
Abstract
Background
Subclinical infection with bovine leukemia virus (BLV) in cows can cause economic losses in milk and meat production in many countries, as BLV-related negative effects. The volatile fatty acids (VFAs) and microbiota present in the digestive tracts of cows can contribute to cow health. Here, we exploratorily investigated the VFAs and microbiota in the rumen and gut with respect to subclinical BLV infection using cows housed at a single farm.
Results
We analyzed a herd of 38 cows kept at one farm, which included 15 uninfected and 23 BLV-infected cows. First, the analysis of the VFAs in the rumen, gut, and blood revealed an absence of statistically significant differences between the uninfected and BLV-infected groups. Thus, BLV infection did not cause major changes in VFA levels in all tested specimens. Next, we analyzed the rumen and gut microbiota. The analysis of the microbial diversity revealed a modest difference between the uninfected and BLV-infected groups in the gut; by contrast, no differences were observed in the rumen. In addition, the investigation of the bacteria that were predominant in the uninfected and BLV-infected groups via a differential abundance analysis showed that no significant bacteria were present in either of the microbiota. Thus, BLV infection possibly affected the gut microbiota to a small extent. Moreover, bacterial associations were compared between the uninfected and BLV-infected groups. The results of this analysis suggested that BLV infection affected the equilibrium of the bacterial associations in both microbiota, which might be related to the BLV-related negative effects. Thus, BLV infection may negatively affect the equilibrium of bacterial associations in both microbiota.
Conclusions
Subclinical BLV infection is likely to affect the rumen and gut microbiota, which may partly explain the BLV-related negative effects.
Similar content being viewed by others


Background
Bovine leukemia virus (BLV) is a viral agent of the Deltaretrovirus genus in the Retroviridae family that infects cattle. BLV is prevalent globally, with the exception of countries in which viral eradication of this agent was achieved. Because about 70% of BLV-infected cattle are asymptomatic, the control of infection with this virus is extremely difficult (LaDronka et al. 2018; Marawan et al. 2021; Murakami et al. 2011). About 30% of BLV-infected cattle develop persistent lymphocytosis, and about 1–5% can develop malignant B-cell lymphosarcoma after a prolonged latency (1–8 years), which is a malignancy that is also known as enzootic bovine leucosis (Aida et al. 2013). Cows diagnosed with this condition are eliminated from commercial distribution, which can cause significant economic damage. Moreover, subclinical BLV infection in cows, which consists of an asymptomatic infection over their lifetime without the development of enzootic bovine leucosis, also appears to cause economic losses, to a greater extent than that expected, as the BLV-related negative effects caused by subclinical infection include increased susceptibility to other infections and a decrease in lifetime milk production, meat production, and reproductive efficiency in cows (Brenner et al. 1989; Nakada et al. 2023; Nekouei et al. 2016; Polat et al. 2017; Schwartz and Levy 1994; Yang et al. 2016). However, the physiological effects of subclinical BLV infection in cows remain unknown, although subclinical BLV infection is believed to cause a systemic impairment of the immune system of these animals (Konnai et al. 2017). Thus, the effects of subclinical BLV infection on the body of cows should be investigated.
The microbiota of digestive tracts, particularly the rumen and gut microbiota, play an essential role in maintaining health in cattle (Cholewinska et al. 2020; Welch et al. 2022). The fermentation of cellulose and other complex plant carbohydrates by the rumen microbiota is essential in cattle because these animals do not possess digestive enzymes to break down these complex carbohydrates. About 70% of the energy requirements of cattle and about 50% of their protein requirements necessitate microbial fermentation in the rumen (Bergman 1990; Pitta et al. 2018). In cattle, rumen microbes become a source of protein, and microbial metabolites, such as volatile fatty acids (VFAs), are a source of energy via gluconeogenesis from acetyl-CoA, as well as a source of fats. VFAs are alternatively termed short-chain fatty acids. The major VFAs produced by rumen fermentation are acetic, propionic, butyric, and valeric acids. The amount and ratio of these VFAs in the rumen are important for maintaining cattle health because, although acetic acid is essential for the biosynthesis of body fat and milk fat and for providing energy, an excessive amount of butyric acid leads to the generation of ketone bodies, thus inducing ketosis (Miettinen and Huhtanen 1996). Moreover, further down the intestinal tract, the hindgut—which is more generally called the gut—is another important intestinal locale. VFAs are also produced by microbes in the gut (Yao et al. 2022), where they not only function as an energy source for gut epithelial cells but also regulate the function of innate immune cells, such as macrophages, neutrophils, and dendritic cells, and regulate the differentiation of T and B cells (Yao et al. 2022). Thus, the rumen and gut microbiota and the VFAs generated by them are important for cattle health.
Infections by some viruses are beginning to be shown to modulate the microbiota of digestive tracts (Yuan et al. 2020). With respect to viruses for which such phenomena have not been examined, exploratory studies, which are required in the early stages of research, are warranted. One exploratory study alone using cows in one farm reported that the gut microbiota of cows can be modulated by BLV infection, even at the subclinical stage (Uchiyama et al. 2020). However, integrative studies of the VFAs and microbiota of the digestive tract with respect to subclinical BLV infection are scarce, to the best of our knowledge. Here, we exploratorily investigated the VFAs in the rumen fluid (rumen), feces (gut), and serum (blood) of uninfected and asymptomatic BLV-infected cows housed at a single farm and examined the rumen and gut microbiota in the two groups using 16S rRNA gene amplicon sequencing.
Results
Enrolled cows
We conducted the study in one commercial farm in Japan, which kept 70 cows, including 42 lactating cows. These 42 lactating cows were housed in the same tie-stall barn. We performed the sampling within 1 day during the autumn season. The cows were fed the same diet. All cows were clinically healthy. We collected the samples within 1 day but were unable to collect samples from four cows because the sampling period coincided with the milking time. Finally, 38 cows were enrolled in the present study.
Among these 38 cows (age, 24–101 months (mean ± standard deviation, 51.7 ± 19.9 months)), 23 were diagnosed as having subclinical BLV infection. The mean ages of the uninfected and BLV-infected cows were not significantly different (46.3 ± 22.9 months and 55.3 ± 16.8 months, respectively; Mann–Whitney U-test, P > 0.05).
VFA status in the rumen, gut, and blood in the uninfected and BLV-infected groups
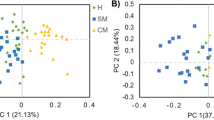
To examine the effect of subclinical BLV infection, we first compared the VFAs in the rumen, gut, and blood of the uninfected group with those of the BLV-infected group using a multivariate analysis of variance (MANOVA) and a principal component analysis (PCA). There were no significant differences in the rumen, gut, and blood between the uninfected and BLV-infected groups (MANOVA, P ≥ 0.05). In addition, the comparison of the levels of each VFA (e.g., acetic acid) between the uninfected and BLV-infected groups revealed an absence of significant differences in the rumen, gut, and blood (Mann–Whitney U-test, P ≥ 0.05; Supplementary Table S1). A nonsignificant difference in the level of isovaleric acid in the gut was observed between the uninfected and BLV-infected groups (Mann–Whitney U-test, P < 0.1; Supplementary Table S1). Moreover, there were no distinct observational differences in the in rumen, gut, and blood PCA score plots between uninfected and BLV-infected cows (Fig. 1).

PCA score plots of VFAs in A rumen, B gut, and C blood. The uninfected and BLV-infected groups are shown as dots of different colors, which are coded in the bottom box. The eclipses with 95% reliability are also plotted on each graph, which are represented in the same color as the dot color
Overview of the rumen and gut microbiota of uninfected and BLV-infected cows
The rumen and gut microbiota of the 38 cows were analyzed using 16S rRNA gene amplicon sequencing. A total of 4,915,395 reads (129,353 ± 44,757 reads/sample) and 5,009,119 reads (131,819 ± 43,641 reads/sample) were obtained from rumen and gut samples, respectively. Read trimming and exclusion of chimeric reads of the rumen and gut samples were performed using the DADA2 software (Callahan et al. 2016), which produced a total of 1,843,202 reads (48,505 ± 19,152 reads/sample) and 1,954,142 reads (51,425 ± 17,723 reads/sample), respectively. The relative abundance of the bacterial community at the phylum level was calculated using these data. In both the rumen and gut microbiota, Bacteroidota and Firmicutes were predominantly present, at about 40‒50% (Fig. 2). The other major phyla, Proteobacteria, Patescibacteria, Actinobacteriota, Planctomycetota, Spirochaetota, and Euryarchaeota (Archaea), were present at a relative abundance of less than about 7% in the rumen microbiota, whereas Verrucomicrobiota, Spirochaetota, and Proteobacteria were present at a relative abundance of less than about 6% in the gut microbiota.

Relative abundance at the phylum level of taxonomy. The average bacterial abundances in all cows are shown in taxa bar plots. A Rumen and B gut. The bacterial taxa are indicated using different colors, as shown on the right of the taxa bar plots
Analyses of microbial diversities and differentially abundant microbes in the rumen and gut microbiota between the uninfected and BLV-infected groups
Microbial diversity was compared between the uninfected and BLV-infected groups using the alpha and beta-diversity metrics (Table 1); the alpha-diversity metrics were evenness and the Shannon and Chao1 indices, whereas the beta-diversity metrics were the weighted and unweighted UniFrac metrics. First, in the rumen analysis, no significant differences in alpha or beta diversity were observed. Similarly, in the fecal analysis, no significant differences were observed for either of the metrics. However, nonsignificant differences were observed in the gut: a lower evenness value was obtained in the BLV-infected vs. the uninfected group (P < 0.1); similarly, there was a difference in unweighted UniFrac values (P < 0.1). Moreover, to gain further insight, we performed a differential abundance analysis of the rumen and gut microbiota at the genus level using an analysis of the composition of microbiome (ANCOM) (Mandal et al. 2015). No bacterial taxa exhibited significant differences in abundance between the uninfected and BLV-infected groups (Supplementary Fig. S1).
Comparison of microbial co-occurrence patterns in the rumen and gut microbiota between the uninfected and BLV-infected groups
The importance of bacterial associations in microbiota has recently been suggested. These associations can be inferred from metataxonomic data via a co-occurrence network analysis (Dai et al. 2019; Matchado et al. 2021). The analysis of the bacterial associations related to BLV infection in the rumen and gut using a co-occurrence network analysis revealed that the overall patterns of the bacterial association networks in the rumen and gut microbiota were topologically different between the uninfected and BLV-infected groups (Supplementary Fig. S2). The comparison of the co-occurrence networks of the uninfected group with those of the BLV-infected group led to the detection of differential bacterial associations. Because of the complexity of the differential networks derived from all detected differential bacterial associations, representative differential networks in the rumen and gut microbiota were simply constructed using the nine differential bacterial associations with the highest weights (Fig. 3).

Differential networks between the uninfected and the BLV-infected groups. A Rumen and B gut. In the networks, each node shows a bacterium at genus level. The types of edges are described on the bottom dotted box. The taxonomic units at genus level (bacterial classifications at family level) shown in A are as follows: Lachnospiraceae_UCG-002 (Lachnospiraceae), horsej-a03 (Oligosphaeraceae), uncultured (Bdellovibrionota), Moraxella (Moraxellaceae), Incertae_Sedis (Ruminococcaceae), Z20 (Oligosphaeraceae), Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium (Rhizobiaceae), Asteroleplasma (Erysipelatoclostridiaceae), Caviibacter (Leptotrichiaceae), Bergeyella (Weeksellaceae), Rikenellaceae_RC9_gut_group (Rikenellaceae), Christensenellaceae_R-7_group (Christensenellaceae), Brevibacterium (Brevibacteriaceae), possible_genus_Sk018 (Lachnospiraceae), Methanomicrobium (Methanomicrobiaceae), Lachnospiraceae_NC2004_group (Lachnospiraceae), and Megasphaera (Veillonellaceae). The taxonomic units (bacterial classifications) shown in Fig. 3B are as follows: Bradymonadales (Bradymonadales), Lysinibacillus (Planococcaceae), Methanocorpusculum (Methanocorpusculaceae), Howardella (Lachnospiraceae), Mogibacterium (Anaerovoracaceae), Megasphaera (Veillonellaceae), Raoultibacter (Eggerthellaceae), Hydrogenoanaerobacterium (Oscillospiraceae), Catenisphaera (Erysipelotrichaceae), uncultured (Verrucomicrobiota), Sanguibacteroides (Marinifilaceae), [Clostridium]_methylpentosum_group (Oscillospirales), [Eubacterium]_xylanophilum_group (Lachnospiraceae), Anaerofustis (Anaerofustaceae), uncultured (Actinobacteriota), Syntrophococcus (Lachnospiraceae), and Kandleria (Erysipelatoclostridiaceae)
In the differential network of the rumen microbiota, seven and two differential bacterial associations were detected that were predominant in the uninfected and the BLV-infected groups, respectively (Fig. 3A). The seven associations that were predominant in the uninfected group included Christensenellaceae–Rikenellaceae, Lachnospiraceae–Oligosphaeraceae, Bdellovibrionota–Moraxellaceae, Ruminococcaceae–Oligosphaeraceae, Rhizobiaceae–Erysipelatoclostridiaceae, Leptotrichiaceae–Weeksellaceae, and Brevibacteriaceae–Lachnospiraceae. The two associations that were predominant in the BLV-infected group included Lachnospiraceae–Methanomicrobiaceae and Lachnospiraceae–Veillonellaceae.
In the differential network of the gut microbiota, five and four differential bacterial associations were predominant in the uninfected and the BLV-infected groups, respectively (Fig. 3B). The five associations that were predominant in the uninfected group included Erysipelotrichaceae–Verrucomicrobiota, Bradymonadales–Planococcaceae, Methanocorpusculaceae–Lachnospiraceae, Anaerovoracaceae–Veillonellaceae, and Eggerthellaceae–Oscillospiraceae. Finally, the four associations that were predominant in the BLV-infected group included Verrucomicrobiota–Marinifilaceae, Oscillospirales–Lachnospiraceae, Anaerofustaceae–Actinobacteriota, and Lachnospiraceae–Erysipelatoclostridiaceae.
Discussion
This was an exploratory study for the integrative analysis of VFAs in the rumen, gut, and blood, as well as the rumen and gut microbiota, with respect to subclinical BLV infection, using cows housed at a single farm. No estimation of sample size was performed prior to sample collection in this study. However, we believe that the herd yielded the appropriate BLV-related results with two reasons. First, the size of the herd used in this study was similar to that of a previous exploratory study (i.e., 22 uninfected and 29 BLV-infected cows) (Uchiyama et al. 2020). Second, the confounding factors were considered to be minimized as much as possible. As described in the previous study (Uchiyama et al. 2020), because factors such as diet, host phenotype, genetics, age, environment, and health status affect the VFAs and microbiota of digestive tracts (Gupta et al. 2020; Kurilshikov et al. 2021; Na and Guan 2022; Wang and LeCao 2020), we collected samples from cows housed in the same environment (i.e., housing style and feeding management) over 1 day. Of note, because older cows generally have more opportunities to be exposed to BLV infection, the age of BLV-infected cows is older than that of uninfected cows. The age of the animals in the two groups was not significantly different, although the average age of the cows in the BLV-infected group was 9 months older than that of those in the uninfected group.
Using the herd, we first compared the VFAs of the rumen, gut, and blood between the uninfected and BLV-infected groups based on statistics and the observation of PCA plots. The uninfected and BLV-infected groups did not differ in the VFAs of the rumen, gut, and blood. Thus, a drastic change in the VFA status was not likely to be triggered by subclinical BLV infection.
We then investigated the rumen and gut microbiota. The composition and abundance of the major bacterial taxa in the rumen and gut microbiota in this study appeared to be similar to those reported for other cows (Li et al. 2021; Uchiyama et al. 2020), which suggests that these cows had typical rumen and gut microbiota. In our subsequent analysis of microbial diversity, the gut microbiota appeared to be minimally altered by BLV infection, whereas the rumen microbiota was hardly altered by BLV infection. The small change observed in the gut microbiota after BLV infection was similar to the results of a previous study (Uchiyama et al. 2020). In addition, we performed a differential abundance analysis with respect to BLV infection in the rumen and gut microbiota. No bacteria in association with BLV infection were detected. Thus, the gut was very slightly affected by BLV infection, whereas the rumen hardly appeared to be affected by it.
Next, the impact of BLV infection on the equilibrium of bacterial associations was examined via a differential network analysis of the rumen and gut microbiota. Because of the detection of differential bacterial associations in both microbiota, subclinical BLV infection was likely to trigger a disequilibrium in the bacterial associations in the rumen and gut microbiota. We predicted the functions of bacterial associations from the bacteria present in the differential networks. First, the differential network of the rumen microbiota was examined. Among the associations that were predominant in the BLV-infected group, the associations between methanogenic bacteria (Methanomicrobiaceae) with cellulose-degrading bacteria (Lachnospiraceae), and between cellulose-degrading bacteria (Lachnospiraceae) and VFA-producing bacteria (Veiollonellaceae), were assessed. Among the associations that were predominant in the uninfected group, cellulose-degrading bacteria and VFA-producing bacteria appeared to form associations. Based on our observation, the cooperation of methane-producing bacteria with cellulose-degrading bacteria may be more likely to occur in the rumen microbiota of BLV-infected groups. The rumen methanogenic bacteria reduce the feed efficiency by releasing some of the energy in the feed as methane (Martinez-Fernandez et al. 2016). Thus, BLV infection may reduce energy production efficiency in the rumen microbiota at an undetectable low level over a long period. Moreover, the differential network of the gut microbiota was examined. Among the associations that were predominant in the BLV-infected or uninfected group, the functions of most bacteria are hardly deducible based solely on bacterial classification. However, cellulose-degrading bacteria (Lachnospiraceae) and VFA-producing bacteria (Veiollonellaceae) were detected in the associations that were predominant in the uninfected group. The VFAs produced by gut microbiota play an important role in host physiology by maintaining homeostasis with the host’s immune system (Welch et al. 2022). Thus, the fermentation in the uninfected group that is required for VFA production may have been imbalanced by BLV infection at an undetectable low level over a long period. Considering these assumptions of differential bacterial associations, we may grasp hints that the disequilibrium of the rumen and gut microbiota caused by BLV infection is related to the BLV-related negative effects associated with subclinical infection.
In this study, we observed that subclinical BLV infection appeared to slightly affect the gut microbiota while having hardly any effect on the rumen microbiota; it did not cause a major change in the VFA composition of blood and digestive tract. Subclinical BLV infection appeared to affect the equilibrium of the bacterial associations in both the rumen and gut microbiota for a long period, particularly the gut microbiota. Considering the importance of the rumen and gut in the health status of cows together with the results of this study, the digestive tract may become a target for the management of BLV-related negative effects. Because this was an exploratory study with low statistical power, our results warrant replication across larger samples based on sample size calculation to confirm the phenomena in the future.
Conclusions
This study investigated not only the VFAs in the rumen, gut, and blood but also the rumen and gut microbiota with respect to subclinical BLV infection. Subclinical BLV infection did not cause major changes in either the VFA levels or the microbiota of the digestive tract. However, the microbiota in the digestive tract, particularly the gut, might be in a slight disequilibrium, which implies a potential relation to BLV-related negative effects. In conclusion, subclinical BLV infection is likely to affect the rumen and gut microbiota, which may partly explain the BLV-related negative effects.
Methods
Animals
One dairy farm located in Tokyo, Japan, which was maintained by one owner, kept 70 cows, including 42 lactating cows. These 42 lactating cows were at different lactation stages and were kept in the same tie-stall barn. These lactating cows were fed a mixture of 39.0% (w/w) roughage, 59.8% concentrate feed, and 1.2% supplements. Veterinarians interviewed farm owners about animals with changes in milk production or abnormal symptoms. After checking the rectal temperature, auscultation was used to check the heart rate, rumen, pinging test, respiratory rate, adventitious sounds, and abnormal respiratory sounds. Other tests were performed to check for external abnormalities, to evaluate the locomotor system in cases of lameness, and to check for abnormalities in the pelvic cavity and female genitalia via rectal examination, to ensure that all cows were in good clinical condition. Cows that were diagnosed as clinically healthy after successful sampling were enrolled in this study.
Samples
Blood samples were collected from the tail veins. In this study, the enrolled cows were commercial production cows, and rumen fluid was not collected by ruminocentesis, to avoid the potential risk of peritonitis. Rumen fluids were collected using a pump after a hose was placed in the mouth of the animal through to the rumen. After 200 mL of the first fluid was discarded, to avoid saliva contamination, 10 mL of rumen fluid was collected into sterilized plastic tubes. Fecal material near the anus was discarded during the rectal palpation, and fecal material in the deep rectum was collected. Immediately after collection, the fecal samples were placed in sterile polypropylene tubes with filter caps (CELLSTAR CELLreactor; Greiner Bio-One, Rainbach im Mühlkreis, Austria) under anaerobic conditions using the AnaeroPack Kenki system (Mitsubishi Gas Chemical Company, Japan). All samplings were performed on September 2, 2019.
Immediately after collection, the samples were temporally stored and transported within a day at 4 °C for blood samples and − 20 °C for rumen fluid and fecal samples. The samples were then stored in the laboratory at 4 °C (blood) and − 80 °C (rumen fluid, feces).
Diagnosis of BLV infection
One day after blood sample collection, sera were prepared from the blood by centrifugation. Genomic DNA was extracted from blood cells using a Wizard Genomic DNA Purification Kit (Promega, Madison, USA). BLV infection was diagnosed using the serum as described previously via both agar gel immunodiffusion assays and detection of BLV proviral genomes (Murakami et al. 2016).
Measurement of VFAs
Ruminal VFAs were analyzed by gas chromatography (GC). After centrifugation of the rumen fluids, 20 μL of 85% phosphoric acid was added to about 1 mL of the supernatant and incubated overnight at 4 °C. After centrifugation (13,000 × g, 10 min, 4 °C) and filtration through an 0.45-μm filter, 2 μL of sample was injected into a 30 m × 0.25-mm FFAP capillary column with a 0.25-μm film thickness (Agilent Technologies, Germany) fitted to a 7820A GC system (Agilent) with flame ionization detection. The GC operation conditions were as follows. Helium was used as the carrier gas with a constant pressure of 57 kPa. Sample injection was carried out in split mode (10:1), with an injection volume of 1 μL and an injector temperature of 250 °C. The initial oven temperature was set to 60 °C and increased to 240 °C in steps of 10 °C/min and then 20 °C/min to 325 °C.
Fecal and serum VFAs were analyzed by gas chromatography–mass spectrometry (GC–MS) after derivatization (tert-butyldimethylsilylation). Fecal samples (50 mg) and serum samples (450 μL) were made to 500 μL with Milli-Q water. To these samples, 5 μL of internal standard (crotonic acid, 200 μg/mL) was added followed by vortexing with 10-μL 50% sulfosalicylic acid to precipitate protein. After centrifugation at 13,000 × g for 10 min, the deproteinized samples were extracted with diethyl ether/hydrochloric acid (1:0.003, v/v) by shaking for 30 min. Subsequently, 8 μL of 1-tert-butyldimethylsilylimidazole was added and allowed to react at 60 °C for 30 min. Two microliters of the mixture were analyzed by GC–MS on a 7890A chromatograph equipped with automated sample injection (MPS from GERSTEL, Germany), a 30 m × 0.25 mm DB-5 ms capillary column with a 0.25-μm film thickness (Agilent), and a 5975B mass spectral detector (Agilent). The mass spectrometer was used in the electrospray ionization mode with an ion source temperature of 250 °C; the temperature of the quadrupole and mass spectral interface was 150 °C and 290 °C, respectively. The mass spectral detector was used in the selected ion monitoring mode, to search for acetic, propionic, butyric, valeric, and crotonic acids, which have m/z values of 159.0, 131.0, 145.0, 159.0, and 143.0, respectively.
Statistical analysis
The JMP Pro 16.2.0 software (SAS Institute, USA) was used for the statistical analysis of cattle age and VFA levels.
DNA extraction and sequencing
DNA was extracted from rumen and fecal samples using an ISOSPIN Fecal DNA Kit (Nippon Gene, Japan). The extracts were subjected to 16S rRNA gene amplicon sequencing, in which the V3–V4 region of the 16S rRNA gene, amplified by polymerase chain reaction, was analyzed using an MiSeq Reagent Kit v3 in 600 cycles in an Illumina MiSeq instrument (Illumina, USA), as described elsewhere (Uchiyama et al. 2022).
Microbiota data analysis
The sequence data were processed using the Quantitative Insights into Microbial Ecology 2 (QIIME2) pipeline v. 2022.2.0 (Bolyen et al. 2019). The DADA2 software package v2021.8.0 incorporated in QIIME 2 was used to correct the amplicon sequence errors and to construct an amplicon sequence variant table (Callahan et al. 2016). The amplicon sequence variant table was rarefied. Microbial taxonomy was assigned using a Naïve-Bayes classifier trained on the SILVA 138 99% database.
Metrics of alpha diversity, including Pielou’s evenness (Evenness) index, Shannon’s (Shannon) index, and the Chao1 index, and those of beta diversity, including unweighted UniFrac and weighted UniFrac, were calculated and statistically analyzed using the QIIME 2 pipeline. A differential abundance analysis was performed using ANCOM implemented in the QIIME 2 pipeline.
Networks were analyzed with the NetCoMi package in the R software version 1.0.2 and in the R software version 4.0.2 using the SPIEC-EASI metric for network construction (Peschel et al. 2021). Associations were estimated via the SPRING approach. The associations in the networks were then compared using the discordant method.
Availability of data and materials
Raw sequence data were deposited in the DNA Data Bank of Japan (DDBJ) sequence read archive (DRA014764) under the BioProject no. PRJDB14276.
Abbreviations
- BLV:
-
Bovine leukemia virus
- VFAs:
-
Volatile fatty acids
- MANOVA:
-
Multivariate analysis of variance
- PCA:
-
Principal component analysis
- ANCOM:
-
Analysis of composition of microbiome
- GC:
-
Gas chromatography
- GC–MS:
-
Gas chromatography–mass spectrometry
- QIIME2:
-
Quantitative Insights into Microbial Ecology 2
References
Aida Y, Murakami H, Takahashi M, Takeshima SN (2013) Mechanisms of pathogenesis induced by bovine leukemia virus as a model for human T-cell leukemia virus. Front Microbiol 4:328. https://doi.org/10.3389/fmicb.2013.00328
Bergman EN (1990) Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev 70:567–590. https://doi.org/10.1152/physrev.1990.70.2.567
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodriguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS 2nd, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vazquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. https://doi.org/10.1038/s41587-019-0209-9
Brenner J, Van-Haam M, Savir D, Trainin Z (1989) The implication of BLV infection in the productivity, reproductive capacity and survival rate of a dairy cow. Vet Immunol Immunopathol 22:299–305. https://doi.org/10.1016/0165-2427(89)90017-2
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Cholewinska P, Czyz K, Nowakowski P, Wyrostek A (2020) The microbiome of the digestive system of ruminants - a review. Anim Health Res Rev 21:3–14. https://doi.org/10.1017/S1466252319000069
Dai W, Chen J, Xiong J (2019) Concept of microbial gatekeepers: positive guys? Appl Microbiol Biotechnol 103:633–641. https://doi.org/10.1007/s00253-018-9522-3
Gupta VK, Kim M, Bakshi U, Cunningham KY, Davis JM 3rd, Lazaridis KN, Nelson H, Chia N, Sung J (2020) A predictive index for health status using species-level gut microbiome profiling. Nat Commun 11:4635. https://doi.org/10.1038/s41467-020-18476-8
Konnai S, Murata S, Ohashi K (2017) Immune exhaustion during chronic infections in cattle. J Vet Med Sci 79:1–5. https://doi.org/10.1292/jvms.16-0354
Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, Le Roy CI, Raygoza Garay JA, Finnicum CT, Liu X, Zhernakova DV, Bonder MJ, Hansen TH, Frost F, Ruhlemann MC, Turpin W, Moon JY, Kim HN, Lull K, Barkan E, Shah SA, Fornage M, Szopinska-Tokov J, Wallen ZD, Borisevich D, Agreus L, Andreasson A, Bang C, Bedrani L, Bell JT, Bisgaard H, Boehnke M, Boomsma DI, Burk RD, Claringbould A, Croitoru K, Davies GE, van Duijn CM, Duijts L, Falony G, Fu J, van der Graaf A, Hansen T, Homuth G, Hughes DA, Ijzerman RG, Jackson MA, Jaddoe VWV, Joossens M, Jorgensen T, Keszthelyi D, Knight R, Laakso M, Laudes M, Launer LJ, Lieb W, Lusis AJ, Masclee AAM, Moll HA, Mujagic Z, Qibin Q, Rothschild D, Shin H, Sorensen SJ, Steves CJ, Thorsen J, Timpson NJ, Tito RY, Vieira-Silva S, Volker U, Volzke H, Vosa U, Wade KH, Walter S, Watanabe K, Weiss S, Weiss FU, Weissbrod O, Westra HJ, Willemsen G, Payami H, Jonkers D, Arias Vasquez A, de Geus EJC, Meyer KA, Stokholm J, Segal E, Org E, Wijmenga C, Kim HL, Kaplan RC, Spector TD, Uitterlinden AG, Rivadeneira F, Franke A, Lerch MM, Franke L, Sanna S, D’Amato M, Pedersen O, Paterson AD, Kraaij R, Raes J, Zhernakova A (2021) Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet 53:156–165. https://doi.org/10.1038/s41588-020-00763-1
LaDronka RM, Ainsworth S, Wilkins MJ, Norby B, Byrem TM, Bartlett PC (2018) Prevalence of bovine leukemia virus antibodies in US dairy cattle. Vet Med Int 2018:5831278. https://doi.org/10.1155/2018/5831278
Li Y, Kreuzer M, Clayssen Q, Ebert MO, Ruscheweyh HJ, Sunagawa S, Kunz C, Attwood G, Amelchanka S, Terranova M (2021) The rumen microbiome inhibits methane formation through dietary choline supplementation. Sci Rep 11:21761. https://doi.org/10.1038/s41598-021-01031-w
Mandal S, Van Treuren W, White RA, Eggesbo M, Knight R, Peddada SD (2015) Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis 26:27663. https://doi.org/10.3402/mehd.v26.27663
Marawan MA, Alouffi A, El Tokhy S, Badawy S, Shirani I, Dawood A, Guo A, Almutairi MM, Alshammari FA, Selim A (2021) Bovine leukaemia virus: current epidemiological circumstance and future prospective. Viruses 13:2167. https://doi.org/10.3390/v13112167
Martinez-Fernandez G, Denman SE, Yang C, Cheung J, Mitsumori M, McSweeney CS (2016) Methane inhibition alters the microbial community, hydrogen flow, and fermentation response in the rumen of cattle. Front Microbiol 7:1122. https://doi.org/10.3389/fmicb.2016.01122
Matchado MS, Lauber M, Reitmeier S, Kacprowski T, Baumbach J, Haller D, List M (2021) Network analysis methods for studying microbial communities: a mini review. Comput Struct Biotechnol J 19:2687–2698. https://doi.org/10.1016/j.csbj.2021.05.001
Miettinen H, Huhtanen P (1996) Effects of the ratio of ruminal propionate to butyrate on milk yield and blood metabolites in dairy cows. J Dairy Sci 79:851–861. https://doi.org/10.3168/jds.S0022-0302(96)76434-2
Murakami K, Kobayashi S, Konishi M, Kameyama K, Yamamoto T, Tsutsui T (2011) The recent prevalence of bovine leukemia virus (BLV) infection among Japanese cattle. Vet Microbiol 148:84–88. https://doi.org/10.1016/j.vetmic.2010.08.001
Murakami H, Uchiyama J, Nikaido S, Sato R, Sakaguchi M, Tsukamoto K (2016) Inefficient viral replication of bovine leukemia virus induced by spontaneous deletion mutation in the G4 gene. J Gen Virol 97:2753–2762. https://doi.org/10.1099/jgv.0.000583
Na SW, Guan LL (2022) Understanding the role of rumen epithelial host-microbe interactions in cattle feed efficiency. Anim Nutr 10:41–53. https://doi.org/10.1016/j.aninu.2022.04.002
Nakada S, Fujimoto Y, Kohara J, Makita K (2023) Economic losses associated with mastitis due to bovine leukemia virus infection. J Dairy Sci 106:576–588. https://doi.org/10.3168/jds.2021-21722
Nekouei O, VanLeeuwen J, Stryhn H, Kelton D, Keefe G (2016) Lifetime effects of infection with bovine leukemia virus on longevity and milk production of dairy cows. Prev Vet Med 133:1–9. https://doi.org/10.1016/j.prevetmed.2016.09.011
Peschel S, Muller CL, von Mutius E, Boulesteix AL, Depner M (2021) NetCoMi: network construction and comparison for microbiome data in R. Brief Bioinform 22:bbaa290. https://doi.org/10.1093/bib/bbaa290
Pitta DW, Indugu N, Baker L, Vecchiarelli B, Attwood G (2018) Symposium review: understanding diet-microbe interactions to enhance productivity of dairy cows. J Dairy Sci 101:7661–7679. https://doi.org/10.3168/jds.2017-13858
Polat M, Takeshima SN, Aida Y (2017) Epidemiology and genetic diversity of bovine leukemia virus. Virol J 14:209. https://doi.org/10.1186/s12985-017-0876-4
Schwartz I, Levy D (1994) Pathobiology of bovine leukemia virus. Vet Res 25:521–536
Uchiyama J, Murakami H, Sato R, Mizukami K, Suzuki T, Shima A, Ishihara G, Sogawa K, Sakaguchi M (2020) Examination of the fecal microbiota in dairy cows infected with bovine leukemia virus. Vet Microbiol 240:108547. https://doi.org/10.1016/j.vetmic.2019.108547
Uchiyama J, Osumi T, Mizukami K, Fukuyama T, Shima A, Unno A, Takemura-Uchiyama I, Une Y, Murakami H, Sakaguchi M (2022) Characterization of the oral and faecal microbiota associated with atopic dermatitis in dogs selected from a purebred Shiba Inu colony. Lett Appl Microbiol 75:1607–1616. https://doi.org/10.1111/lam.13828
Wang Y, LeCao KA (2020) Managing batch effects in microbiome data. Brief Bioinform 21:1954–1970. https://doi.org/10.1093/bib/bbz105
Welch CB, Ryman VE, Pringle TD, Lourenco JM (2022) Utilizing the gastrointestinal microbiota to modulate cattle health through the microbiome-gut-organ axes. Microorganisms 10:1391. https://doi.org/10.3390/microorganisms10071391
Yang Y, Fan W, Mao Y, Yang Z, Lu G, Zhang R, Zhang H, Szeto C, Wang C (2016) Bovine leukemia virus infection in cattle of China: association with reduced milk production and increased somatic cell score. J Dairy Sci 99:3688–3697. https://doi.org/10.3168/jds.2015-10580
Yao Y, Cai X, Fei W, Ye Y, Zhao M, Zheng C (2022) The role of short-chain fatty acids in immunity, inflammation and metabolism. Crit Rev Food Sci Nutr 62:1–12. https://doi.org/10.1080/10408398.2020.1854675
Yuan L, Hensley C, Mahsoub HM, Ramesh AK, Peng R, Zhou P (2020) Microbiota in viral infection and disease in humans and farm animals. In: Sun J (ed) Progress in Molecular Biology and Translational Science, vol 171. Elsevier. Cambridge, MA, USA, pp 15–60
Acknowledgements
We thank the undergraduate students, School of Veterinary Medicine, Azabu University, for experimental assistance. We especially thank Anicom Pafe, Inc. for the next-generation sequencing and online English for English proofreading.
Funding
This study is funded by Grants-in-Aid for the Promotion of the Livestock Industry from the Japan Racing Association in 2021.
Author information
Authors and Affiliations
Contributions
TS, HM, JU, and RS designed the study. TS, HM, JU, RS, IT, and MO collected the samples. TS, HM, JU, and IT performed the experiments, analyzed the data, and wrote the manuscript. All authors reviewed and critically revised the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study farm was willing to cooperate with us. Our care and handling of the animals followed the Azabu University Animal Experiment Guidelines, which are in accordance with the ARRIVE guidelines. All experiments complied with the guidelines of the Ethics Committee of Azabu University, which reviewed and approved this study (approval number: 17113–3).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
ANCOM volcano plots. The centered log ratio (clr) on the horizontal axis is the clr representative of the difference in the abundance of a given taxonomical unit at the genus level between the uninfected and the BLV-infected groups. The W value on the vertical axis indicates the value of the statistical test corresponding to the number of times the null hypothesis was rejected for each taxonomical unit. The positive and negative values on the horizontal axis indicate the abundance of bacteria in the uninfected and BLV-infected groups, respectively. (A) Rumen and (B) gut microbiota. Each dot represents one bacterial taxonomy unit at the genus level; dots with W = 1 are labeled. Fig. S2. Microbial co-occurrence network of (A) rumen and (B) gut microbiota. The 150 genera with the highest variance were selected from each group, and the co-occurrence networks of the uninfected and BLV-infected groups were constructed using the 100 edges with the highest weights. The networks of uninfected and BLV-infected groups are shown on the left and right, respectively. Each node is a bacterial taxonomic unit at genus level. The node positions in the left and right networks are the same, and nodes in the same position indicate the same bacterial species. The clusters were inferred using the Louvain method, which are shown by different node colors. All the edges shown in the networks are positive associations, shown by green lines. Table S1. Measurement of metabolites in rumen, gut and blood. Supplementary protocol 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Suzuki, T., Murakami, H., Uchiyama, J. et al. Exploratory study of volatile fatty acids and the rumen-and-gut microbiota of dairy cows in a single farm, with respect to subclinical infection with bovine leukemia virus. Ann Microbiol 73, 31 (2023). https://doi.org/10.1186/s13213-023-01737-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13213-023-01737-4