
Abstract
Background
YKL-40 (Chitinase 3-like I) is increased in CSF of Alzheimer’s disease (AD) and frontotemporal lobar degeneration (FTLD) patients and is therefore considered a potential neuroinflammatory biomarker. Whether changed YKL-40 levels in the CSF reflect dysregulation of YKL-40 in the brain is not completely understood yet. We aimed to extensively analyze YKL-40 levels in the brain of AD and different FTLD pathological subtypes. The direct relationship between YKL-40 levels in post-mortem brain and ante-mortem CSF was examined in a small set of paired brain-CSF samples.
Method
YKL-40 was analyzed in post-mortem temporal and frontal cortex of non-demented controls and patients with AD and FTLD (including FTLD-Tau and FTLD-TDP) pathology by immunohistochemistry (temporal cortex: 51 controls and 56 AD and frontal cortex: 7 controls and 24 FTLD patients), western blot (frontal cortex: 14 controls, 5 AD and 67 FTLD patients), or ELISA (temporal cortex: 11 controls and 7 AD and frontal cortex: 14 controls, 5 AD and 67 FTLD patients). YKL-40 levels were also measured in paired post-mortem brain and ante-mortem CSF samples from dementia patients (n = 9, time-interval collection: 1.4 years) by ELISA.
Results
We observed that YKL-40 post-mortem brain levels were similar between AD, FTLD, and controls as shown by immunohistochemistry, western blot, and ELISA. Interestingly, strong YKL-40 immunoreactivity was observed in AD cases with cerebral amyloid angiopathy (CAA; n = 6). In paired CSF-brain samples, YKL-40 concentration was 8-times higher in CSF compared to brain.
Conclusion
Our data suggest that CSF YKL-40 changes may not reflect YKL-40 changes within AD and FTLD pathological brain areas. The YKL-40 reactivity associated with classical CAA hallmarks indicates a possible relationship between YKL-40, neuroinflammation, and vascular pathology.
Similar content being viewed by others

Background
Compelling evidence from clinical and preclinical studies indicate that neuroinflammatory processes are relevant mediators in the development of different neurodegenerative dementias such as Alzheimer’s disease (AD) and frontotemporal lobar degeneration (FTLD) [1, 2]. Chronic activation of glial cells has shown to exacerbate AD and FTLD pathology and phenotypes [3,4,5] and modulate disease progression [2, 6, 7]. Moreover, causal and susceptibility genes associated with AD or FTLD (e.g., TREM-2, CD33, GRN) are related to neuroinflammation [8, 9]. Biomarkers that accurately reflect such neuroinflammatory processes in vivo could aid in the development of tools tracking disease progression and disease-modifying therapies [10, 11].
YKL-40 (or chitinase-3-like protein 1) is a glycoprotein involved in different mechanisms such as extracellular matrix remodeling [12, 13] or endothelial dysfunction [14]. It is also involved in the innate immune system [15] and has shown to be secreted by activated macrophages [16]. Several independent studies have shown that YKL-40 levels are increased in the cerebrospinal fluid (CSF) of AD [17,18,19,20,21,22,23,24,25] and FTLD patients [20, 21, 23, 26,27,28,29,30,31]. In brain, YKL-40 immunoreactivity has mainly been detected in astrocytes from controls and different tauopathies (AD and FTLD-Tau), whereby it correlated positively with tau pathology [22, 32] and surrounded amyloid-beta plaques in AD [17, 22, 32]. YKL-40 is also present in brain tissue from different neurological conditions (e.g., multiple sclerosis; MS, Creutzfeldt-Jakob disease; CJD, and bacterial meningitis) [22, 33, 34]. In MS, YKL-40 was detected in both astrocytes and microglial cells which correlated to the high inflammatory state of the disease [34]. These CSF and post-mortem brain data suggest that YKL-40 is a promising biomarker reflecting the neuroinflammatory process in different neurodegenerative dementias.
Several studies have analyzed the cellular location YKL-40 in the brain of AD, FTLD, and MS cases [17, 22, 32, 34]. However, few studies have analyzed YKL-40 levels using semi-quantitative approaches [22, 32], which revealed that the number of YKL-40 positive astrocytes was overall relatively limited, representing less than 10% of all GFAP-positive astrocytes [32]. Also, the correlation between YKL-40 levels in brain and CSF is rather unknown. Here, we aimed to further understand YKL-40 expression in brain and its relation with ante-mortem CSF. Thus, we analyzed YKL-40 levels in a large sample set of post-mortem human brain of non-demented controls and dementia cases with different underlying pathologies (AD, FTLD-Tau, and FTLD-TDP). Furthermore, we also examined the association between the levels of YKL-40 in post-mortem brain and ante-mortem CSF in a small set of paired brain-CSF samples from dementia patients.
Methods
Human samples
Post-mortem brain tissue
Temporal cortex of AD patients (n = 56) and non-demented controls (NDC, n = 51) were obtained from the Netherlands Brain Bank (Amsterdam). Six of these AD cases had severe cerebral amyloid angiopathy (CAA) type-1 and no or low numbers of parenchymal amyloid deposits [35]. Frontal cortex of cases with AD (n = 4) and FTLD (n = 67) pathology and NDC (n = 13) were obtained from the Netherlands Brain Bank (FTLD n = 21 and NDC = 5), CIEN Foundation Brain bank (AD n = 4, FTLD n = 23, and NDC n = 4; BT-CIEN, Madrid, Spain) and Emory University (FTLD n = 22 and NDC n = 4). The FTLD group included cases with pathological diagnosis of FTLD-Tau (n = 33) and FTLD-TDP (n = 34). FTLD-Tau group consisted of different forms of tauopathies such as progressive supranuclear palsy (PSP, n = 11), corticobasal degeneration (CBD, n = 3), and Pick’s disease (PiD, n = 3). Familial cases with MAPT mutation were also included (n = 16) [36]. The FTLD-TDP group also included familial cases with C9orf72 (n = 7) or GRN (n = 6) mutations [37, 38]. Pathological examination was performed following consensus guidelines described previously [39,40,41,42,43]. As far as stated in the autopsy report, controls with no clinically relevant inflammatory-related diseases were excluded. Written informed consent for brain autopsy and the use of medical records for research purposes was given by all donors or their next of kin. An overview of patient details including age, sex, and post-mortem delay is summarized in Table 1. Additional specific information per case is provided in Supplementary Table 1.
For immunohistochemistry, paraffin-embedded brain tissue was obtained. Sections of 5-μm thick were mounted on tissue slides (Superfrost® plus, Menzel Glaser, Braunschweig, Germany) and dried overnight at 37°C. For enzyme-linked immunosorbent assay (ELISA) and western blot analysis, frozen brain tissue blocks were provided and homogenized using Tissue Protein Extraction Reagent (T-PER, 0.1 g/mL, Thermo Scientific, Waltham, USA) containing EDTA-free protease inhibitor cocktail (1:25, Roche, Basel, Switzerland) and phosphatase inhibitor (1:10, Roche, Basel, Switzerland). Tissue homogenates were centrifuged at 10.000 g at 4°C for 5 min. Thereafter, total protein content for the supernatant was measured using the Pierce™ BCA Protein Assay Kit (Thermo Scientific, Waltham, USA) following the manufacturer's instructions. Brain lysates were aliquoted and stored at -80°C until further analysis.
Paired ante-mortem CSF sand post-mortem brain samples
Paired ante-mortem CSF and post-mortem frontal cortex samples (n = 9) were obtained from Emory University which included different types of autopsy-confirmed dementias (FTLD-Tau = 3, FTLD with amyotrophic lateral sclerosis; FTLD-ALS = 5, and dementia with Lewy body; DLB = 1). FTLD-ALS associates with TDP-43 pathology [44]. Neuropathology for TDP and DLB was determined following international guidelines [40, 45]. CSF material was obtained by standard lumbar puncture and collected in polypropylene (PP) tubes. CSF was aliquoted and stored at −80°C prior to analysis. CSF total protein content was measured using the Pierce™ BCA Protein Assay Kit. Informed consent was provided from subjects or their next of kin. The demographic data of cases included in this study are summarized in Supplementary Table 2.
Immunohistochemistry
Brain sections (n = 134) were deparaffinized and immersed for 30 min in 0.3% H202 to quench endogenous peroxide activity. Antigen retrieval was performed using sodium citrate buffer (10 mmol/L, pH 6.0) for 20 min. Sections were incubated overnight at 4°C in a humid environment with Goat anti-human Chitinase 3-like 1 Antibody (1:50, Cat. No AF2599, R&D Systems, Minneapolis, USA) in antibody diluent (Immunologic, Duiven, The Nederlands). Mouse anti-human YKL-40 antibody (1:500, Quidel Corporation, San Diego, USA) was also tested for comparison purposes and to support specificity. Both antibodies showed similar staining patterns, though the former showed stronger YKL-40 immunoreactivity (Supplementary Fig. 1). After incubation with primary antibody, sections were washed with PBS and incubated for 30 min with biotin-conjugated rabbit anti-goat antibody at room temperature (1:400, DAKO, Glostrup, Denmark). Then sections were washed with PBS and a 60-min incubation with HRP-conjugated streptavidin antibody (1:300, DAKO) was performed. Sections were subsequently stained with 3,3-diaminobenzidine tetrahydrochloride dihydrate (DAB; 0.1 mg/mL, 0.02% H2O2, DAKO) for 10 min, rinsed in tap water and counterstained with hematoxylin for 1 min. After extensive washing in tap water, sections were dehydrated in a series of ethanol and xylene baths and mounted with Quick-D mounting medium (Klinipath, Duiven, The Nederlands). First, different dilutions of the primary and secondary antibodies were tested (Supplementary Fig. 2A-B). Thereafter, the specificity of the antibody for immunohistochemistry was tested by pre-absorption. The anti-Chitinase 3-like 1 antibody was pre-absorbed for 4 h at room temperature with 45 molar excess of recombinant protein (aa22–aa383, R&D), thereafter, sections were incubated with the pre-absorbed antibody overnight at 4°C (Supplementary Fig. 2C-D).
Positive or negative YKL-40 immunoreactivity was determined within either the temporal or frontal cortex region. Staining patterns were considered positive if YKL-40 immunoreactivity was present in two or more cell clusters within each section (Supplementary Fig. 3A). Staining patterns were considered negative if YKL-40 immunoreactivity was absent or present in only one cell per section (Supplementary Fig. 3B). In a subset of cases (temporal cortex from NDC = 6 and AD = 6), YKL-40 immunoreactivity was quantified by analyzing the DAB positive pixels with the QuPath software (thresholds: down-sample factor = 4, Gaussian sigma = 2, hematoxylin threshold (negative) = 0.1, DAB threshold (positive) = 0.3) as previously described [46]. Examination of the staining patterns was performed by a researcher who was blinded for diagnosis or other patient characteristics.
Western blot
Human frontal cortex lysates (n = 104, 10 μg) were prepared in sample buffer (0.03 M Tris, 2% SDS, 10% glycerol, 50mM DTT, 0.1 mM bromophenol blue) and heated for 5 min at 95°C. Electrophoresis was performed using 1.5 mm NuPAGE Novex 4-12% Bis-Tris Protein Gels (Thermofisher Scientific, Waltham, USA) and proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, USA). Subsequently, PVDF membranes were blocked for 30 min with blocking buffer containing 5% (w/v) milk powder with 0.5% PBS Tween 20 (PBS-T) and incubated overnight at 4°C in blocking buffer containing either goat anti-human chitinase 3-like 1 antibody (1:200, R&D systems). After washing with wash buffer (PBS-T containing 0.05% (w/v) milk powder), PVDF membranes were incubated 1 h at room temperature with corresponding secondary antibodies (polyclonal rabbit anti-goat IgG/HRP (1:2000, DAKO, Glostrup, Denmark) or goat anti-mouse IgG/HRP (1:1000, DAKO)) in blocking buffer. Proteins were detected with the ECLTM Western Blotting detection kit (GE Healthcare, Amersham, UK). After YKL-40 protein detection, PVDF membranes were incubated with stripping buffer (0.08M Tris-base, 2% SDS, 0.8% β-Mercaptoethanol; Sigma-Aldrich, Saint Louis, USA) for 30 min at 50°C to ensure removal of YKL-40 antibody. PVDF membranes were then re-incubated with mouse anti-human actin antibody (1:5000, clone AC-40, Sigma-Aldrich) following western blot procedure as described above. The protein signal was quantified using the ImageLabTM software version 3.0 (Bio-Rad, Hercules, USA). A total of 8 blots were analyzed, all including a reference sample (frontal cortex) that was used to correct potential differences across blots (Supplementary Fig. 4). Actin reactivity was used as a protein loading control.
Enzyme-linked immunosorbent assay
YKL-40 levels were measured in post-mortem brain lysates and CSF using MicroVue YKL-40 enzyme immunoassay (Quidel Corporation) following the manufacturer’s instructions. This kit was previously validated for analysis in CSF with intra- and inter-assay coefficient variations of < 4% and < 11% [27]. Additional analysis indicated that this kit could measure YKL-40 in post-mortem tissue with intra- and inter-assay variabilities < 8% and < 12%. Brain lysates and CSF samples were diluted 3-fold in sample dilution buffer and YKL-40 levels were corrected for dilution factor. Brain and CSF YKL-40 levels were normalized against the total protein content to analyze YKL-40 differences between pathological groups in brain lysates or between the paired CSF and brain samples.
Statistical analysis
Statistical analysis was performed with IBM SPSS statistics (version 26, IBM, Armonk, NY). Normal distribution of the data was assessed using the Shapiro-Wilk test. Skewed data were normalized using Templeton’s two-step method if applicable [47]. The influence of different demographic variables on YKL-40 levels was determined by linear regression analysis, Student’s t-test or Mann–Whitney test. Differences in YKL-40 levels between groups were analyzed either by Pearson’s chi-square, Kruskal-Wallis test, or Analysis of covariance including center and age as covariates followed by Bonferonni post hoc analysis. YKL-40 levels in AD temporal and frontal cortex and FTLD frontal cortex were compared to controls from corresponding areas. Correlation between paired ante-mortem CSF and post-mortem brain was assessed using a Pearson correlation test. p values < 0.05 were considered significant.
Results
YKL-40 immunoreactivity and protein levels remain similar in post-mortem temporal and frontal cortex from AD and non-demented controls
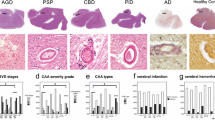
YKL-40 immunoreactivity was mainly present in glial cells but immunoreactivity was also detected in neuronal cells in the temporal cortex of both AD and controls (Fig. 1A). We observed that the number of YKL-40 positive cells was similar across groups (Fig. 1A). In a subset of cases, we also quantified the intensity of the YKL-40 positive immunoreactivity within a given cell and this did not differ either across pathological groups (Fig. 1A). We did not observe an association between YKL-40 immunoreactivity and the degree of Amyloid or Tau pathology as measured by Thal and Braak stages (Supplementary Fig. 5). Of note, strong YKL-40 immunoreactivity was detected around the cerebral vessels and in areas resembling amyloid plaques in AD cases with CAA pathology (AD-CAA, Fig. 1A), although differences did not reach statistical significance (NDC vs AD-CAA, p = 0.07, Fig. 1A). ELISA and western blot analysis indicate that the levels of brain YKL-40 in either the temporal or frontal cortex was similar between AD and controls (Fig. 1B, C). We confirmed that the ELISA employed could still detect differences in CSF YKL-40 concentration using a small CSF cohort of AD and controls (p = 0.015, Supplementary Fig. 6).

YKL-40 immunoreactivity and protein levels remain similar in post-mortem temporal and frontal cortex from AD and non-demented controls. A Representative images of paraffin sections from temporal cortex stained with the anti-YKL-40 antibody. YKL-40 immunoreactivity is present in glial and neuronal cells in NDC and AD cases. In AD-CAA sections, YKL-40 was detected around cerebral vessels and in structures resembling amyloid plaques. Scale bars represent 50μM. Semi-quantitation of YKL-40 immunoreactivity was performed by grouping cases into negative (i.e., zero or 1 positive cell) or positive (i.e., 2 or more positive cell groups). Stacked bar plots represent the percentage of cases with either negative (−, white area) or positive (+, black area) YKL-40 immunoreactivity in NDC (n = 51), AD (n = 52), and AD-CAA cases (n = 6). Box-dot plots show quantification of YKL-40 immunoreactivity by DAB+ pixel count in NDC (n = 6, Braak stage < I, Thal stage = 0) and AD (n = 6, Braak stage > V, Thal stage = 3) cases. B Representative immunoblot of YKL-40 in frontal cortex from NDC and AD patients. Actin was used as a loading control. Box-dot plot depict YKL-40 immunoblot reactivity corrected for actin protein loading in NDC (n = 4) and AD cases (n = 5). (C) YKL-40 levels were quantified by ELISA and corrected for total protein concentration in temporal cortex (NDC = 14 and AD = 6) and frontal cortex (NDC = 4 and AD = 5). Overall, no significant differences across groups were identified. Box represents median ± interquartile range with bars showing the lowest to highest points. Abbreviations: NDC, non-demented control; AD, Alzheimer’s disease; CAA, cerebral amyloid angiopathy; n.s., non-significant difference between groups
YKL-40 immunoreactivity and protein levels remain similar in post-mortem frontal cortex from FTLD and non-demented controls
In line with the findings described above, YKL-40 immunoreactivity was present in the frontal cortex from both FTLD cases and non-demented controls. We observed that YKL-40 immunoreactivity was mainly present in glial cells but it was also detected in neuronal cells (Fig. 2A). YKL-40 levels were similar between FTLD and non-demented controls as observed by either immunohistochemistry (Fig. 2A), western blot (Fig. 2B), or ELISA (Fig. 2C). No differences in brain YKL-40 levels were observed neither when the main FTLD pathological subtypes were analyzed separately (FTLD-Tau and FTLD-TDP; Fig. 2A–C) or between their sub-classifications (i.e., PSP, CBD, PiD, MAPT, C9orf72, GRN; Supplementary Fig. 7A-B).

YKL-40 immunoreactivity and protein levels remain similar in post-mortem frontal cortex from FTLD and non-demented controls. A Representative images of paraffin sections from the frontal cortex stained with the anti-YKL-40 antibody. YKL-40 immunoreactivity is present in FTLD and control cases in glial and neuronal cells. Scale bars represent 50μM. Semi-quantitation of YKL-40 immunoreactivity was performed by grouping cases into either negative (i.e., zero or 1 positive cell) or positive (i.e., 2 or more positive cell groups). Stacked bar plots represent the percentage of cases with either negative (−, white area) or positive (+, black area) YKL-40 immunoreactivity in NDC (n = 7) and FTLD cases (n = 24; FTLD-Tau = 19 and FTLD-TDP = 5). B Representative immunoblot of YKL-40 in frontal cortex lysates. Actin was used as a loading control. Box-dot plots depict YKL-40 immunoblot reactivity corrected for actin in NDC (n = 14) and FTLD cases (n = 67; FTLD-Tau = 33, FTLD-TDP = 34). C YKL-40 levels were quantified by ELISA and corrected for total protein concentration in NDC (n = 14) and FTLD cases (n = 67; FTLD-Tau = 33 and FTLD-TDP = 34). Overall, no significant differences across groups were identified. Box represents median ± interquartile range with bars showing the lowest to highest points. Abbreviations: NDC, non-demented control; FTLD, frontal temporal lobar degeneration; n.s., non-significant difference between groups
YKL-40 levels in ante-mortem CSF are higher and inversely associated with YKL-40 levels in post-mortem brain of dementia patients
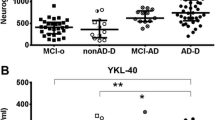
To further investigate the relationship between YKL-40 levels in brain and CSF, we analyzed the levels of YKL-40 in a small set of ante-mortem CSF samples paired with post-mortem frontal cortex from cases with FTLD and DLB pathology. The CSF YKL-40 concentration in this autopsy cohort ranged from 227 to 653 pg/mL, thereby covering the full range of CSF YKL-40 usually detected in other CSF studies. YKL-40 levels were 8-times higher in CSF compared to frontal cortex (p = 0.001, Fig. 3A). Though correlation data should be interpreted with caution considering the limited sample size, we observed an inverse, albeit non-significant, association between YKL-40 levels in the frontal cortex and the CSF (r = − 0.49, p = 0.18, Fig. 3B). A similar pattern was observed upon removing two outliers (r = − 0.448, p = 0.3, Supplementary Fig. 8).

YKL-40 levels in ante-mortem CSF are higher and inversely associated with YKL-40 levels in post-mortem brain. A YKL-40 was quantified by ELISA and corrected for total protein concentration in paired ante-mortem CSF and post-mortem frontal cortex (n = 9). YKL-40 was 8-times higher in CSF compared to post-mortem frontal cortex (B). Scatter plot depicts a correlation between the two matrices; however, this did not reach significance (r = −0.49, p = 0.18). Box represents a median ± interquartile range with bars showing the lowest to highest points, *** p ≤ 0.001. Abbreviations: CSF, cerebrospinal fluid; tot prot. conc, total protein concentration
Discussion
In this study, we observed that the CSF YKL-40 changes detected in AD or FTD patients in this and previous studies were not detected in AD and FTLD pathological areas. We observed that YKL-40 levels are remarkably higher in CSF compared to brain. Interestingly, we found increased YKL-40 immunoreactivity in cases with AD-CAA pathology, suggesting a relationship of brain YKL-40 with CAA pathophysiology and/or vascular pathology.
CSF biomarker levels often reflect pathophysiological changes in the brain, as is the case for the classical AD CSF biomarkers: Aβ and hyperphosphorylated tau that reflect brain amyloidosis and tangle formation, respectively [48, 49]. However, AD is a multifaceted disorder in which multiple processes beyond amyloid and Tau are known to contribute to disease pathogenesis [50], including immunity [51]. Additional markers that reflect the neuroinflammatory changes underlying dementia might be useful not only to better define individual patients’ phenotype [52] but also to monitor treatment responses of anti-inflammatory drugs [11]. YKL-40 is involved in the immune system response [16, 33] and human neuropathological studies have shown an astrocytical YKL-40 immunoreactivity in cases with different neurological disorders [17, 22, 32, 34, 53]. Thus, we hypothesized that the elevated levels of YKL-40 in CSF of AD [17,18,19,20,21,22,23,24] and FTLD [20, 21, 23, 26,27,28,29] patients reflect neuroinflammatory changes in the brain areas that are typically affected in these dementia types. In agreement with previous studies, we observed that YKL-40 immunoreactivity was mainly found in clusters of glial cells (likely astrocytes) [17, 22, 32], and to some extent also in neurons. We observed that YKL-40 immunoreactivity was overall low with many cases (>50%) showing none or few YKL-40 positive cells. This is in line with a previous report showing that only 10% of all GFAP-astrocytes were positive for YKL-40 [32]. In agreement with a previous study, we also observed a prominent YKL-40 immunoreactivity around the cerebral vessels in AD cases with CAA pathology, suggesting a role of YKL-40 with vascular function [22]. However, while previous studies reported increased YKL-40 immunoreactivity in AD or FTLD cases [22, 32], we observed, using complementary methods, that the YKL-40 levels were similar in AD and FTLD compared to non-demented controls. Regarding AD, previous studies focused on frontal cortex areas while here we examined mainly the temporal cortex. The different regions analyzed could partly explain the discrepancies observed, especially considering that YKL-40 expression levels show some regional differences within the brain [22, 53]. Still, we did not observe any tendency in the small set of samples from AD frontal cortex. Noteworthy, our AD cohort (temporal cortex) is considerably large including more than 50 AD cases and controls, which increases the statistical power of our study. Regarding FTD, previous studies included only FTLD-Tau cases [32], while here we additionally included FTLD cases with TDP-43 pathology. However, we did not detect any differences when the FTLD pathological subtypes (i.e., FTLD-Tau and FTLD-TDP) were analyzed separately. It is unlikely that the observed immunohistochemically discrepancies are explained by the sample size as the FTLD group was comparable to previous studies. Considering the overall low expression pattern of YKL-40, the semi-quantitative methods applied in the different studies may partly explain the discrepancies across studies. We here analyzed the complete slide and dichotomized the outcomes into either positive or negative, and thus we might have missed subtle changes of YKL-40 immunoreactivity. Quantification of the intensity of YKL-40 in a subset of cases also showed similar results. Furthermore, we employed additional quantitative technologies, including the same immunoassay that is widely used for CSF analysis, which showed again no differences on YKL-40 levels between AD or FTLD and controls post-mortem brain. It is worth noting that a recent study showed no difference in CHI3L1 mRNA post-mortem tissue from early-onset AD cases, which was attributed to the younger age of these patients [54]. In line with our findings, a recent mass-spectrometry-based study detected YKL-40 changes in the CSF of AD patients but not in post-mortem tissue [55]. Interestingly, using paired CSF-brain samples, we observed that CSF YKL-40 levels are 8-times higher than those detected in post-mortem frontal cortex. Strikingly, we observed a non-significant but moderate inverse association of YKL-40 between these two matrices but considering the low sample size of the paired samples (n = 9), these correlation findings should be interpreted with caution.
Our data thus overall suggest that the YKL-40 changes observed in CSF of dementia patients does not reflect changes of this neuroinflammatory protein in the brain areas that are typically affected. Interestingly, similar results have been observed previously for α-synuclein which was increased in CSF of CJD patients but not in post-mortem brain tissue [56]. CSF protein levels are dynamic and may change over time depending on the disease stage, as previously observed in longitudinal CSF biomarker studies performed in familial AD patients [57]. Thus, it could be possible that the YKL-40 changes observed in ante-mortem CSF in various reports might not be observed at the end stage of the disease within brain tissue. However, longitudinal studies have shown that CSF YKL-40 levels increased continuously with disease progression, suggesting that normalization of CSF YKL-40 levels with advancing stage may not explain the lack of differences in tissue [58, 59]. We neither observed an association of YKL-40 immunoreactivity with advanced pathological stages in brain tissue (i.e., Thal or Braak stages). The absence of YKL-40 changes in AD and FTLD post-mortem tissue together with the prominent reactivity of YKL-40 observed in AD with CAA pathology might indicate that CSF YKL-40 changes could be associated with the peripheral blood compartment. This is, however, less likely since YKL-40 levels in the blood are lower and do not correlate with those in CSF and its levels remain unchanged in the blood of dementia patients [27, 60]. YKL-40 is a protein secreted by various cell types including astrocytes [33, 61]. Thus, YKL-40 may quickly diffuse from cells to the extracellular space and the CSF, which may ultimately hamper its detection in post-mortem brain. The origin of the increased CSF YKL-40 levels consistently observed in AD and FTD patients is thus still not clear. CSF YKL-40 changes may originate from an alternative area not analyzed in the current study, such as the choroid plexus, which is involved in the production and regulation of CSF and has shown to express YKL-40 at least during brain development [62]. The choroid plexus could play an important role in facilitating the inflammatory process in the central nervous system. This is supported by others that show the presence of immune cells [63, 64] and an up-regulation of pro-inflammatory cytokines and chemokines in the choroid plexus of AD patients [65], suggesting an involvement of this area within the neuroinflammatory process associated with aging and AD [66, 67].
This study is not without limitations. The sample size of AD-CAA cases, as well as those with paired CSF-brain tissue, was small, and thus, such data should be confirmed in larger cohorts. In addition, our paired CSF-brain samples did not include AD patients or non-demented controls, and thus we can not exclude that the correlation of YKL-40 in brain and CSF might be different in these groups. However, the CSF YKL-40 values of the paired samples covered a wide concentration range (200–700 ng/mL) including YKL-40 values comparable to those previously detected in the control or AD groups. Furthermore, our study is limited to the frontal and temporal cortex, and thus other brain areas (e.g., choroid plexus) may contribute to the strong YKL-40 changes observed in CSF. We also acknowledge that we could not compare YKL-40 across different diseases due to the difference in brain regions. The strengths of our study are the use of complementary methods ensuring reliable measurements of YKL-40 protein and the high number of cases analyzed.
Conclusion
The extensive analysis of YKL-40 performed here, using three different (semi-)quantitative technologies, suggests that the pathophysiological correlates underlying the increased CSF YKL-40 changes do not come from typically affected areas in AD or FTD. Understanding the origin of CSF YKL-40 changes is not trivial, considering its potential as a biomarker tracking ongoing neuroinflammation in different dementia types [10, 11, 68] or even as a novel potential therapeutic target [69, 70]. Interestingly, the prominent YKL-40 reactivity related to CAA pathology in the vessel wall suggests a potential involvement of brain YKL-40 levels in CAA pathophysiology which should be investigated in future studies.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- CAA:
-
Cerebral amyloid angiopathy
- CBD:
-
Corticobasal degeneration
- CJD:
-
Creutzfeldt-Jakob disease
- DAB:
-
Diaminobenzidine tetrahydrochloride dihydrate
- DLB:
-
Dementia with Lewy body
- ELISA:
-
Enzyme-linked immunosorbent assay
- FTLD:
-
Frontotemporal lobar degeneration
- FTLD-ALS:
-
FTLD with amyotrophic lateral sclerosis
- MS:
-
Multiple sclerosis
- NDC:
-
Non-demented controls
- PiD:
-
Pick’s disease
- PSP:
-
Progressive supranuclear palsy
- PVDF:
-
Polyvinylidene difluoride
- T-PER:
-
Tissue Protein Extraction Reagent
References
Heneka M, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's Disease. Lancet Neurol. 2015;14:4.
Bright F, Werry EL, Dobson-Stone C, Piguet O, Ittner LM, Halliday GM, et al. Neuroinflammation in frontotemporal dementia. Nat Rev Neurol. 2019;15(9):540–55.
Nordengen K, Kirsebom BE, Henjum K, Selnes P, Gisladottir B, Wettergreen M, et al. Glial activation and inflammation along the Alzheimer's disease continuum. J Neuroinflammation. 2019;16(1):46.
Oeckl P, Weydt P, Steinacker P, Anderl-Straub S, Nordin F, Volk AE, et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry. 2019;90(1):4–10.
Lant SB, Robinson AC, Thompson JC, Rollinson S, Pickering-Brown S, Snowden JS, et al. Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2014;40(6):686–96.
Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer's disease. Immunol Cell Biol. 2020;98(1):28–41.
Zhang J, Velmeshev D, Hashimoto K, Huang YH, Hofmann JW, Shi X, et al. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature. 2020;588(7838):459–65.
Efthymiou AG, Goate AM. Late onset Alzheimer's disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12(1):43.
Milanesi E, Bonvicini C, Alberici A, Pilotto A, Cattane N, Premi E, et al. Molecular signature of disease onset in granulin mutation carriers: a gene expression analysis study. Neurobiol Aging. 2013;34(7):1837–45.
Boxer AL, Gold M, Feldman H, Boeve BF, Dickinson SL, Fillit H, et al. New directions in clinical trials for frontotemporal lobar degeneration: Methods and outcome measures. Alzheimers Dement. 2020;16(1):131–43.
Hampel H, Caraci F, Cuello AC, Caruso G, Nistico R, Corbo M, et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer's Disease. Front Immunol. 2020;11:456.
Prakash M, Bodas M, Prakash D, Nawani N, Khetmalas M, Mandal A, et al. Diverse pathological implications of YKL-40: answers may lie in 'outside-in' signaling. Cell Signal. 2013;25(7):1567–73.
Pouyafar A, Heydarabad MZ, Mahboob S, Mokhtarzadeh A, Rahbarghazi R. Angiogenic potential of YKL-40 in the dynamics of tumor niche. Biomed Pharmacother. 2018;100:478–85.
Malinda KM, Ponce L, Kleinman HK, Shackelton LM, Millis AJ. Gp38k, a protein synthesized by vascular smooth muscle cells, stimulates directional migration of human umbilical vein endothelial cells. Exp Cell Res. 1999;250(1):168-73. https://doi.org/10.1006/excr.1999.4511.
Rehli M, Niller HH, Ammon C, Langmann S, Schwarzfischer L, Andreesen R, et al. Transcriptional regulation of CHI3L1, a marker gene for late stages of macrophage differentiation. J Biol Chem. 2003;278(45):44058–67.
Baeten D, Boots AM, Steenbakkers PG, Elewaut D, Bos E, Verheijden GF, Berheijden G, Miltenburg AM, Rijnders AW, Veys EM, De Keyser F. Human cartilage gp-39+,CD16+ monocytes in peripheral blood and synovium: correlation with joint destruction in rheumatoid arthritis. Arthritis Rheum. 2000;43(6):1233-43. https://doi.org/10.1002/1529-0131(200006)43:6<1233::AID-ANR6>3.0.CO;2-9.
Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry. 2010;68(10):903–12.
Mattsson N, Tabatabaei S, Johansson P, Hansson O, Andreasson U, Mansson JE, et al. Cerebrospinal fluid microglial markers in Alzheimer's disease: elevated chitotriosidase activity but lack of diagnostic utility. NeuroMolecular Med. 2011;13(2):151–9.
Olsson B, Hertze J, Lautner R, Zetterberg H, Nagga K, Hoglund K, et al. Microglial markers are elevated in the prodromal phase of Alzheimer's disease and vascular dementia. J Alzheimers Dis. 2013;33(1):45–53.
Janelidze S, Hertze J, Zetterberg H, Landqvist Waldo M, Santillo A, Blennow K, et al. Cerebrospinal fluid neurogranin and YKL-40 as biomarkers of Alzheimer's disease. Ann Clin Transl Neurol. 2016;3(1):12–20.
Baldacci F, Toschi N, Lista S, Zetterberg H, Blennow K, Kilimann I, et al. Two-level diagnostic classification using cerebrospinal fluid YKL-40 in Alzheimer's disease. Alzheimers Dement. 2017;13(9):993–1003.
Llorens F, Thune K, Tahir W, Kanata E, Diaz-Lucena D, Xanthopoulos K, et al. YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol Neurodegener. 2017;12(1):83.
Alcolea D, Irwin DJ, Illan-Gala I, Munoz L, Clarimon J, McMillan CT, et al. Elevated YKL-40 and low sAPPbeta:YKL-40 ratio in antemortem cerebrospinal fluid of patients with pathologically confirmed FTLD. J Neurol Neurosurg Psychiatry. 2019;90(2):180–6.
Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91(9):e867–e77.
Bos I, Vos S, Verhey F, Scheltens P, Teunissen C, Engelborghs S, et al. Cerebrospinal fluid biomarkers of neurodegeneration, synaptic integrity, and astroglial activation across the clinical Alzheimer's disease spectrum. Alzheimers Dement. 2019;15(5):644–54.
Alcolea D, Carmona-Iragui M, Suarez-Calvet M, Sanchez-Saudinos MB, Sala I, Anton-Aguirre S, et al. Relationship between beta-Secretase, inflammation and core cerebrospinal fluid biomarkers for Alzheimer's disease. J Alzheimers Dis. 2014;42(1):157–67.
Teunissen CE, Elias N, Koel-Simmelink MJ, Durieux-Lu S, Malekzadeh A, Pham TV, et al. Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimers Dement (Amst). 2016;2:86–94.
Alcolea D, Vilaplana E, Suárez-Calvet M, Illán-Gala I, Blesa R, Clarimón J, et al. CSF sAPPb, YKL-40, and neurofilament light in frontotemporal lobar degeneration. Neurology. 2017;89:10.
Del Campo M, Galimberti D, Elias N, Boonkamp L, Pijnenburg YA, van Swieten JC, et al. Novel CSF biomarkers to discriminate FTLD and its pathological subtypes. Ann Clin Transl Neurol. 2018;5(10):1163–75.
Woollacott IOC, Nicholas JM, Heller C, Foiani MS, Moore KM, Russell LL, et al. Cerebrospinal Fluid YKL-40 and Chitotriosidase Levels in Frontotemporal Dementia Vary by Clinical, Genetic and Pathological Subtype. Dement Geriatr Cogn Disord. 2020;49(1):56–76.
Abu-Rumeileh S, Steinacker P, Polischi B, Mammana A, Bartoletti-Stella A, Oeckl P, et al. CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimers Res Ther. 2019;12(1):2.
Querol-Vilaseca M, Colom-Cadena M, Pegueroles J, San Martin-Paniello C, Clarimon J, Belbin O, et al. YKL-40 (Chitinase 3-like I) is expressed in a subset of astrocytes in Alzheimer's disease and other tauopathies. J Neuroinflammation. 2017;14(1):118.
Bonneh-Barkay D, Bissel SJ, Kofler J, Starkey A, Wang G, Wiley CA. Astrocyte and macrophage regulation of YKL-40 expression and cellular response in neuroinflammation. Brain Pathol. 2012;22(4):530–46.
Canto E, Tintore M, Villar LM, Costa C, Nurtdinov R, Alvarez-Cermeno JC, et al. Chitinase 3-like 1: prognostic biomarker in clinically isolated syndromes. Brain. 2015;138(Pt 4):918–31.
Hondius DC, Eigenhuis KN, Morrema THJ, van der Schors RC, van Nierop P, Bugiani M, et al. Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer's disease. Acta Neuropathol Commun. 2018;6(1):46.
Mann DMA, Snowden JS. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 2017;27(6):723–36.
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–9.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68.
Lund T. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry. 1994;57:2.
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5–22.
Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer's disease. Neurobiol Aging. 1997;18(4 Suppl):S85-8. https://doi.org/10.1016/s0197-4580(97)00062-6.
Greenberg SM, Vonsattel J-PG. Diagnosis of Cerebral Amyloid Angiopathy. Stroke. 1997;28(7):1418–22.
Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Del Tredici K, Braak H. Two Types of Sporadic Cerebral Amyloid Angiopathy. J Neuropathol Exp Neurol. 2002;61(3):11.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130-3. https://doi.org/10.1126/science.1134108.
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, Bayston A, Beach TG, Blanc F, Bohnen N, Bonanni L, Bras J, Brundin P, Burn D, Chen-Plotkin A, Duda JE, El-Agnaf O, Feldman H, Ferman TJ, Ffytche D, Fujishiro H, Galasko D, Goldman JG, Gomperts SN, Graff-Radford NR, Honig LS, Iranzo A, Kantarci K, Kaufer D, Kukull W, Lee VMY, Leverenz JB, Lewis S, Lippa C, Lunde A, Masellis M, Masliah E, McLean P, Mollenhauer B, Montine TJ, Moreno E, Mori E, Murray M, O'Brien JT, Orimo S, Postuma RB, Ramaswamy S, Ross OA, Salmon DP, Singleton A, Taylor A, Thomas A, Tiraboschi P, Toledo JB, Trojanowski JQ, Tsuang D, Walker Z, Yamada M, Kosaka K. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88-100. https://doi.org/10.1212/WNL.0000000000004058.
Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878.
Templeton GF. A Two-Step Approach for Transforming Continuous Variables to Normal: Implications and Recommendations for IS Research. Commun Assoc Inf Syst. 2011;28. https://doi.org/10.17705/1CAIS.02804.
Irwin DJ, Lleo A, Xie SX, McMillan CT, Wolk DA, Lee EB, et al. Ante mortem cerebrospinal fluid tau levels correlate with postmortem tau pathology in frontotemporal lobar degeneration. Ann Neurol. 2017;82(2):247–58.
Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, Pirttilä T. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66(3):382-9. https://doi.org/10.1001/archneurol.2008.596.
Aisen PS, Cummings J, Jack CR Jr, Morris JC, Sperling R, Frolich L, et al. On the path to 2025: understanding the Alzheimer's disease continuum. Alzheimers Res Ther. 2017;9(1):60.
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–72.
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535–62.
Sanfilippo C, Malaguarnera L, Di Rosa M. Chitinase expression in Alzheimer's disease and non-demented brains regions. J Neurol Sci. 2016;369:242–9.
Molina-Martinez P, Corpas R, Garcia-Lara E, Cosin-Tomas M, Cristofol R, Kaliman P, et al. Microglial Hyperreactivity Evolved to Immunosuppression in the Hippocampus of a Mouse Model of Accelerated Aging and Alzheimer's Disease Traits. Front Aging Neurosci. 2020;12:622360.
Higginbotham L, Ping L, Dammer EB, Duong DM, Zhou M, Gearing M, Hurst C, Glass JD, Factor SA, Johnson ECB, Hajjar I, Lah JJ, Levey AI, Seyfried NT. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer's disease. Sci Adv. 2020;6(43):eaaz9360. https://doi.org/10.1126/sciadv.aaz9360.
Llorens F, Zafar S, Ansoleaga B, Shafiq M, Blanco R, Carmona M, et al. Subtype and regional regulation of prion biomarkers in sporadic Creutzfeldt-Jakob disease. Neuropathol Appl Neurobiol. 2015;41(5):631–45.
Fagan AM, Xiong C, Jasielec MS, Bateman RJ, Goate AM, Benzinger TL, et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer's disease. Sci Transl Med. 2014;6(226):226ra30.
Schindler SE, Li Y, Todd KW, Herries EM, Henson RL, Gray JD, et al. Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer's disease. Alzheimers Dement. 2019;15(5):655–65.
Lleo A, Alcolea D, Martinez-Lage P, Scheltens P, Parnetti L, Poirier J, et al. Longitudinal cerebrospinal fluid biomarker trajectories along the Alzheimer's disease continuum in the BIOMARKAPD study. Alzheimers Dement. 2019;15(6):742–53.
Villar-Pique A, Schmitz M, Hermann P, Goebel S, Bunck T, Varges D, et al. Plasma YKL-40 in the spectrum of neurodegenerative dementia. J Neuroinflammation. 2019;16(1):145.
Renkema GH, Boot RG, Au FL, Donker-koopman WE, Strijland A, Muijsers AO, et al. Chitotriosidase, a chitinase, and the 39-kDa human cartilage glycoprotein, a chitin-binding lectin, are homologues of family 18 glycosyl hydrolases secreted by human macrophages. Eur J Biochem. 1998;251:5.
Bjornbak C, Brochner CB, Larsen LA, Johansen JS, Mollgard K. Brain barriers and a subpopulation of astroglial progenitors of developing human forebrain are immunostained for the glycoprotein YKL-40. J Histochem Cytochem. 2014;62(5):369–88.
Strominger I, Elyahu Y, Berner O, Reckhow J, Mittal K, Nemirovsky A, et al. The Choroid Plexus Functions as a Niche for T-Cell Stimulation Within the Central Nervous System. Front Immunol. 2018;9:1066.
Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J. 2014;33(1):7–22.
Stopa EG, Tanis KQ, Miller MC, Nikonova EV, Podtelezhnikov AA, Finney EM, et al. Comparative transcriptomics of choroid plexus in Alzheimer's disease, frontotemporal dementia and Huntington's disease: implications for CSF homeostasis. Fluids Barriers CNS. 2018;15(1):18.
Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346(6205):89–93.
Steeland S, Gorlé N, Vandendriessche C, Balusu S, Brkic M, Van Cauwenberghe C, Van Imschoot G, Van Wonterghem E, De Rycke R, Kremer A, Lippens S, Stopa E, Johanson CE, Libert C, Vandenbroucke RE. Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer's disease. EMBO Mol Med. 2018;10(4):e8300. https://doi.org/10.15252/emmm.201708300.
Baldacci F, Lista S, Palermo G, Giorgi FS, Vergallo A, Hampel H. The neuroinflammatory biomarker YKL-40 for neurodegenerative diseases: advances in development. Expert Rev Proteomics. 2019;16(7):593–600.
Lananna BV, McKee CA, King MW, Del-Aguila JL, Dimitry JM, Farias FHG, Nadarajah CJ, Xiong DD, Guo C, Cammack AJ, Elias JA, Zhang J, Cruchaga C, Musiek ES. Chi3l1/YKL-40 is controlled by the astrocyte circadian clock and regulates neuroinflammation and Alzheimer's disease pathogenesis. Sci Transl Med. 2020;12(574):eaax3519. https://doi.org/10.1126/scitranslmed.aax3519.
Choi JY, Yeo IJ, Kim KC, Choi WR, Jung JK, Han SB, et al. K284-6111 prevents the amyloid beta-induced neuroinflammation and impairment of recognition memory through inhibition of NF-kappaB-mediated CHI3L1 expression. J Neuroinflammation. 2018;15(1):224.
Acknowledgements
We acknowledge L. Boonkamp from the Neurochemistry Laboratory department of Clinical Chemistry at the VU University Medical Centers for her technical assistants.
Funding
This project was funded by the memorable project PRODIA (project number 733050206) which is supported by ZonMw (The Netherlands).
Author information
Authors and Affiliations
Contributions
YSH contributed to the study design and was responsible for acquisition, statistical analysis, and interpretation of the data. All authors participated in data acquisition or interpretation. YSH drafted the manuscript. All authors critically revised the manuscript. JJMH, CET, and MC conducted the study concept and design, data acquisition, and interpretation, critically revised the manuscript, and supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethical review boards of each center. Informed consent was obtained from all subjects or their authorized representative.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1
. Additional information for the individual cases included in this study.
Additional file 2: Supplementary Table 2
. Demographic data of paired ante-mortem CSF and post-mortem tissue samples. Supplementary Table 3. Demographic details of CSF samples. Supplementary figure 1. Different antibodies show similar YKL-40 staining patterns. Supplementary figure 2. Antibody characterization. Supplementary figure 3. Representative image of the YKL-40 immunoreactivity semi-quantification. Supplementary figure 4. Full Western blot showing YKL-40 and actin protein bands in post-mortem frontal cortex from AD, FTLD and controls.
Additional file 3: Supplementary figure 5
. YKL-40 immunoreactivity in post-mortem temporal cortex tissue does not correlate to pathology stages. Supplementary figure 6. YKL-40 levels are increased in CSF of AD patients. Supplementary figure 7. YKL-40 protein levels remain similar in post-mortem frontal cortex between the FTLD subclassifications and non-demented controls. Supplementary figure 8. YKL-40 levels in ante-mortem CSF are inversely associated with YKL-40 levels in post-mortem brain.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hok-A-Hin, Y.S., Hoozemans, J.J.M., Hu, W.T. et al. YKL-40 changes are not detected in post-mortem brain of patients with Alzheimer’s disease and frontotemporal lobar degeneration. Alz Res Therapy 14, 100 (2022). https://doi.org/10.1186/s13195-022-01039-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-022-01039-y