
Abstract
Purpose
Anabaena sp. PCC7120 is a genetically tractable model organism for nitrogen fixation and photosynthesis research. The importance of small regulatory RNAs (sRNAs) as mediators of a number of cellular processes in bacteria has begun to be recognized. Bacterial sRNA binds to target genes through base pairing, and play a regulatory role. Many studies have shown that bacterial sRNA can regulate cell stress response, carbon and nitrogen fixation, and so on. However, little is known about sRNAs in Anabaena sp. PCC 7120 regarded to nitrogen fixation under later steady state.
Methods
To provide a comprehensive study of sRNAs in this model organism, the sRNA (< 200 nt) extracted from Anabaena sp. PCC 7120 under nitrogen step-down treatment of 12 days, together with the sRNA from the control, was analyzed using deep RNA sequencing. Possible target genes regulated by all identified putative sRNAs were predicted by IntaRNA and further analyzed for functional categorizations for biological pathways.
Result
Totally, 14,132 transcripts were produced from the de novo assembly. Among them, transcripts that are located either in the intergenic region or antisense strand were kept, which resulted in 1219 sRNA candidates, for further analysis. RPKM-based differential expression analysis showed that 418 sRNAs were significantly differentially expressed between the samples from control (nitrogen addition, N+) and nitrogen depletion, (N−). Among them, 303 sRNAs were significantly upregulated, whereas 115 sRNAs were significantly downregulated. RT-PCR of 18 randomly chosen sRNAs showed a similar pattern as RNA-seq result, which confirmed the reliability of the RNA-seq data. In addition, the possible target genes regulated by unique sRNAs of Anabaena sp. PCC 7120 under nitrogen addition (N+) condition or that under nitrogen depletion (N−) condition were analyzed for functional categorization and biological pathways, which provided the evidences that sRNAs were indeed involved in many different metabolic pathways.
Conclusion
The information from the present study provides a valuable reference for understanding the sRNA-mediated regulation of the nitrogen fixation in Anabaena PCC 7120 under steady state conditions.
Similar content being viewed by others


Introduction
Cyanobacteria are a group of photoautotrophs originated nearly 3.5 billion years ago, during which they have evolved a diverse array of metabolic capabilities (Carr and Whitton 1973; Herrero and Flores 2008; Kaushik et al. 2016; Srivastava et al. 2017). They have been studied as prokaryotic hosts for sustainable biofuel production due to their high photosynthetic efficiency, genetic manipulability, and diverse metabolic pathways (Dismukes et al. 2008; Ducat et al. 2011; Halfmann et al. 2014b; Halfmann et al. 2014a; Chen et al. 2015; Burnat et al. 2018; Singh et al. 2019; Ishikawa et al. 2019). Some filamentous cyanobacteria, including Anabaena PCC strain 7120, are able to differentiate heterocyst to fix atmospheric dinitrogen in response to nitrogen deficiency (Flores and Herrero 2010; Kumar et al. 2010; Xu et al. 2015; Esteves-Ferreira et al. 2018; Olmedo-Verd et al. 2019). Understanding of the underlying mechanisms of nitrogen fixation and diazotrophic growth in cyanobacteria will shed light on basic mechanisms of bacterial genetic regulation and physiology character. Besides, it can help the catalog of genetically modified cyanobacterial strains to increase and produce renewable chemicals and biofuels.
In Anabaena PCC 7120, 5 to 10% vegetative cells along the filament differentiate into heterocysts in a semi-regular pattern after deprivation of combined nitrogen (Maldener and Muro-Pastor 2001). Metabolite exchange occurs between the heterocysts and neighboring vegetative cells, that is, vegetative cells supply fixed carbon, such as sucrose, to adjacent heterocysts, while heterocysts provide vegetative cells with fixed nitrogen in the form of amino acids (López-Igual et al. 2010; Vargas et al. 2010; Brenes-Álvarez et al. 2019). It is known that the heterocysts differentiate from vegetative cells, involving substantial changes in cell morphology and physiology that result in a micro-oxic environment for the expression and function of oxygen-sensitive nitrogenase (Wolk et al. 2004; Flores and Herrero 2010). The development of heterocysts is mainly regulated by two transcription factors, NtcA and HetR (Buikema and Haselkorn 1991; Herrero et al. 2004; Zhao et al. 2010; Chen et al. 2011; Herrero et al. 2013). Deprivation of combined nitrogen leads to the accumulation of 2-oxogluatarate which is the backbone for nitrogen assimilation (Laurent et al. 2005). The increased cellular levels of 2-oxoglutarate in consequence activates NtcA to initiate the heterocysts differentiation (Zhao et al. 2010). The hetR gene is required for an early step of heterocyst differentiation, whenever under N− conditions or N+ conditions (Buikema and Haselkorn 1991).
Small regulatory RNAs (sRNAs) are known to be key genetic regulators in many organisms including bacteria, plants, and animals (Babski et al. 2014; Morris and Mattick 2014; Higo et al. 2017; Lambrecht et al. 2018). They perform many important regulatory functions, such as in stress response, regulation of virulence genes, carbon source uptake, and metabolism (Gottesman 2005; Gottesman and Storz 2011). In bacteria, sRNAs typically range from 50 to 200 nt in length (Storz et al. 2011), and are often encoded in intergenic regions in trans to their target genes or in cis on the opposite strand of their target gene (Babski et al. 2014). The former ones usually interact with their target mRNAs near the ribosomal binding site (RBS) through imperfect base-pairing. The latter ones have full complementarity to their target mRNA. The interactions between sRNA and its targeted mRNA can result in gene translational repression by masking the RBS or activation by making the RBS accessible, as well as affecting mRNA degradation and mRNA stability (Storz et al. 2011; Babski et al. 2014).
sRNAs in cyanobacteria are frequently implicated in photosynthesis, stress responses, and nitrogen fixation (Nakamura et al. 2007; Hernández et al. 2010; Ionescu et al. 2010; Gong and Xu 2012; Georg et al. 2014; Muro-Pastor 2014; Klähn et al. 2015; de Porcellinis et al. 2016; Higo et al. 2018). For example, in Synechocystis sp. PCC 6803, the small RNA PsrR1 is a regulatory factor controlling photosynthetic functions. In filamentous cyanobacterium Anabaena PCC 7120, furA, the gene for the ferric uptake regulator, is interfered by a cis-acting antisense RNA whose knockout mutation results in an iron deficiency phenotype (Hernández et al. 2006; Hernández et al. 2010). In unicellular cyanobacterium Synechocystis, the antisense RNA IsrR regulates the expression of iron stress-induced protein IsiA in a co-degradation mechanism under iron stress (Dühring et al. 2006).
Examples of sRNA involved in nitrogen fixation in cyanobacterium are nitrogen stress-induced RNAs (NsiR). NsiR1 is the first known bacterial non-coding RNA specifically upregulated in response to nitrogen step-down, and its expression requires NtcA and HetR (Ionescu et al. 2010; Mitschke et al. 2011a), it has been suggested to be an early marker for cells undergoing differentiation (Muro-Pastor 2014). NsiR2 and NsiR3 that were identified in a differential RNA-seq analysis to Anabaena PCC 7120 annotated an unknown function (Mitschke et al. 2011a). NsiR4 was firstly found to be strongly upregulated in Synechocystis sp. PCC 6803 under nitrogen depletion (Mitschke et al. 2011b; Kopf et al. 2014). Later, the ortholog in Anabaena PCC 7120 was found to play an important role in the regulation of glutamine synthetase, a key enzyme in biological nitrogen assimilation (Klähn et al. 2015). Recently, using the Northern blot, a study showed that NsiR8 and NsiR9 were strongly induced in the WT control, but not in a HetR mutant subjected to a 24-h nitrogen step-down treatment, suggesting they could be related to heterocysts differentiation or function (Brenes-Álvarez et al. 2016).
Although many sRNAs related with nitrogen fixation have been identified in Anabaena PCC 7120 using RNA sequencing-based approaches (Flaherty et al. 2011; Mitschke et al. 2011a). They were all identified from cells at the early stage (1–24 h) of nitrogen depletion. However, up to now, there has not been any report on the characterization of the sRNA regulation profiles in Anabaena PCC 7120 under steady state after nitrogen step-down. In this study, we have employed the RNA-seq method to analyze the sRNA transcripts (< 200 nt) in Anabaena PCC 7120 after 12 d treatment of nitrogen step-down. By applying this approach, 1219 sRNA candidates were identified. Among them, 1124 were shared by the control of nitrogen addition (N+) and the treatment of nitrogen depletion (N−); 46 ones were unique in the N− group and 49 in the control group. Possible target genes regulated by all identified putative sRNAs were predicted by IntaRNA and further analyzed for functional categorizations for biological pathways. Overall, our results provide a new insight towards understanding the complex regulatory network of sRNAs in Anabaena sp. PCC 7120 after combined nitrogen depletion under steady state conditions.
Materials and methods
Strains, growth conditions, and sample preparations
Anabaena PCC 7120 was grown in AA/8 medium with KNO3 and NaNO3 as the combined nitrogen source (Allen and Arnon 1955) and incubated at 30 °C with shaking at 120 rpm in a shaker under continuous white-light (ca. 50 μE m−2 s−1) illumination. To conduct the nitrogen step-down treatment, Anabaena PCC 7120 grown in AA/8(N+) medium were collected by centrifugation at 5000 × g for 10 min and washed 3 times in AA/8 nitrate free medium AA/8(N−). Then cells were resuspended and grown in 300-ml AA/8(N−) medium until OD700 was around 0.5. The nitrogen step-down treatments were repeated 4 times. After culturing 12 d, cells were harvested by centrifugation at 5000 × g for 10 min, cell pellets were quickly frozen in liquid nitrogen and stored at − 80 °C for later use. The control group was prepared similarly as the nitrogen-deprived samples except grown in AA/8(N+) medium.
RNA isolation and sequencing
Total RNA from Anabaena sp. PCC 7120 strain samples cultured on media of AA/8(N+) and AA/8(N−) were separately extracted using TRIzol® reagent (INVITROGEN, Carlsbad, CA, USA). The Anabaena sp. PCC 7120 strain samples above cultured on media of AA/8(N+) and AA/8(N−) were mixtures of three repeated samples. RNA quantity and quality including concentration, RIN value, and 23S/16S ratio were assessed using Agilent 2100 Bioanalyzer (Agilent Technologies). Samples with high quality of 260/280 and 260/230 ratios were selected for Illumina sequencing library construction. RNA samples were treated with DNase I (Promega), and 3 μg of total RNA from each sample was electrophoresed by agarose gel electrophoresis for RNA fractionation, then the electrophoretic bands of 18–200 nt RNA segments were excised from the PAGE gel. The library was constructed according to Illumina TruSeqTM Small RNA Sample Preparation protocol. The sequencing run was conducted on an Illumina HiSeq™ 2000 platform at the Beijing Genome Institute, Shenzhen, China. The deep sequencing data have been submitted to the NCBI Sequence Read Archive with the accession number SUB1682364.
Sequence data processing, assembly, and annotation
The raw reads generated from the high-throughput sequencing were firstly cleaned by removing adaptor sequences. Then sequences were filtered off based on low quality value (Q20 ratio > 40%) and the high N numbers in the reads (> 10%).Then, all the clean reads from all replications were merged and subjected to de novo assembly using the Trinity program (Haas et al. 2013). The assembled transcripts were mapped onto the Anabaena sp. PCC 7120 genome and its six megaplasmids using BWA (Li and Durbin 2009). For the annotation of candidate sRNAs, we searched databases of sRNAMap (Huang et al. 2009), sRNATarBase (Cao et al. 2010), SIPHI (http://newbio.cs.wisc.edu/sRNA/), and BSRD (Li et al. 2012) based on the similarity of sequences using Blast (e value < 0.00001).
Identification of differentially expressed sRNA
The RPKM method (reads per kilo base per million mapped reads) was used for length normalization and calculation of the transcript expression levels (Mortazavi et al. 2008). An FDR (false discovery rate) of < 0.001 was used as the threshold p value in multiple tests to judge the degree of differences in gene expression (Reiner et al. 2003). In this study, the sRNA differential expression between two groups was considered when the p value was less than 0.001 and the expression level was at least a two-fold change between the two groups.
Target prediction
To obtain the target information, we searched the target genes of all the putative sRNAs by using the program IntaRNA (Busch et al. 2008; Wright et al. 2014) to take the sRNAs as query with default parameters. GO annotations with the default parameters were performed with the functional annotation tool WEIGO (Ye et al. 2006). All putative sRNA/target pairs were subsequently filtered by correlation analysis of the sRNA and transcript expression. Only sRNA/target pairs with strong negative correlation were retained as putatively sRNA regulated transcripts.
qRT-PCR validation
Total RNA was treated with RNase-free DNase I (FERMENTAS, Life Sciences, Germany), then it was used as a template to synthesize first-strand cDNA using a One Step SYBR PrimeScript RT-PCR kit (TAKARA) following the manual. We selected 18 differentially expressed sRNA for qRT-PCR analysis to evaluate our Illumina sequencing result. The sRNA primers were designed using Primer 5 and listed in Table 1. The expression level of the 18 sRNA was analyzed by using the comparative CT method (2–ΔΔCT method). The housekeeping gene rrn16Sa in Anabaena sp. PCC7120 was used as the internal control for normalization (Pinto et al., 2012), and used the Mix of AceQTM qPCR SYBR® Green Master to establish the qRT-PCR reaction system. qRT-PCR reactions were carried out by using BIO-RAD CFX96 in three biological repeats for every treatment. Primers, reaction systems, and programs are listed in Additional file. GraphPad Prism 6 was applied to analysis and construction.
Results
Strain growth observations on different media
When cultured on different media, filament cells of Anabaena sp. PCC 7120 showed different growth, heterocysts differentiated directly on AA/8(N−) medium (Fig. 1a, c) after 3 d culturing; however, only the vegetative cells grown on AA/8(N+) medium (Fig. 1b, d). It is obvious that nitrogen step-down of the medium induced the heterocysts differentiation.

Filamentous cells of Anabaena sp. PCC 7120 on different media after 3 d culturing. a and c Show the cells on AA/8(N−) medium, arrow heads indicate the heterocysts. b and d Show the vegetative cells on AA/8(N+) medium. Bars = 10 μm
sRNA assembly and functional annotation
To comprehensively identify sRNA involved in nitrogen fixation at a steady state, we compared the sRNAs profile (18–200 nt) of Anabaena sp. PCC 7120 grown on media of AA/8(N+) and AA/8(N−) after 12 d nitrogen step-down treatment. A total of 13,219,856 clean reads were obtained from the control group (N+), and 13,778,584 reads from the nitrogen-depleted group (N−); while Q ≥ 20. Totally, 14,132 transcripts were produced from the de novo assembly. These assembled transcripts were then mapped onto the Anabaena sp. PCC 7120 genome (92.96%) and its 6 megaplasmids (7.04%). The mapped transcripts were classified into 4 groups: group I contains sRNAs that were partially overlapped to mRNA (PM, 6.68%); group II comprises intergenic sRNAs that were mapped to an intergenic region (IGR, 24.24%); group III were referred to as antisense sRNAs which were located antisense to known genes (AM, 29.76%); and group IV were those located within mRNAs (IM, 39.32%) (Fig. 2). Transcripts that were placed into either group II or III, with a total expression both in N+ and N− media bigger than 20 were kept as candidate sRNAs; thus, a total of 1219 candidate sRNAs were selected for further analysis (supplementary Table 1). It should be noted that 157 candidate sRNAs were matched to the registered sRNAs by searching the databases of sRNAMap (Huang et al. 2009), sRNATarBase (Cao et al. 2010), SIPHI (http://newbio.cs.wisc.edu/sRNA/), and BSRD (Li et al. 2012), indicating the reliability of our method. As more as 83.1% of the candidate sRNAs was 20–80 nt; the size distribution of the candidate sRNAs is shown in Supplementary Fig. 1. Among all the sRNA candidates, 1124 were shared by the nitrogen-depleted group and the control group; 46 were unique in the nitrogen-depleted group and 49 in the control group.

a Illustration diagram of the four classes of assembled transcripts. PM partially overlap to mRNA, represented by nc-01, nc-02; IGR intergenic region, represented by nc-03; IM included in mRNA, represented by nc-04; AM antisense to mRNA, represented by nc-05, nc-06. b Pie chart clarifying the sRNA transcripts. The percentages of all the deep sequencing transcripts mapped to the previously annotated or non-annotated regions of the Anabaena sp. PCC 7120 genome
sRNA differential expression
We identified differentially expressed sRNAs between the control (N+) and nitrogen depletion treatment (N−) groups by comparing the relative transcript abundance using the RPKM-based method (supplementary Table 2). Principally, any sRNA that has a value of (|log2Ratio| ≥ 1 and FDR ≤ 0.001) was kept as significantly differentially expressed sRNA. Totally, 418 sRNA were found to be differentially expressed between the control (N+) and nitrogen-depleted (N−) samples (supplementary Table 3), with 303 (72.49%) being significantly upregulated and 115 (27.51%) downregulated. Among them, 323 sRNA were differentially expressed in both samples; 46 sRNA were only expressed in control (N+) sample; and 49 sRNA were only expressed in nitrogen-depleted (N−) sample.
By adding different adaptors to the 3′ and 5′ ends of each RNA molecule in the sample prior to cDNA synthesis, RNA-seq analysis here enabled us to distinguish the direction of each transcription. In total, 110 antisense RNAs were found and 29 of them were significantly differentially expressed between the (N−) and (N+) groups (Table 2). In addition, 1109 intergenic region sRNAs were found and 389 were significantly differentially expressed (Table 3). We further characterized these antisense sRNAs and intergenic region sRNAs by retrieving putative target gene function information from the cyanobase (Table 2 and Table 3).
RT-PCR validation
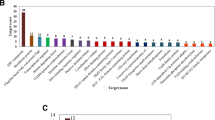
For validation of the Illumina sequencing results, a number of 18 sRNA were randomly chosen for qRT-PCR analysis. The qRT-PCR results showed in Fig. 3, 11 sRNA showed significant differences in the relative expression between N+ and N− groups, and 7 sRNAs were in a significantly upregulated trend in N− group compared with N+ group, while 4 sRNAs were significantly downregulated in N− group. Similar trends were observed for the expression of these 18 sRNA analyzed by the 2 methods, the biggest discrepancy was around 7-fold difference in sRNA1133, and the lowest was around 0.5-fold difference in sRNA0703 (Fig. 3). However, the relative expression results from these 2 analyses did not match perfectly, perhaps due to sequencing biases or different normalization controls.

Comparison of the expression profiles of selected genes as determined by Illumina Hiseq TM 2000 sequencing (grey) and qRT-PCR (black). Error bars indicated standard deviations of averages from three replicates. sRNAs (1–18) represent sRNA0246, sRNA0320, sRNA0411, sRNA0423, sRNA0468, sRNA0530, sRNA0580, sRNA0645, sRNA0703, sRNA1133, sRNA0092, sRNA0188, sRNA0410, sRNA0699, sRNA0987, sRNA1073, sRNA1153, and sRNA1169, respectively
sRNA target gene prediction
To better understand the role of sRNAs in different biological modules, sRNA-regulating target genes were predicted using IntaRNA (Busch et al. 2008; Wright et al. 2014). Totally, 6043 target genes, which covering 97.11% of ORFs in Anabaena 7120, were found from the prediction using the 418 differentially expressed sRNAs as queries (supplementary Table 3). This indicates that single sRNA could target multiple genes. To further explore the differences of biological modules between the (N+) and (N−) groups, target genes that were predicted using the 49 (N+) unique sRNAs and 46 (N–) unique sRNAs as queries were subjected to functional categorization using the Gene ontology (GO) database (Consortium 2015). In total, 1674 target genes were predicted from the (N+) unique sRNAs, and 1811 were predicted from the (N−) unique sRNAs, while 630 were predicted in both. Functional annotation tool WEIGO (Ye et al. 2006) was used to analyze the target GO annotation terms. As shown in Fig. 4, there is no major difference for the GO analysis between these 2 groups. The GO terms were distributed widely with regard to their respective biological processes, from metabolic process to translation or transcription regulation and signal transducing.

GO analysis of target genes that are predicted to be involved in biological processes, molecular functions, and their cellular components
Discussion
With the development of high-through sequencing methods such as RNA-seq, small RNAs have been increasingly recognized as major modulators of gene expression in bacteria (Prévost et al. 2011; Eisenhut et al. 2012; Sakurai et al. 2012; Yan et al. 2013; Behrens et al. 2014; Papenfort et al. 2015 ; Luo et al. 2019). However, only a few studies have sought to identify the global profile of sRNAs in cyanobacteria (Voß et al. 2007; Georg et al. 2009; Voß et al. 2009; Mitschke et al. 2011b; Bi et al. 2018). Flaherty et al. (2011) used the directional RNA-seq to analyze the Anabaena transcriptome during nitrogen step-down. Their RNA-seq data provided information on transcript abundance and boundaries, including detection of operons and the length of the untranslated region (UTR) of each transcript. Totally, they found that 434 and 396 genes were substantially upregulated at 12 h and 21 h, respectively. In contrast, only 32 and 35 genes were downregulated at 12 h and 21 h, respectively (Flaherty et al. 2011). Using the differential RNA sequencing method, Mitschke et al. (2011a, 2011b) addressed the differential use of transcriptional start sites (TSS) in Anabaena PCC 7120 after nitrogen depletion in the wild type strain and a hetR mutant which was unable to differentiate heterocysts (Buikema and Haselkorn 1991; Mitschke et al. 2011a). They compared RNA profiles between these 2 strains grown in medium with or without combined nitrogen for 8 h. They identified > 900 putative TSS with a minimal 8-fold change in response to nitrogen deficiency. Among them, 209 were not induced in the hetR mutant indicating their involvement in heterocyst differentiation (Mitschke et al. 2011a). No study has been carried out to characterize the transcriptome in Anabaena PCC 7120 at later steady stage after combined nitrogen deprivation. In this study, we utilized the deep RNA-seq method for the identification of sRNAs in the model cyanobacterium Anabaena sp. strain PCC 7120 under steady state after 12-day nitrogen step-down. We identified 14,132 transcripts in total, which was comparable to transcripts (13,705) identified by Mitschke et al. (2011a, 2011b). Generally, sRNAs are transcribed from the non-coding regions, such as the antisense region and 5′ or 3′ UTR region (Gottesman and Storz 2011; Tsai et al. 2015). So we kept 1219 sRNAs that were located either on the antisense strand or intergenic region for further analysis (supplementary Table 2). Among them, 29 antisense sRNAs and 389 intergenic sRNAs were significantly differently expressed between the (N−) and (N+) samples (supplementary Table 3).
The 2 studies based on deep sequencing mentioned above have shown the presence of multiple non-coding RNA and antisense transcripts (Flaherty et al. 2011; Mitschke et al. 2011a). For example, Mitschke et al. (2011a, 2011b) detected 1414 intergenic transcripts and 2412 antisense RNAs, which were 17% and 39% of all genes in the genome, respectively. However, we detected 5557 intergenic transcripts (39.32%) and 4205 antisense RNA (29.76%), the discrepancy might come from the different preparation of RNA samples (< 200 nt vs whole RNA) or different culture condition.
Our RNA-seq data showed sRNAs throughout the Anabaena transcriptome. For example, we identified an antisense transcript sRNA0414 covering 21 nt of 3′ end of alr1690 and 73-nt downstream sequence. It partially overlapped with a previously identified cis-acting antisense RNA, α-furA, which was co-transcribed with alr1690 and could modulate ferric uptake regulation protein FurA (All1691) expression (Hernández et al. 2006; Hernández et al. 2010). Also, we found 8 transcripts around a heterocyst regulatory gene alr3546 (hetF). One of them, sRNA0787, matched the non-coding RNA NsiR1 (Ionescu et al. 2010; Muro-Pastor 2014). This indicates that the NsiR1 might reach its expression peak around 12 h (Ionescu et al. 2010; Muro-Pastor 2014), then maintains a low abundance to regulate target genes at later steady state. Regulation of nitrogen fixation genes is the culminating event of heterocyst differentiation. Our RNA-seq data showed that strong upregulation of sRNA3661 and sRNA3671 that target intergenic region of all1517 (nifB)-all1518 and all1455 (nifH)-all1456 (nifU) after nitrogen step-down (Table 3). These transcripts might be the degradation of long mRNA of these genes, which indicates that nitrogenase enzymes still maintain active functions at later steady state. However, other well-characterized nitrogen fixation genes, such as heterocyst exopolysaccharide synthesis (hep) and heterocyst glycolipid synthesis (hgl) were not observed to be regulated by differently expressed sRNAs here. This indicates that the mature heterocyst might not require an upregulation of these genes at later steady state or not regulated by sRNA presented in this study.
Conclusion
We have comprehensively analyzed sRNA expression profile after nitrogen step-down using Anabaena PCC 7120 and detected 418 differentially expressed sRNAs. Though the targets of sRNAs are currently unknown and need to be further analyzed, the antisense transcripts and intergenic RNAs perhaps play important roles in regulation during the response to combined nitrogen deprivation. This study provides more information about the regulatory network of sRNAs and will advance our understanding of sRNA functions in cyanobacteria.
References
Allen MB, Arnon DI (1955) Studies on nitrogen-fixing blue-green algae. I. Growth and nitrogen fixation by Anabaena Cylindrica Lemm. Plant Physiology 30:366–372
Babski J, Maier LK, Heyer R, Jaschinski K, Prasse D, Jäger D, Randau L, Schmitz RA, Marchfelder A, Soppa J (2014) Small regulatory RNAs in Archaea. RNA Biology 11:484–493
Behrens S, Widder S, Mannala GK, Qing X, Madhugiri R, Kefer N, Mraheil MA, Rattei T, Hain T (2014) Ultra deep sequencing of Listeria monocytogenes sRNA transcriptome revealed new antisense RNAs. PLoS One 9:e83979
Bi YQ, Pei GS, Sun T, Chen ZX, Chen L, Zhang WW (2018) Regulation mechanism mediated by trans-encoded sRNA Nc117 in short chain alcohols tolerance in Synechocystis sp PCC 6803. Front Microbiol 9:863
Brenes-Álvarez M, Mitschke J, Olmedo-Verd E, Georg J, Hess WR, Vioque A, Muro-Pastor AM (2019) Elements of the heterocyst-specific transcriptome unravelled by co-expression analysis in Nostoc sp. PCC 7120. Environ Microbiol 21:2544–2558
Brenes-Álvarez M, Olmedo-Verd E, Vioque A, Muro-Pastor AM (2016) Identification of conserved and potentially regulatory small RNAs in heterocystous cyanobacteria. Front Microbiol 7:48
Buikema WJ, Haselkorn R (1991) Characterization of a gene controlling heterocyst differentiation in the cyanobacterium Anabaena 7120. Genes Dev 5:321–330
Burnat M, Li B, Kim SH, Michael AJ, Flores E (2018) Homospermidine biosynthesis in the cyanobacterium Anabaena requires a deoxyhypusine synthase homologue and is essential for normal diazotrophic growth. Mol Microbiol 109:763–780
Busch A, Richter AS, Backofen R (2008) IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 24:2849–2856
Cao Y, Wu J, Liu Q, Zhao Y, Ying X, Cha L, Wang L, Li W (2010) sRNATarBase: a comprehensive database of bacterial sRNA targets verified by experiments. RNA 16:2051–2057
Carr NG, Whitton BA (1973) The biology of blue-green algae. University of California Press
Chen K, Xu X, Gu L, Hildreth M, Zhou R (2015) Simultaneous gene inactivation and promoter reporting in cyanobacteria. Appl Microbiol Biotechnol 99:1779–1793
Chen YF, Motteux O, Bédu S, Li YZ, Zhang CC (2011) Characterization of two critical residues in the effector-binding domain of NtcA in the cyanobacterium Anabaena sp. Strain PCC 7120. Curr Microbiol 63:32–38
Consortium TGO (2015) Gene Ontology Consortium: going forward. Nucleic Acids Res 43:D1049–D1056
de Porcellinis AJ, Klähn S, Rosgaard L, Kirsch R, Gutekunst K, Georg J, Hess WR, Sakuragi Y (2016) The non-coding RNA Ncr0700/PmgR1 is required for photomixotrophic growth and the regulation of glycogen accumulation in the cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol 57:2091–2103
Dismukes GC, Carrieri D, Bennette N, Ananyev GM, Posewitz MC (2008) Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol 19:235–240
Ducat DC, Way JC, Silver PA (2011) Engineering cyanobacteria to generate high-value products. Trends Biotechnol 29:95–103
Dühring U, Axmann IM, Hess WR, Wilde A (2006) An internal antisense RNA regulates expression of the photosynthesis gene isiA. Proc Nat Acad Sci 103:7054–7058
Eisenhut M, Georg J, Klähn S, Sakurai I, Mustila H, Zhang P, Hess WR, Aro EM (2012) The antisense RNA As1_flv4 in the cyanobacterium Synechocystis sp. PCC 6803 prevents premature expression of the flv4-2 operon upon shift in inorganic carbon supply. J Biol Chem 287:33153–33162
Esteves-Ferreira AA, Inaba M, Fort A, Araujo WL, Sulpice R (2018) Nitrogen metabolism in cyanobacteria: metabolic and molecular control, growth consequences and biotechnological applications. Crit Rev Microbiol 44:541–560
Flaherty BL, Van Nieuwerburgh F, Head SR, Golden JW (2011) Directional RNA deep sequencing sheds new light on the transcriptional response of Anabaena sp. strain PCC 7120 to combined-nitrogen deprivation. BMC Genomics 12:1–10
Flores E, Herrero A (2010) Compartmentalized function through cell differentiation in filamentous cyanobacteria. Nat Rev Micro 8:39–50
Georg J, Dienst D, Schürgers N, Wallner T, Kopp D, Stazic D, Kuchmina E, Klähn S, Lokstein H, Hess WR, Wilde A (2014) The small regulatory RNA SyR1/PsrR1 controls photosynthetic functions in cyanobacteria. Plant Cell 26:3661–3679
Georg J, Voß B, Scholz I, Mitschke J, Wilde A, Hess WR (2009) Evidence for a major role of antisense RNAs in cyanobacterial gene regulation. Mol Syst Biol 5:305
Gong Y, Xu X (2012) A small internal antisense RNA (aftsH) of all3642 (ftsH) in Anabaena sp. PCC 7120. Chin Sci Bull 57:756–761
Gottesman S (2005) Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet 21:399–404
Gottesman S, Storz G (2011) Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harbor Perspect Biol 3:a003798
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protocols 8:1494–1512
Halfmann C, Gu L, Gibbons W, Zhou R (2014b) Genetically engineering cyanobacteria to convert CO2, water, and light into the long-chain hydrocarbon farnesene. Appl Microbiol Biotechnol 98:9869–9877
Halfmann C, Gu L, Zhou R (2014a) Engineering cyanobacteria for the production of a cyclic hydrocarbon fuel from CO2 and H2O. Green Chem 16:3175–3185
Hernández JA, Alonso I, Pellicer S, Luisa Peleato M, Cases R, Strasser RJ, Barja F, Fillat MF (2010) Mutants of Anabaena sp. PCC 7120 lacking alr1690 and α-furA antisense RNA show a pleiotropic phenotype and altered photosynthetic machinery. J Plant Physiol 167:430–437
Hernández JA, Muro-Pastor AM, Flores E, Bes MT, Peleato ML, Fillat MF (2006) Identification of a furA cis antisense RNA in the Cyanobacterium Anabaena sp. PCC 7120. J Mol Biol 355:325–334
Herrero A, Flores FG (2008) The cyanobacteria: molecular biology, genomics, and evolution. Caister Academic Press
Herrero A, Muro-Pastor AM, Valladares A, Flores E (2004) Cellular differentiation and the NtcA transcription factor in filamentous cyanobacteria. FEMS Microbiol Rev 28:469–487
Herrero A, Picossi S, Flores E (2013) Gene expression during heterocyst differentiation. Genomics Cyanobacteria 65:281–329
Higo A, Isu A, Fukaya Y, Hisabori T (2017) Designing synthetic flexible gene regulation networks using RNA devices in cyanobacteria. ACS Synthetic Biol 6:55–61
Higo A, Isu A, Fukaya Y, Hisabori T (2018) Spatio-temporal gene induction systems in the heterocyst-forming multicellular cyanobacterium Anabaena sp PCC 7120. Plant Cell Physiol 59:82–89
Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13
Ionescu D, Voss B, Oren A, Hess WR, Muro-Pastor AM (2010) Heterocyst-specific transcription of NsiR1, a non-coding RNA encoded in a tandem array of direct repeats in cyanobacteria. J Mol Biol 398:177–188
Ishikawa Y, Miyagi A, Ishikawa T, Nagano M, Yamaguchi M, Hihara Y, Kaneko Y, Kawai-Yamada M (2019) One of the NAD kinases, sll1415, is required for the glucose metabolism of Synechocystis sp. PCC 6803. Plant J 98:654–666
Kaushik MS, Singh P, Tiwari B, Mishra AK (2016) Ferric uptake regulator (FUR) protein: properties and implications in cyanobacteria. Ann Microbiol 66:61–75
Klähn S, Schaal C, Georg J, Baumgartner D, Knippen G, Hagemann M, Muro-Pastor AM, Hess WR (2015) The sRNA NsiR4 is involved in nitrogen assimilation control in cyanobacteria by targeting glutamine synthetase inactivating factor IF7. Proc Nat Acad Sci 112:E6243–E6252
Kopf M, Klähn S, Scholz I, Matthiessen JKF, Hess WR, Voß B (2014) Comparative analysis of the primary transcriptome of Synechocystis sp. PCC 6803. DNA Res 21:527–539
Kumar K, Mella-Herrera RA, Golden JW (2010) Cyanobacterial heterocysts. Cold Spring Harbor Perspect Biol 2:a000315
Lambrecht SJ, Wahlig JML, Steglich C (2018) The GntR family transcriptional regulator PMM1637 regulates the highly conserved cyanobacterial sRNA Yfr2 in marine picocyanobacteria. DNA Res 25:489–497
Laurent S, Chen H, Bédu S, Ziarelli F, Peng L, Zhang CC (2005) Nonmetabolizable analogue of 2-oxoglutarate elicits heterocyst differentiation under repressive conditions in Anabaena sp. PCC 7120. Proc Nat Acad Sci U S A 102:9907–9912
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760
Li L, Huang D, Cheung MK, Nong W, Huang Q, Kwan HS (2012) BSRD: a repository for bacterial small regulatory RNA. Nucleic Acids Res 41:D233–D238
López-Igual R, Flores E, Herrero A (2010) Inactivation of a heterocyst-specific invertase indicates a principal role of sucrose catabolism in heterocysts of Anabaena sp. J Bacteriol 192:5526–5533
Luo X, Li JX, Chang TL, He HY, Zhao Y, Yang X, Zhao YW, Xu Y (2019) Stable reference gene selection for RT-qPCR analysis in Synechococcus elongatus PCC 7942 under abiotic stresses. BioMed Res Int https://doi.org/10.1155/2019/7630601
Maldener I, Muro-Pastor AM (2001) Cyanobacterial heterocysts. In eLS. Wiley
Mitschke J, Georg J, Scholz I, Sharma CM, Dienst D, Bantscheff J, Voß B, Steglich C, Wilde A, Vogel J, Hess WR (2011b) An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Nat Acad Sci 108:2124–2129
Mitschke J, Vioque A, Haas F, Hess WR, Muro-Pastor AM (2011a) Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc Nat Acad Sci 108:20130–20135
Morris KV, Mattick JS (2014) The rise of regulatory RNA. Nat Rev Genet 15:423–437
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth 5:621–628
Muro-Pastor AM (2014) The heterocyst-specific NsiR1 small RNA is an early marker of cell differentiation in cyanobacterial filaments. mBio 5:e01079–e01014
Nakamura T, Naito K, Yokota N, Sugita C, Sugita M (2007) A cyanobacterial non-coding RNA, Yfr1, is required for growth under multiple stress conditions. Plant Cell Physiol 48:1309–1318
Olmedo-Verd E, Brenes-Alvarez M, Vioque A, Muro-Pastor AM (2019) A heterocyst-specific antisense RNA contributes to metabolic reprogramming in Nostoc sp. PCC 7120. Plant Cell Physiol 60:1646–1655
Papenfort K, Förstner KU, Cong JP, Sharma CM, Bassler BL (2015) Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc Nat Acad Sci 112:E766–E775
Pinto F, Pacheco CC, Ferreira D, Moradas-Ferreira P, Tamagnini P (2012) Selection of suitable reference genes for RT-qPCR analyses in cyanobacteria. PLoS One 7(4):e34983.
Prévost K, Desnoyers G, Jacques JF, Lavoie F, Massé E (2011) Small RNA-induced mRNA degradation achieved through both translation block and activated cleavage. Genes Dev 25:385–396
Reiner A, Yekutieli D, Benjamini Y (2003) Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19:368–375
Sakurai I, Stazic D, Eisenhut M, Vuorio E, Steglich C, Hess WR, Aro EM (2012) Positive regulation of psbA gene expression by cis-encoded antisense RNAs in Synechocystis sp. PCC 6803. Plant Physiol 160:1000–1010
Singh DK, Lingaswamy B, Koduru TN, Nagu PP, Jogadhenu PSS (2019) A putative merR family transcription factor Slr0701 regulates mercury inducible expression of MerA in the cyanobacterium Synechocystis sp. PCC6803. Microbiol Open:e838
Srivastava A, Brilisauer K, Rai AK, Ballal A, Forchhammer K, Tripathi AK (2017) Down-regulation of the alternative sigma factor SigJ confers a photoprotective phenotype to Anabaena PCC 7120. Plant Cell Physiol 58:287–297
Storz G, Vogel J, Wassarman Karen M (2011) Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43:880–891
Tsai CH, Liao R, Chou B, Palumbo M, Contreras LM (2015) Genome-wide analyses in bacteria show small-RNA enrichment for long and conserved intergenic regions. J Bacteriol 197:40–50
Vargas WA, Nishi CN, Giarrocco LE, Salerno GL (2010) Differential roles of alkaline/neutral invertases in Nostoc sp. PCC 7120: Inv-B isoform is essential for diazotrophic growth. Planta 233:153–162
Voß B, Georg J, Schön V, Ude S, Hess WR (2009) Biocomputational prediction of non-coding RNAs in model cyanobacteria. BMC Genomics 10:123
Voß B, Gierga G, Axmann IM, Hess WR (2007) A motif-based search in bacterial genomes identifies the ortholog of the small RNA Yfr1 in all lineages of cyanobacteria. BMC Genomics 8:375
Wolk CP, Ernst A, Elhai J (2004) Heterocyst metabolism and development. In: Bryant D (ed) In the molecular biology of cyanobacteria. Springer, Netherlands, pp 769–823
Wright PR, Georg J, Mann M, Sorescu DA, Richter AS, Lott S, Kleinkauf R, Hess WR, Backofen R (2014) CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res 42:W119–W123
Xu X, Gu L, He P, Zhou R (2015) Characterization of five putative aspartate aminotransferase genes in the N2-fixing heterocystous cyanobacterium Anabaena sp. strain PCC 7120. Microbiology 161:1219–1230
Yan Y, Su S, Meng X, Ji X, Qu Y, Liu Z, Wang X, Cui Y, Deng Z, Zhou D, Jiang W, Yang R, Han Y (2013) Determination of sRNA expressions by RNA-seq in Yersinia pestis grown in vitro and during infection. PLoS One 8:e74495
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:W293–W297
Zhao MX, Jiang YL, He YX, Chen YF, Teng YB, Chen Y, Zhang CC, Zhou CZ (2010) Structural basis for the allosteric control of the global transcription factor NtcA by the nitrogen starvation signal 2-oxoglutarate. Proc Nat Acad Sci 107:12487–12492
Acknowledgements
The authors are very grateful to Professor Ruanbao Zhou (Department of Biology and Microbiology, South Dakota State University, Brookings, USA) for his valuable advice and the supply of Anabaena PCC 7120 strain.
Funding
This study was funded by the CNRS and a grant from the ANR (ANR-14-CE02–0009). Funding agencies contributed strictly financially to the performed research.
Author information
Authors and Affiliations
Contributions
PH, KC, XF: Conceived and designed the experiments. PH, XC, KC: Performed the experiments. PH, XC, KC, XF: Analyzed the data. PH, XC, KC: Contributed reagents/materials/analysis tools. PH, KC, XF: Wrote the paper. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not involving human participants and/or animals
Consent for publication
All relevant personnel had been informed.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Supplementary figure 1. Length distribution of sRNA candidates.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
He, P., Cai, X., Chen, K. et al. Identification of small RNAs involved in nitrogen fixation in Anabaena sp. PCC 7120 based on RNA-seq under steady state conditions. Ann Microbiol 70, 4 (2020). https://doi.org/10.1186/s13213-020-01557-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13213-020-01557-w