum sampling and urine collection (both over the 24-h period) were repeated at Visit 5 (end of treatment). Blood samples were also collected to determine FF plasma levels at Visit 5 (pre-dose and 30 min post-dose).
The study protocol was approved by an ethics committee or institutional review board at each study site and conducted in accordance with the International Conference for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Good Clinical Practice (GCP) and applicable country-specific requirements, and the ethical principles of the Declaration of Helsinki. Protocol amendments after study initiation included clarifying the lower reference range and removing the upper reference range for the randomization exclusion criterion for SC. Informed written consent was obtained from at least one parent/caregiver along with assent from the subject. This study is registered in ClinicalTrials.gov (NCT02483975). Anonymized individual participant data and study documents can be requested for further research from http://www.clinicalstudydatarequest.com.
Study population
Eligible children were males and premenarchal females, aged 5–11 years (inclusive) and weighing ≥ 17 kg, with a documented diagnosis of asthma for at least 6 months prior to Visit 1 (run-in period) and a Childhood Asthma Control Test score of > 19. They had been receiving SABA alone or in combination with non-corticosteroid controller medication(s) for asthma management for at least 4 weeks prior to Visit 1. Exclusion criteria are provided in the online only content (Additional file 1: Methods S1).
Subjects were assigned to FF 50 QD or placebo at Visit 2 in accordance with a randomization schedule generated prior to study start using validated internal software. Randomization schedules were concealed until database freeze was achieved. Children were eligible for randomization if they generated a peak expiratory flow > 75% of their personal best measured in the last 7 days of the run-in period and completed 24-h SC assessments and a 24-h urine collection prior to randomization at Visit 2. Further details on treatment eligibility and criteria for withdrawal are provided in the online only content (Additional file 1: Methods S1).
Endpoints
The primary endpoint was the change from baseline (expressed as a ratio) in 0–24 h weighted mean SC at the end of the treatment period. Secondary endpoints were the change from baseline (expressed as a ratio) at the end of the treatment period in: area under the curve 0–24-h (AUC0–24) SC; 24-h urinary cortisol (UC) excretion; and 24-h 6-β hydroxycortisol excretion. Other endpoints included the change from baseline in trough SC at the end of the 6-week treatment period and FF plasma concentrations. The incidence of adverse events (AEs) was monitored throughout the study.
Pharmacodynamic assessments
Samples for SC and UC (free cortisol and 6-β hydroxycortisol) analysis were taken over 24-h periods at Visit 2 (start time between 7:00 am and 9:00 am) and Visit 5 (start time before 10:00 am). For SC, 2 mL blood samples were taken at 0, 2, 4, 8, 12, 16, and 24-h time points relative to sampling start time (24-h sampling was completed prior to administration of first dose at Visit 2; sampling was post-dose at Visit 5). SC was measured via an approved, validated high-performance liquid chromatography (HPLC) system in combination with a tandem mass spectrometer (MS/MS). The lower limit of quantification (LLQ) of the assay was 1 μg/dL (28 nmol/L). Further details of SC and UC assays are provided in the online only content (Additional file 1: Methods S1).
Pharmacokinetic assessments
Blood samples (2 mL) were taken for pharmacokinetic (PK) analysis at Visit 5, pre-dose and at 0.5-h post-dose. Plasma samples were analyzed for FF using solid phase extraction followed by HPLC–MS/MS analysis with the LLQ for FF 10 pg/mL.
Populations and statistical analyses
The intent-to-treat (ITT) population comprised all subjects randomized to treatment and who received at least one dose of study drug and was the primary population for the safety analyses. The SC population included all subjects in the ITT population with no protocol violations considered to affect the SC endpoint and whose serum samples were not considered to have confounding factors that would affect interpretation of the results. The decision to exclude a subject from the SC population was made prior to unblinding of the study. Analysis of the primary endpoint was based on the SC population, with the ITT population used for supportive analyses (with equal weighting). The UC population included all subjects who had no protocol violations that were considered to affect the UC endpoint and was the primary population for UC analyses. This population was also determined prior to unblinding of the study. The PK population comprised subjects in the ITT population for whom a PK sample was obtained and analyzed.
The SC weighted mean (0–24-h) was calculated by dividing the AUC0–24 by the time period. The ratio from baseline of the 0–24-h weighted mean SC concentration was loge transformed prior to analysis. The loge transformed ratio was compared between treatment groups (SC and ITT populations) using an analysis of covariance (ANCOVA) model, allowing for the effects of baseline (loge transformed), age, and sex. Treatment ratios for each comparison were calculated by back-transforming the difference between the least square means. Using the pooled estimate of variance, the 95% confidence interval (CI) was calculated for the difference and then back-transformed. Non-inferiority would be demonstrated if the lower limit of the (geometric mean ratio of FF 50 QD and placebo) two-sided 95% CI was greater than 0.80. Further information on study populations and statistical analyses is provided in the online only content (Additional file 1: Methods S1).
Results
Demographics and baseline characteristics
Of 156 subjects screened, 111 were randomized and received study drug (FF 50 QD, n = 56; placebo, n = 55; ITT population). Of these, 107 (96%) subjects completed the study (between 9 October, 2015 and 21 June, 2016) (Additional file 2: Figure S1), 104 (94%) subjects were included in the SC population, 108 (97%) subjects were included in the UC population and 106 (95%) were included in the PK population. Four (4%) subjects withdrew prematurely from the study treatment, primarily due to being lost to follow-up. Seven subjects were excluded from the SC population due to missing 24-h SC concentrations (n = 4) or study treatment compliance deviations (n = 3). Three subjects were excluded from the UC population owing to study treatment compliance deviations.
Demographics and baseline characteristics were generally comparable between groups (Table 1) and were similar between the SC and ITT populations. The mean age of the subjects was 8.4 years and 55% of the subjects were male (Table 1).
Exposure and treatment compliance
The mean exposure to study treatment (ITT population) was similar between groups (placebo: 42.4 days; FF 50 QD: 40.9 days). Most subjects received study treatment for > 35–≤ 42 days (FF 50 QD: 61%; placebo: 55%); the majority of the remainder of subjects were exposed to study treatment for > 42 days (FF 50 QD: 36%; placebo: 44%).
Mean compliance (according to the dose counter on the inhaler device; ITT population), was comparable between groups: placebo 106.7%; FF 50 QD 103.0%. Most subjects were within the compliance range of ≥ 80–≤ 120%, with only one subject in the FF 50 QD group demonstrating compliance below 80%.
Pharmacodynamics
Primary analysis
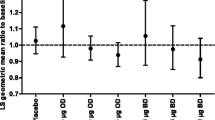
At week 6, the geometric mean for the derived SC weighted mean (0–24-h) was similar to the respective baseline values in both treatment groups (FF 50 QD group: baseline, 153.20 nmol/L and week 6, 157.52 nmol/L; placebo group: baseline, 177.22 nmol/L and week 6, 176.83 nmol/L; Additional file 3: Table S1). Analysis of the SC weighted mean (0–24-h) in the SC population demonstrated that FF 50 QD was non-inferior to placebo (ratio = 0.93 [95% CI 0.8096, 1.0620]; Table 2) since the lower limit of the 95% CI for the geometric mean treatment ratio of FF 50 QD versus placebo was greater than 0.80 (Fig. 2a). Findings from the ITT population were similar to those for the SC population and showed non-inferiority of FF 50 QD versus placebo (Table 2 and Additional file 3: Table S1). The geometric mean SC concentration—24-h time profiles were similar between treatment groups at baseline and week 6 (Fig. 2b).

a Adjusted treatment ratios for SC weighted mean (0–24 h) at week 6; b geometric mean (95% CI) SC concentration–time profile by treatment group and visit (SC population). CI confidence interval, FF 50 QD fluticasone furoate 50 µg once daily, h hours, SC serum cortisol
Secondary endpoints
The geometric mean SC AUC0–24 was similar at baseline and week 6 within both treatment groups (Additional file 3: Table S1). Based on the statistical analysis, the ratio for the change from baseline in SC AUC0–24 for FF 50 QD over placebo was 0.93 (95% CI 0.81, 1.06).
The geometric mean 24-h UC excretion and 6-β hydroxycortisol excretion (geometric mean) were also similar at baseline and week 6 within both treatment groups (Additional file 3: Table S1). The ratio of 24-h UC excretion for FF 50 QD over placebo was 0.79 (95% CI 0.61, 1.03) and that of 6-β hydroxycortisol excretion for FF 50 QD over placebo was 0.88 (95% CI 0.72, 1.07).
Other endpoints
At week 6, the geometric mean SC trough drug (24-h planned time point) was 315.51 nmol/L for the FF 50 QD group and 296.64 nmol/L for the placebo group (Additional file 4: Table S2). Based on the statistical analysis, the ratio of SC trough drug (24-h) for FF 50 QD over placebo from baseline was 1.10 (95% CI 0.90, 1.34).
A post hoc summary of the peak SC values observed over the 24-h period at baseline and week 6 determined the ratio for the change from baseline to be 1.03 in the FF 50 QD group and 1.10 in the placebo group (Additional file 4: Table S2).
Pharmacokinetics
Fifty-three subjects provided blood samples, with data available for 52 subjects at Visit 5. Plasma concentrations of FF were quantifiable in 4/52 subjects pre-dose and 35/52 subjects at 30 min post-dose. The median FF concentration was 12.05 pg/mL at 30 min post-dose (tmax; time to peak plasma concentration) and ranged from 0 to 41.7 pg/mL with a variance of 88%.
Safety
The proportion of subjects experiencing an AE (on- or post-treatment) in the FF 50 QD and placebo groups was 13% and 18%, respectively. While on treatment, the most commonly reported AEs were infections and infestations (7% of subjects in each group), followed by gastrointestinal disorders (no cases in the FF 50 QD group and 7% of subjects in placebo group). One case of tonsillitis (post-treatment) was reported in the FF 50 QD group. Only one drug-related AE was reported (gastritis; placebo group), and no serious AEs (SAEs) were reported during the study.
Three AEs of special interest were reported: one case of oropharyngeal pain in the FF 50 QD group and two cases (of oropharyngeal pain and allergic sinusitis, respectively) in the placebo group; all were deemed unrelated to the study drug.
Discussion
While ICS are established as an effective anti-inflammatory treatment for all severities of persistent asthma [1], their use is associated with a dose-related risk of HPA axis suppression [10, 11]. Although rare, HPA axis suppression may occur in children taking standard doses of corticosteroids [11, 12]. Diminished cortisol secretion by the adrenal cortex is an early sign of adrenal suppression and can occur within hours or days of high-dose corticosteroid administration [13,14,15]. Loss of the effects of adrenocorticotropic hormone (ACTH) on the adrenal gland in response to chronic use of exogenous corticosteroids results in adrenal atrophy and loss of function [16]. Evaluation of endogenous cortisol secretions is complicated due to the presence of circadian rhythms, with peak concentrations in the morning and trough concentrations around midnight [17]. Therefore, single time point measures are considered insensitive measures of HPA axis function [18]. The 24-h SC concentration time curve is the most sensitive method for detecting changes in cortisol production and diurnal variation after taking exogenous glucocorticoids [18].
The current study investigated the effects of FF inhalation powder on the HPA axis system of children, assessed through the pharmacodynamics (PD) of cortisol secretion. No clinical decrease in derived weighted mean SC was observed from baseline to week 6 in the FF 50 QD group; FF 50 QD was also non-inferior when compared with placebo. A suppressive effect on the HPA axis would have led to reduced cortisol secretion; therefore, the high peak SC values observed in a post hoc summary in this study suggest normal adrenal function in children undergoing treatment with FF 50 QD. Although not evaluated in this study, a potential suppressive effect of long-term administration of FF 100 QD on the HPA axis cannot be ruled out; however, this dose is not currently indicated for use in children aged 5–11 years [5].
The risk of adrenal insufficiency was shown in previous studies to increase with ICS dose and treatment duration [10], as the adrenal gland needs prolonged suppression (i.e., several weeks or months) before losing functionality [18]. The use of a standard ICS dose in the present study and the 6-week treatment period mean that the adrenal response to stress was likely unaffected, even though this does not exclude the risk of long-term adrenal failure after a normal ACTH test. However, given the sensitivity of cortisol secretion to exogenous steroids, the finding of undiminished cortisol secretion after 6 weeks of FF 50 QD is reassuring and suggests that the risk of adverse adrenal effects may not increase with long-term use of FF. However, it is acknowledged that there may be inter-individual variations in the sensitivity of the adrenal gland; this is a well-established class effect, and thus it is recommended that one should remain vigilant, especially in children receiving long-term treatment.
A wealth of recent data has shown that prolonged use (several months or years) of even low-to-medium ICS doses can lead to adrenal suppression [19,20,21,22]. The assessment of adrenal insufficiency was usually based on a single point morning cortisol or ACTH test [20,21,22]. As noted above, a single morning cortisol test is not considered sufficiently sensitive for measuring HPA axis function. In addition, some subjects may have taken oral corticosteroids (OCS), thus confounding the results. It is also possible that a minority of more susceptible individuals experienced an early suppressive effect. To our knowledge, there are no published studies showing the 24-h cortisol profiles of subjects with a documented normal profile after short-term ICS who underwent long-term treatment. Therefore, it is not clear whether the HPA axis response to low-dose ICS can become more sensitive over time, leading to loss of functionality.
In the current study, plasma FF concentrations were measured using pre-dose and 30 min post-dose blood samples, since the median tmax following inhaled administration of FF or FF/VI is around 0.5 h [5, 23]. An examination of the maximum concentration of FF achieved after dosing (Cmax) from other pediatric studies [7,8,9] indicated that the average Cmax obtained in the current study (~ 12 pg/mL) was dose proportional with the values reported in the literature. The median Cmax observed in this study was also consistent with the predicted average Cmax in the pediatric population PK analysis (11.6 pg/mL for FF 50 μg) [7].
By extrapolation, in the current study the predicted FF systemic exposure in terms of AUC was around 100 pg · h/mL. Established PK/PD models using nine studies of healthy subjects and adults and adolescents with asthma suggested that an AUC0–24 of 1000 pg h/mL would be required to reduce 24-h SC excretion by 20% and 24-h UC excretion by 17% [24]. Based on these data, no clinically relevant effects on cortisol levels were anticipated for the FF 50 μg dose, even in the smallest children and using the most conservative assumptions. The absence of an effect on serum cortisol in the present study confirmed these predictions, and based on these findings, FF 50 μg was anticipated to have an acceptable safety profile in children with persistent asthma aged 5–11 years.
The 24-h urine collection method is also reflective of cortisol production [18, 25] and it is widely used to assess HPA axis suppression in patients with asthma due to its non-invasive nature. However, UC measurements are mainly suitable for diagnosing oversecretion of cortisol (i.e., Cushing syndrome) rather than adrenal suppression for which assessment of SC is a more sensitive parameter. This is due to the variability of urine secretion as well as the fact that in adrenal suppression the lower levels of urinary free cortisol concentrations are not contributory to adrenal insufficiency [26].
In the present study, the FF 50 QD group had numerically lower UC levels compared with the placebo group. The ratio of FF 50 QD over placebo was 0.79 (95% CI 0.61, 1.03) for 24-h UC excretion and 0.88 (95% CI 0.72, 1.07) for 6-β hydroxycortisol excretion. Given the caveats associated with UC for the diagnosis of adrenal suppression, these results should not override the findings from SC measurements, which showed little difference between the FF 50 QD group and placebo. Furthermore, in this study, urine samples were only collected for 24-h rather than for 3 successive days, and the study was not statistically powered for assessing UC. However, as with all ICS, prescribers should remain vigilant in children treated with long-term FF.
Overall, FF 50 QD was well tolerated with no reported SAEs (fatal or non-fatal) or AEs leading to study withdrawal. There were no AEs consistent with adrenal insufficiency, and none of the three AEs of special interest reported were deemed related to study drug.
Strengths of the present study include the use of a robust methodology for assessing HPA axis function, high levels of exposure and compliance to study treatment, and a high number of complete 24-h profiles available. Domiciled clinic visits at the beginning and end of treatment were incorporated to standardize, control, and monitor the collection of blood and plasma parameters over 24 h. As part of the study protocol, peak SC values were captured in the morning in all children. Subject compliance with blinded inhaled medication was assessed by reviewing the dose counter on the inhaler.
The study has limitations. The 24-h SC concentration–time curve is not a good assessment of adrenal gland stress response, which should be measured using a dynamic function test such as the ACTH or the insulin tolerance test [27]. However, the gold standard insulin tolerance test is not usually carried out in pediatric patients due to safety considerations. The high-dose ACTH test is highly sensitive, although a positive result does not fully exclude the risk of adrenal failure. The low-dose ACTH test is more commonly used/preferred in clinical practice and is considered reliable for assessing the risk of adrenal crisis [28].
In addition, this study did not include an OCS (such as prednisolone) as a positive control due to ethical concerns related to corticosteroid administration in children younger than 12 years old at doses high enough to cause HPA axis suppression. This is consistent with other similar trials of ICS and intranasal corticosteroids in children. High peak SC levels may be explained by the study conditions (inpatient stay in a non-familiar setting) combined with the stress related to blood sampling in young children. Furthermore, the number of blood samples was limited in young children to assess more precisely the 24-h diurnal profile. Nevertheless, 24-h SC profiles were usually consistent between periods for individual subjects, with no cases of individual profiles showing a significant decrease in cortisol secretion at 6 weeks after treatment with FF by comparison with the baseline.
Conclusions
The results of this study indicate that 6 weeks of treatment with the therapeutic pediatric dose of FF 50 QD was non-inferior to placebo on the HPA axis function, as assessed by weighted mean 24-h SC production. These data do not show evidence of a clinically significant suppressive effect of FF 50 QD on the HPA axis after 6 weeks of treatment in children aged 5–11 years. However, given the differences in sensitivity to ICS between patients, prescribers should remain vigilant in regard to adrenal function, especially in young children on long-term treatment with FF.
Availability of data and materials
Anonymized individual participant data and study documents can be requested for further research from http://www.clinicalstudydatarequest.com.
Abbreviations
- ACTH:
-
adrenocorticotropic hormone
- AE:
-
adverse event
- ANCOVA:
-
analysis of covariance
- AUC:
-
area under the curve
- CI:
-
confidence interval
- Cmax :
-
maximum concentration
- CV:
-
coefficient of variation
- FF:
-
fluticasone furoate
- FF 50 QD:
-
FF 50 µg once daily
- GINA:
-
Global Initiative for Asthma
- GCP:
-
Good Clinical Practice
- h:
-
hour
- HPA:
-
hypothalamic–pituitary–adrenocortical
- HPLC:
-
high-performance liquid chromatography
- ICH:
-
International Conference for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
- ICS:
-
inhaled corticosteroid
- ITT:
-
intent-to-treat
- LLQ:
-
lower limit of quantification
- LS:
-
least squares
- Max.:
-
maximum
- Min.:
-
minimum
- MS/MS:
-
tandem mass spectrometer
- OCS:
-
oral corticosteroid
- PD:
-
pharmacodynamic
- PK:
-
pharmacokinetic
- QD:
-
once daily
- SABA:
-
short-acting β2-agonist
- SAE:
-
serious adverse event
- SC:
-
serum cortisol
- SD:
-
standard deviation
- tmax :
-
time to peak plasma concentration
- UC:
-
urinary cortisol
- VI:
-
vilanterol
References
Global Initiative for Asthma. Global strategy for asthma management and prevention. Updated 2018. https://ginasthma.org/wp-content/uploads/2019/01/2018-GINA.pdf. Accessed Jan 2020.
Zollner EW. Hypothalamic–pituitary–adrenal axis suppression in asthmatic children on inhaled corticosteroids: part 1. Which test should be used? Pediatr Allergy Immunol. 2007;18:401–9.
Smith RW, Downey K, Gordon M, Hudak A, Meeder R, Barker S, et al. Prevalence of hypothalamic–pituitary–adrenal axis suppression in children treated for asthma with inhaled corticosteroid. Paediatr Child Health. 2012;17:e34–9.
Todd GR, Acerini CL, Ross-Russell R, Zahra S, Warner JT, McCance D. Survey of adrenal crisis associated with inhaled corticosteroids in the United Kingdom. Arch Dis Child. 2002;87:457–61.
GSK. ARNUITY_ELLIPTA Prescribing Information. 2018. https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Arnuity_Ellipta/pdf/ARNUITY-ELLIPTA-PI-PIL-IFU.PDF. Accessed Aug 2018.
Allen A, Schenkenberger I, Trivedi R, Cole J, Hicks W, Gul N, et al. Inhaled fluticasone furoate/vilanterol does not affect hypothalamic–pituitary–adrenal axis function in adolescent and adult asthma: randomised, double-blind, placebo-controlled study. Clin Respir J. 2013;7:397–406.
Oliver AJ, Covar RA, Goldfrad CH, Klein RM, Pedersen SE, Sorkness CA, et al. Randomized trial of once-daily fluticasone furoate in children with inadequately controlled asthma. J Pediatr. 2016;178(246–53):e242.
Oliver A, Allen A, VanBuren S, Hamilton M, Tombs L, Kempsford R, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of fluticasone furoate, a novel inhaled corticosteroid, in children aged 5–11 years with persistent asthma: a randomized trial. Clin Pharmacol Drug Dev. 2014;3:144–50.
Oliver A, VanBuren S, Allen A, Hamilton M, Tombs L, Inamdar A, et al. Tolerability of fluticasone furoate/vilanterol combination therapy in children aged 5 to 11 years with persistent asthma. Clin Ther. 2014;36(928–39):e921.
Lipworth BJ. Systemic adverse effects of inhaled corticosteroid therapy: a systematic review and meta-analysis. Arch Intern Med. 1999;159:941–55.
Rao Bondugulapati LN, Rees DA. Inhaled corticosteroids and HPA axis suppression: how important is it and how should it be managed? Clin Endocrinol. 2016;85:165–9.
Kapadia CR, Nebesio TD, Myers SE, Willi S, Miller BS, Allen DB, et al. Endocrine effects of inhaled corticosteroids in children. JAMA Pediatr. 2016;170:163–70.
Henzen C, Suter A, Lerch E, Urbinelli R, Schorno XH, Briner VA. Suppression and recovery of adrenal response after short-term, high-dose glucocorticoid treatment. Lancet. 2000;355:542–5.
Mortimer KJ, Tata LJ, Smith CJ, West J, Harrison TW, Tattersfield AE, et al. Oral and inhaled corticosteroids and adrenal insufficiency: a case–control study. Thorax. 2006;61:405–8.
Chrousos G, Pavlaki AN, Magiakou MA. Glucocorticoid therapy and adrenal suppression. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, editors. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000.
De Groot LJ. Endocrine glands. Philadelphia: W. B. Saunders Company; 2001.
Raff H, Sharma ST, Nieman LK. Physiological basis for the etiology, diagnosis, and treatment of adrenal disorders: Cushing's syndrome, adrenal insufficiency, and congenital adrenal hyperplasia. Compr Physiol. 2014;4:739–69.
Lipworth BJ, Seckl JR. Measures for detecting systemic bioactivity with inhaled and intranasal corticosteroids. Thorax. 1997;52:476–82.
Zollner EW, Lombard CJ, Galal U, Hough FS, Irusen EM, Weinberg E. Hypothalamic–pituitary–adrenal axis suppression in asthmatic school children. Pediatrics. 2012;130:e1512–9.
Sim DW, Choi IS, Kim S-H. Suppressive effects of long-term treatment with inhaled steroids on hypothalamic–pituitary–adrenal axis in asthma. Allergy Asthma Respir Dis. 2014;2:285–92.
Choi IS, Sim DW, Kim SH, Wui JW. Adrenal insufficiency associated with long-term use of inhaled steroid in asthma. Ann Allergy Asthma Immunol. 2017;118(66–72):e61.
Woods CP, Argese N, Chapman M, Boot C, Webster R, Dabhi V, et al. Adrenal suppression in patients taking inhaled glucocorticoids is highly prevalent and management can be guided by morning cortisol. Eur J Endocrinol. 2015;173:633–42.
GSK. BREO_ELLIPTA Prescribing Information. 2017. https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Breo_Ellipta/pdf/BREO-ELLIPTA-PI-PIL-IFU.PDF. Accessed Aug 2018.
Allen A. The relationship between fluticasone furoate systemic exposure and cortisol suppression. Clin Pharmacokinet. 2013;52:885–96.
Brown PH, Blundell G, Greening AP, Crompton GK. Screening for hypothalamo-pituitary-adrenal axis suppression in asthmatics taking high dose inhaled corticosteroids. Respir Med. 1991;85:511–6.
Charmandari E, Nicolaides NC, Chrousos GP. Adrenal insufficiency. Lancet. 2014;383:2152–67.
Agwu JC, Spoudeas H, Hindmarsh PC, Pringle PJ, Brook CG. Tests of adrenal insufficiency. Arch Dis Child. 1999;80:330–3.
Tordjman K, Jaffe A, Trostanetsky Y, Greenman Y, Limor R, Stern N. Low-dose (1 microgram) adrenocorticotrophin (ACTH) stimulation as a screening test for impaired hypothalamo-pituitary-adrenal axis function: sensitivity, specificity and accuracy in comparison with the high-dose (250 microgram) test. Clin Endocrinol. 2000;52:633–40.
Acknowledgements
Medical writing and editorial assistance in the form of developing an initial draft based on author input and editorial assistance were provided by Madhuri Vemuri and Katy Tucker of Fishawack Indicia Ltd, UK, which was funded by GlaxoSmithKline (GSK).
Funding
This study (NCT02483975, Protocol number 107118) was funded by GSK.
Author information
Authors and Affiliations
Contributions
PB, ST, VI, RM, and SK were involved in the study conception or design, and the analysis or interpretation of data. MT and KD were involved in the acquisition of data. All authors have revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by an ethics committee or institutional review board at each study site (Pharma-Ethics, Centurion, Gauteng, South Africa; Schulman Associates IRB Inc., Cincinnati, OH, USA, and Research Triangle Park, NC, USA). The study was conducted in accordance with the International Conference for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Good Clinical Practice (GCP) and applicable country-specific requirements, and the ethical principles of the Declaration of Helsinki. Informed written consent was obtained from at least one parent/caregiver along with assent from the subject. This study is registered in ClinicalTrials.gov (NCT02483975).
Consent for publication
Not applicable.
Competing interests
PB, ST, VI, RM, and SK are employees of and hold shares in GSK. KD and MT have no competing interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Additional methods.
Additional file 2: Figure S1.
Subject disposition.
Additional file 3: Table S1.
Derived SC weighted mean (0–24 h) (SC and ITT population), geometric mean SC AUC0–24 (SC population), geometric mean 24-h UC excretion (UC population), and geometric mean 6-β hydroxycortisol excretion (UC population).
Additional file 4: Table S2.
Change from baseline in peak SC (post hoc summary) and trough SC (24-h planned time point) at week 6 (SC population).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bareille, P., Tomkins, S., Imber, V. et al. A randomized, double-blind, placebo-controlled, parallel-group study of once-daily inhaled fluticasone furoate on the hypothalamic–pituitary–adrenocortical axis of children with asthma. Allergy Asthma Clin Immunol 16, 11 (2020). https://doi.org/10.1186/s13223-020-0406-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13223-020-0406-6