
Abstract
Background
Many Alzheimer’s disease (AD) genetic association studies disregard age or incorrectly account for it, hampering variant discovery.
Methods
Using simulated data, we compared the statistical power of several models: logistic regression on AD diagnosis adjusted and not adjusted for age; linear regression on a score integrating case-control status and age; and multivariate Cox regression on age-at-onset. We applied these models to real exome-wide data of 11,127 sequenced individuals (54% cases) and replicated suggestive associations in 21,631 genotype-imputed individuals (51% cases).
Results
Modeling variable AD risk across age results in 5–10% statistical power gain compared to logistic regression without age adjustment, while incorrect age adjustment leads to critical power loss. Applying our novel AD-age score and/or Cox regression, we discovered and replicated novel variants associated with AD on KIF21B, USH2A, RAB10, RIN3, and TAOK2 genes.
Conclusion
Our AD-age score provides a simple means for statistical power gain and is recommended for future AD studies.
Similar content being viewed by others

Background
Genetics plays an important role in the onset of Alzheimer’s disease (AD) with an estimated heritability ranging from 58 to 79% [1]. Over the last decade, genome-wide association studies (GWAs) of AD have identified over 40 susceptibility loci [2,3,4,5], by meta-analyzing genotype-imputed data from numerous cohorts genotyped on various single nucleotide polymorphism (SNP) arrays. With each updated GWA, the increasing sample sizes and improved imputation quality of low frequency variants have enabled additional discoveries. A complementary approach is to use next generation sequencing to directly genotype every variant, alleviating the need for imputation and enabling rare variant discoveries. To this aim, the Alzheimer’s Disease Sequencing Project (ADSP) undertook whole-exome sequencing (WES) of 10,836 individuals (53% cases) which led to the discovery of novel AD risk genes [6, 7]. The ADSP individuals were part of existing AD cohorts and were selected based on a risk score accounting for APOE ε2 and APOE ε4 alleles, sex, and age at onset (AAO) for cases and age at last exam or death for controls [6]. This design promoted the inclusion of controls least likely to develop AD by age 85 years and was shown to maximize statistical power compared to other approaches such as using age-matched cases/controls [6].
Across prior AD GWAs, the common approach to association testing was to perform case-control logistic regression analyses adjusted for age. Theoretically, this adjustment should account for increasing AD prevalence with age in the population, independently of genetic factors [8, 9]. However, most AD cohorts include the AAO for cases and last known age without cognitive impairment for controls. This common design leads to the average age of cases being lower than the average age of controls. If one performs a case-control logistic regression with a traditional age adjustment, the model will infer that age has a negative effect on AD risk, meaning that younger individuals are more likely to develop AD. Since advanced age is the greatest risk factor for AD [9], it appears essential to correctly account for age. The latter conundrum is particularly relevant to the ADSP where, by design, the average age of controls is 10 years greater than that of cases.
In this work, we aimed to improve on prior AD GWA studies by evaluating and implementing models that inherently, correctly account for age effects on AD. To this aim, we estimated the statistical power of different models on simulated data, reflecting various age differences between cases and controls as found in AD cohorts. These models included logistic regression on AD case-control status adjusted and not adjusted for age, linear regression on a newly designed score which weights case-control status by age, and multivariate Cox regression on AAO, which models cumulative conversion risk across the life span. We then applied these models to exome-wide AD data with a next generation sequenced discovery sample (5075 controls and 6052 cases) and replicated suggestive associations in an independent sample of genotype-imputed individuals (10,539 controls and 11,092 cases).
Methods
Power simulations
We performed power simulation studies to evaluate the performance of different AD genetic association models (R.v3.5.1). We first simulated population level data that mimics population AD prevalence estimates at ages 60–100 across a range of age-related risk effect estimates (OR 1.01–1.25) [10, 11]. The age effect estimate on AD status (OR 1.16) served as a reference to evaluate power for AD GWA studies [12]. We then simulated AD case-control datasets by random sampling of cases and controls from the population level data. To simulate realistic AD case-control datasets [13,14,15], subjects’ mean age was centered on 75 years following a binomial distribution with a standard deviation of 8 years. Simulated subjects were restricted to the age range of 60–100, after which cases and controls were randomly drawn abiding by model conditions. To evaluate how age differences between cases and controls affect power for variant discovery, subjects were further sampled to three conditions: (1) no mean age difference between cases and controls, (2) cases’ mean age is 5 years younger than in controls, and (3) cases’ mean age is 10 years younger than in controls. These conditions, particularly condition 2, are similar to those observed for common AD GWAS cohorts [13,14,15], while condition 3 mimics the design of the ADSP WES study. The power was calculated based on 1000 simulation replicates, and the linear regression on the AD-age score was estimated with bootstrap-based inference (100 resamplings). Each replicate included either 1000 cases and 1000 controls, or 5000 cases and 5000 controls, respectively testing for a significance level of α = 0.05 or α = 5 × 10−7 (i.e., exome-wide significance). These parameters respectively mimic common AD GWA cohorts and the ADSP WES study [7]. We evaluated power for a range of realistic effect sizes (OR 1.05, 1.10, 1.20, 1.50) and common minor allele frequency (MAF) 0.01, 0.05–0.45 (at 0.05 increments) Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD).
Participants
All samples were available from publicly released AD-related cohorts, with phenotype and genotype ascertainment described elsewhere [3, 6, 13, 16,17,18,19,20,21,22,23,24,25,26].
The European individuals in ADSP WES [6, 7], ADSP whole-genome sequencing (WGS) [21, 25], and the Accelerating Medicine Partnership in AD (AMP-AD) WGS [22, 24, 26] cohorts comprise our discovery sample and were mega-analyzed (Table 1 and Table S1). The ADSP WES selection criteria have already been introduced; the selection scheme led to a 10 years’ average age difference between cases and controls [6, 7]. For AMP-AD, the reported age for cases was not always AAO; thus, the average age of controls was only 2 years greater than that of cases (Fig. 1).

Proposed AD-age score visualization and its distribution in the discovery and replication samples. a Illustration of the proposed AD-age scores with a linear and a piecewise definition of the weight (age) function (see the “Methods” section). b, c Cases (AD) and controls (CN) age distribution in the discovery, composed of next generation sequencing data, and in the replication, composed of SNP-array imputed data, and d, e their respective AD-age score distributions
As a replication sample, we mega-analyzed 34 cohorts, each corresponding to a specific SNP array applied to an AD case/control dataset [3, 16,17,18,19,20,21,22,23,24]. Some of these cohorts correspond to the same AD study but individuals were genotyped on different platforms. These cohorts are heterogeneous in terms of age reported and are extensively described elsewhere [3, 13] (Table 1 and Table S2). When multiple ages were available for a given subject, the order of priority for which age to use was AAO then age at examination then age at death in affected individuals, and age at death then age at last examination in control participants [13]. We removed any duplicated individuals across these cohorts and the discovery sample.
Genetic quality control
For each cohort included in our analysis, we first determined the ancestry of each individual with SNPWeights v2.1 [27] using reference populations from the 1000 Genomes Consortium [28]. Prior to ancestry determination, variants were filtered based on genotyping rate (< 95%), MAF < 1% and Hardy-Weinberg equilibrium (HWE) in controls (p < 10−6). By applying an ancestry percentage cut-off > 75%, the samples were stratified into five super populations: South-Asians, East-Asians, Americans, Africans and Europeans, and an admixed group composed of individuals not passing cut-off in any single ancestry. Since most individuals were Europeans and to avoid spurious associations, we focused on European ancestry individuals.
Carriers of known pathogenic mutations on APP, PSEN1, PSEN2, and MAPT were excluded from our analysis. Discordant pathology cases, defined as any clinically diagnosed AD individual with Braak stage below III or neuritic plaques level below moderate, were excluded from our analysis.
The joint called set of exome variants in the ADSP WES is composed of 1,524,414 SNPs [6, 7]. We restricted downstream analysis to these variants, meaning that variants called only in ADSP WGS or AMP-AD were not included. To remove potential sequencing artifacts, we applied several quality control (QC) steps to each dataset. First, SNPs were checked for consistency with the Haplotype reference consortium (HRC) panel [29]. This check included flipping SNPs reported on incorrect strand and excluding SNPs with more than 10% MAF difference with the HRC panel. Second, we removed SNPs that deviated from HWE in controls (p < 10−6) or that had a genotyping rate below 95%. Third, we removed any variants which had a flag different than PASS in gnomADv3 [30].. Following these QC steps, 905,341 variants remained. For analysis, we considered 124,679 variants with minor allele count above 10, to ensure a minimum number of carriers.
In each cohort of the replication sample, SNPs with less than 95% genotyping rate or deviating from HWE in controls (p < 10−6) were excluded. Then, we used the gnomAD database [30] to filter out SNPs that met one of the following criteria: (i) located in low complexity region, (ii) located within common structural variants (MAF > 1%), (iii) multiallelic SNPs with MAF > 1% for at least two alternate alleles, (iv) located within a common Ins/Del (insertion/deletion), (v) having any flag different than PASS in gnomAD, and (vi) having potential probe polymorphisms [31]. The latter are defined as SNPs for which the probe may have variable affinity due to the presence of other SNP(s) within 20 bp and with MAF > 1%. Individuals with more than 5% genotype missingness were excluded. Imputation was performed on the Michigan imputation server using the TOPMed reference panel [32, 33]. Per cohort, only variants with sufficient imputation quality (r2 > 0.3) were included in the replication analysis (Table S3).
Identity-by-descent was run to determine the relatedness between all individuals using PLINKv1.9 [34]. In the discovery sample, we kept only one version of duplicated individuals and removed first degree relatives keeping AD relatives over controls, and when both had a concordant diagnosis, we kept the younger case or older control. In the replication sample, we removed any individuals already present in the discovery, and for duplicate subjects, we kept the copy from the SNP array with the highest genome coverage.
On the subset of remaining individuals, we computed genetic principal components to account for population stratification [35] in both the discovery and replication samples, separately.
Statistics, association models, and AD-age score
We considered four main models: logistic regression on AD diagnosis adjusted for age, logistic regression on AD diagnosis, linear regression on a score integrating case-control status and age, and multivariate Cox regression on AAO. When AAO was not available, the first known age with AD diagnosis was used. Our analyses removed individuals younger than 60 and censored maximum age at 100. We considered controls below 60 as uninformative and cases below 60 as early onset AD potentially due to a causal mutation.
For the third model, we defined the AD-age score as follows:
-
log(1-weight (age)) − 0.5 for controls
-
−log (weight (age)) + 0.5 for cases
The score was designed to abide by the following rules: cases and controls should be clearly separated (maximum value for controls − 0.5 and minimum value for cases + 0.5, ensuring that the minimum difference between cases/controls is greater than 1), younger cases should have higher scores compared to older cases, and older controls should have lower scores than younger ones. This ensured that younger cases and older controls were at opposite extremes of the score spectrum and assumed these individuals influenced genetic associations the most.
We defined two weight (age) functions:
-
A.
A linear definition: weight (age) = (age-59.5)/(100.5–59.5);
-
B.
A piecewise continuous definition:
○ 60 and below: weight (age) = 5/320
○ > 60 to 65: weight (age) = (age−55)/320
○ > 65 to 75: weight (age) = 4*(age−55)/320–3/32
○ > 75 to 80: weight (age) = 10*(age−55)/320–15/32
○ > 80 to 90: weight (age) = 16*(age−55)/320–30/32
○ > 90 to 100: weight (age) = 6*(age−55)/320 + 5/32
(A) corresponds to a linear effect of age between 60 and 100 and (B) accounts for the changes in AD prevalence slope in this age range [8] (Fig. 1).
For the analysis of exome-wide data, all models had two subversions: (1) adjusted for sex and 10 first principal components of population structure and (2) additionally adjusted for APOE ε2 and APOE ε4 alleles.
The associations for the first three models were estimated with PLINKv2.0 [36] using the –glm flag, which performs a logistic regression for case/control phenotype and a linear regression for quantitative phenotype. The Cox regression associations were estimated with gwasurvir [37].
We calculated the number of independent variants with PLINKv1.9 [34] (option –indep-pairwise 1000 50 0.1), which identified 87,034 linkage disequilibrium blocks covering the 124,679 considered variants. Thus, the exome-wide threshold was set at p < 5 × 10−7 (0.05/87034, Bonferroni correction) and the suggestive threshold at p < 1 × 10−5 (1/87304). A 1 Mb region around the APOE locus was excluded from the reported results due to its well-established association with AD. We did not correct for the number of tested models due to their high correlation (cf. Results), nor for the two versions of adjustment (APOE ε2 and APOE ε4 alleles adjusted or not), as in Bis et al. [7], since these were similarly highly correlated.
Thirty-one variants passing the suggestive threshold in the discovery were evaluated in the replication sample. We disentangled spurious and true associations based on their associations in the replication dataset. SNVs with discordant direction of effect were considered to be spurious associations. Variants which had a concordant direction of effect and p < 1.6 × 10−3 (0.05/31, Bonferroni correction) in the same regression model, allowing different covariate adjustment, were considered significant, while those with p < 0.05 were considered to replicate nominally.
For more robust and powerful inference with the AD-age score, which is not normally distributed, we performed bootstrapping (100 resamplings) consistent with what was done in power simulations. To limit the computational burden, we only computed the bootstrap-based inference for the set of replicated variants, which allowed us to compare the significance of the linear regression on AD-age score with the Cox regression for true associations.
Last, we performed a fixed-effect meta-analysis using the metafor package in R [38] to estimate the significance of the replicated variants in the combined discovery and replication samples .
Gene and variant annotations
Each variant consequence was annotated with the Ensembl Variant Effect Predictor toolset [39]. Non-synonymous variants, such as missense or frameshift variants, may lead to loss or gain of function that may affect the enzymatic activity, stability, and/or interaction properties at the protein level. Synonymous variants, by contrast, do not typically directly affect protein function; however, they can influence protein expression both at the transcriptional and translational level [40].
To disentangle the role of the synonymous common variants as potential expression quantitative trait loci (eQTL), we queried the largest brain cis-eQTL meta-analysis which included 1433 post-mortem brain samples from the AMP-AD and CommonMind Consortium [41].
Lastly, for mapped genes harboring significant variants, we queried the AMP-AD fixed-effect meta-analysis of gene differential expression between AD and control individuals across brain tissues [22, 24, 26].
Results
Age-informed AD risk estimation increases power for genetic association testing
Power outcomes for specific illustrations of simulation analyses, considering a range of age-related risk effect estimates, are presented in Fig. 2 and Figure S1. An overview of power differences between different association models for all simulations’ conditions, varying the AD risk associated with age, is provided in Figure S2. In simulations where the mean age of cases was younger than in controls, adjustment for age in logistic regression analyses compared to not adjusting for age led to critical power loss (Fig. 2), amounting to as much as 90% power loss in some conditions (Figure S2 A-D). The AD-age score model performed best overall across all four models, displaying power increases regardless of age differences between cases and controls, particularly for the estimated age effect on AD status [12] corresponding to the vertical gray line on Fig. 2, S2. Power gain of the AD-age score with regard to logistic regression not adjusted for age was on average 5%, up to 10% in some scenarios (Figure S2 C-D). The Cox regression on AAO performed worse than the unadjusted logistic regression when the cases and controls were age matched and better when the age difference increased (Fig. 2e, f). Power gain of the AD-age score with regard to Cox regression was between 5 and 10% in some scenarios, notably when cases controls were age matched (Figure S2 G-H). When age difference is 10 years, the AD-age score and Cox regression performs similarly with some scenarios showing 1% increased power for AD-age while others showed 0–2% power gain for the Cox regression. Figure S3 shows that all models have the same type I error control under our simulation paradigm.

Power of different association models for two specific simulation outcomes. a–c A common variant with moderate effect size, evaluated in 1000 cases and 1000 controls at a significance level of α = 0.05, mimicking the condition of common AD cohorts genotyped on SNP arrays. d–f An uncommon variant with large effect size, evaluated in 5000 cases and 5000 controls at a significance level of α = 5 × 10−7, mimicking the condition of ADSP WES which allows exploration of uncommon and rare variant associations. Panels show power on the y-axis and age-related effect estimates on the x-axis. Outcomes for four models are shown (cf. legend) and the age-related effect estimate for AD [OR (Age-AD)] is marked by a vertical gray dotted line. From left to right, panels show simulation results for increasing mean age differences between cases and controls (cases being younger than controls where applicable)
Exome-wide association
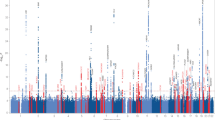
Exome-wide association with AD in the discovery sample for all four models and their subversions are shown in (Figure S4-S7). QQ plots for each exome-wide association show no inflation (λ < 1.1), except for the Cox regression adjusted for APOE ε2 and APOE ε4 allele dosages (λ = 1.19) (Table S4, Figure S8-S11). The logistic regression adjusted for age showed no associations above the suggestive threshold outside of the APOE region (Figure S4). Across the three other models, a total of 31 variants passed suggestive significance, including 5 known AD risk loci [7]. The parameter estimates of these models: (i) OR (odd ratio) for logistic regression, (ii) exp(β) for the linear regression, and (iii) HR (hazard ratio) for the Cox regression were found to be highly correlated (Figure S12), with (i–ii) Pearson correlation: r2 = 0.80 (p = 3 × 10−12), (i–iii) r2 = 0.84 (p = 4 × 10−14), and (ii–iii) r2 = 0.97 (p < 2 × 10−16). The known TREM2 missense single nucleotide variant (SNV) (rs75932628) was exome-wide significant in the three models. Other known associations included synonymous SNVs on PILRA (rs2405442), MS4A6A (rs12453), NSF (rs199533, lead SNV of a locus also encompassing MAPT and KANSL1), and a frameshift deletion on ABCA7 (rs547447016) (Fig. 3, Tables 2 and 3). The association on PILRA was exome-wide significant in the AD-age score linear regression and suggestive in the Cox regression but did not reach the suggestive threshold in the logistic regression. Similarly, the association on ABCA7 was suggestive in both AD-age score and Cox regressions, but not in the logistic regression. On the contrary, the association on MS4A6A was suggestive in the logistic regression and in the AD-age score and just below significance in the Cox regression. The association on NSF/MAPT/KANSL1 was suggestive in all three models. In addition to these 5 known exonic associations, associations on 26 other exonic loci were at least suggestive in one of the three models (Table S5). Logistic regression (Figure S5) produced one spurious association on ETV3L, the AD-age score linear regression led to three spurious associations on TACR3, PCDHA7, and the one on ETV3L, while the Cox regression (Figure S6) had 16 spurious associations including the one on TACR3. The logistic regression model showed no novel suggestive association. The AD-age score linear regression, prior to bootstrap (Figure S7), produced two novel suggestive-level associations: one USH2A missense SNV (rs111033333) and one RIN3 missense SNV (rs150221413), which replicated nominally. The Cox regression produced several exome-wide significant associations in the discovery with concordant direction of effect in the replication including NAV2 (rs11828836), RAB10 (rs149622307), and the USH2A and RIN3 associations, also found in the AD-age score linear regression. Among suggestive associations in the Cox regression, two significantly replicated: RAB10 synonymous SNV (rs149622307) and TAOK2 synonymous SNV (rs4077410), and three nominally replicated: KIF21B synonymous SNV (rs2297911), and the previous missenses on USH2A and RIN3. NAV2 synonymous SNV (rs11828836) did not reach nominal significance (p = 0.17), but it was imputed with sufficient quality in only 9235 individuals (less than 50% of imputed individuals). CDKL1 intronic SNV (rs61981931) did not reach nominal significance (p = 0.09).

Manhattan plots of exome wide associations in the four main models excluding the APOE region. The age-adjusted logistic regression has no suggestive association (dashed line, p < 1 × 10−5). The main causal variant on TREM2 is exome-wide significant (solid line, p < 5 × 10−7) in the other three models. Among suggestive associations, (i) known AD associations are in red and (ii) novel associations which replicate (p < 0.05) in an independent dataset are in blue (cf Table 3). Colored dots were bootstrapped in the AD-age score model (see the “Methods” section). The minimum p value from the adjustment models for each main model is displayed as in [7]
For the set of replicated variants (Table 2), we meta-analyzed the discovery and independent replication results. Seven out of the ten exonic variants were most significant in the linear regression on the AD-age score, while only two performed best in the Cox regression, those on KIF21B and TAOK2, and one in the logistic regression, on MS4A6A (Figure S13). After meta-analysis, the variants located on RAB10, TREM2, PILRA, MS4A6A, and RIN3 were exome-wide significant (p < 5 × 10−7) (Table S6).
Functional annotation
Among the mapped genes (Table 3), the synonymous variants on PILRA and KANSL1 were significantly associated with the expression of their respective mapped gene (false discovery rate (FDR) corrected). At the nominal significance level, TAOK2 and KIF21B synonymous variants were also associated with the expression of their respective genes. Among nearby genes with FDR-significant eQTL association, PVRIG was the strongest association at the PILRA locus, KANSL1-AS1 at the NSF/MAPT/KANSL1 locus, and INO80E at the TAOK2 locus (Table S6).
In the meta-analysis of differential gene expression across brain tissues in AMP-AD, TREM2, KANSL1, RAB10, MS4A6A, and RIN3 were found to be significantly upregulated in AD compared to control individuals, while TAOK2 was significantly downregulated (reported associations were FDR-significant, Table S7).
Discussion
In the AD data simulation, we showed that incorrectly adjusting for age led to critical power loss and that weighting the known effect of age on AD risk in the phenotype increased statistical power for variant discovery. Testing these models on real AD data confirmed our simulation observations and enabled the discovery of novel variants modulating AD risk.
Previous literature
The main prior AD WES study aimed to address the age adjustment conundrum in the ADSP WES data by implementing three different logistic regression models: the main one being unadjusted for age, while the other two were age adjusted [7]. However, given that cases were on average younger than controls, the age adjustment was in the opposite direction of the true age effect on AD risk. It is perhaps unsurprising, therefore, that there were no replicated findings from the two age-adjusted models (only associations from the main age-unadjusted model in the ADSP discovery were replicated) [7].
An alternative approach has been to use Cox regression on AAO for improved power compared to logistic regression that only considers case-control status. Cox regression has proven successful in predicting an individual’s AD conversion risk by calculating a polygenic hazard score [42, 43]. However, it needs to abide by several assumptions, including proportional hazards across age. Several studies have shown that Cox regression performs better than logistic regression on case-control data when AAO is available [44, 45], but it has not been applied to the ADSP WES data. Cox regression was previously applied to AD GWA, using genotype-imputed data overlapping partially with the ADSP sample used here, and led to the discovery of novel associations [46]. Alternative approaches have been proposed when Cox regression’s assumptions are violated as in AD GWA, including age stratification [47] and generalized Cox regression [48]. Our proposed AD-age score offers additional flexibility without these assumptions and it can accommodate age information other than AAO such as age-at-study and age-at-death. Unlike Cox regression models, the AD-age score can be flexibly incorporated as a quantitative outcome into conventional tools (e.g., PLINK) for GWAS and new methods (e.g., BOLT-LMM, SAIGE) for analysis of large/biobank scale genetic data with related samples. Additionally, the linear and logistic regressions are faster than Cox regression and thus more advantageous for larger datasets [44].
Oversampling cases with early AAO and controls with late censoring time for exome sequencing is an efficient design because it directs limited study resources towards subjects that are most useful for discovering the genetic associations of AD in the original cohorts [49, 50]. We proposed the AD-age score for improved power in the discovery stage and validated the findings using an independent replication sample. Although the hypothesis testing is appropriate in the discovery stage with extreme sampling, it is worth noting that the estimated genetic effect/odds ratio may not represent that in the whole population [51]. To obtain unbiased genetic effect estimations of AD risk in the whole population, it may be advisable to turn to more advanced methods that can explicitly address the biased sampling design (e.g., [49, 52]).
Potential disease mechanisms
The novel variants identified through our exome-wide association, with the exception of the USH2A SNV, are located on genes previously linked to AD, re-enforcing our confidence in these associations (Table S8).
Our main finding is a rare variant on RAB10 passing the exome-wide threshold in discovery and surviving Bonferroni correction in the replication. RAB proteins are key regulators of vesicular trafficking and play a major role in the endolysosomal and retromer pathways known to be linked to AD [53]. Another rare RAB10 SNV was shown to segregate with AD resilience in pedigrees at risk for AD and RAB10 was shown to be upregulated in AD brains [54], a finding corroborated in our study. RAB10 knockdown significantly decreased Aβ42 and Aβ42/Aβ40 ratio in neuroblastoma cells [54]. Silencing of RAB10 decreased β-amyloid peptides (Aβ) and increased soluble ectodomain of APP β (sAPPβ) [55], supporting a role of RAB10 in either γ-secretase cleavage of APP and the degradation of Aβ. Moreover, phosphorylated Rab10 was prominent in neurofibrillary tangles in the hippocampus of AD individuals but scarce in controls [56]. Mechanistically, the JNK-interactin protein 1 (JIP1) mediates the anterograde transport of Rab10-positive cargo to axonal tips which promotes axonal growth and is critical for proper neuronal function [57]. JIP1 also regulates anterograde and retrograde transport of APP along axons [58]. These molecular mechanisms suggest that Rab10 could play a role in APP trafficking along axons.
Additionally, our exome-wide analysis identified a missense variant on Rab interactor 3 (RIN3). Common variants in a locus near RIN3 and SLC24A4 were reported to be associated with AD susceptibility [2]. Increased RIN3 expression in APP/PS1 mouse models was shown to correlate with endosomal dysfunction and altered axonal trafficking and processing of APP [59]. For these reasons, the Rab-related proteins involved in the endolysosomal and retromer pathways have been considered as promising therapeutic targets for AD [53].
Two common exonic variants, on TAOK2 and KIF21B, were identified as suggestive in our discovery analysis and replicated (Bonferroni corrected and nominally, respectively). Previous AD GWAS summary statistics show a concordant direction of effect with our analysis [2, 3] with the SNVs p values on TAOK2 and KIF21B in those studies equal to 0.05 and 10−5, respectively (Table S8). TAOK2 was shown to be phosphorylated in AD and frontotemporal lobar degeneration brains. Its expression was colocalized with tangles and its inhibition reduced tau phosphorylation [60]. Further, KIF21B is involved in neuronal and synaptic signaling and increased KIF21B expression levels were associated with more severe AD pathology [61].
Limitation
For common synonymous variants, the regulated gene and true causal variant remain uncertain because our study focused on exomes and we cannot perform a genome-wide colocalization analysis. The causal variant may be intergenic and in linkage disequilibrium with a common synonymous variant identified in our analysis. Thus, future genome-wide studies are warranted to help disentangle which nearby genes are regulated, notably for the novel common loci encompassing KIF21B and TAOK2.
Conclusion
Correctly accounting for the risk-increasing effect of age on AD is an efficient means of increasing statistical power. Thus, our AD-age score should prove useful in future AD genetic association studies to enable the discovery of additional novel variants.
Availability of data and materials
The summary statistics of the discovery analysis, the simulation code, and a snippet to compute the AD-age scores are available at:
https://github.com/YannLeGuen/AD-age_score
All samples were available from publicly released AD-related cohorts, with phenotype and genotype ascertainment described elsewhere [3, 6, 13, 16,17,18,19,20,21,22,23,24,25,26].
https://www.synapse.org/#!Synapse:syn2580853/files/
https://dss.niagads.org/datasets/ng00067/
References
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–74. https://doi.org/10.1001/archpsyc.63.2.168.
Lambert J-C, Sorbi S, Matthews F, Moebus S, Beekly D, Owen MJ, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8. https://doi.org/10.1038/ng.2802.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30. https://doi.org/10.1038/s41588-019-0358-2.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, Voyle N, Proitsi P, Witoelar A, Stringer S, Aarsland D, Almdahl IS, Andersen F, Bergh S, Bettella F, Bjornsson S, Brækhus A, Bråthen G, de Leeuw C, Desikan RS, Djurovic S, Dumitrescu L, Fladby T, Hohman TJ, Jonsson PV, Kiddle SJ, Rongve A, Saltvedt I, Sando SB, Selbæk G, Shoai M, Skene NG, Snaedal J, Stordal E, Ulstein ID, Wang Y, White LR, Hardy J, Hjerling-Leffler J, Sullivan PF, van der Flier WM, Dobson R, Davis LK, Stefansson H, Stefansson K, Pedersen NL, Ripke S, Andreassen OA, Posthuma D. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–13. https://doi.org/10.1038/s41588-018-0311-9.
Andrews SJ, Fulton-Howard B, Goate A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020;19(4):326–35. https://doi.org/10.1016/S1474-4422(19)30435-1.
Beecham GW, Bis JC, Martin ER, Choi S-H, DeStefano AL, van Duijn CM, et al. The Alzheimer’s disease sequencing project: study design and sample selection. Neurol Genet. 2017;3(5):e194. https://doi.org/10.1212/NXG.0000000000000194.
Bis JC, Jian X, Kunkle BW, Chen Y, Hamilton-Nelson KL, Bush WS, et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-associated variants involved in immune response and transcriptional regulation. Mol Psychiatry. 2020;25(8):1859–75. https://doi.org/10.1038/s41380-018-0112-7.
van der Lee SJ, Wolters FJ, Ikram MK, Hofman A, Ikram MA, Amin N, et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol. 2018;17:434–44.
Guerreiro R, Bras J. The age factor in Alzheimer’s disease. Genome Med. 2015;7 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4617238/. [cited 2020 Aug 1].
Nichols E, Szoeke CEI, Vollset SE, Abbasi N, Abd-Allah F, Abdela J, Aichour MTE, Akinyemi RO, Alahdab F, Asgedom SW, Awasthi A, Barker-Collo SL, Baune BT, Béjot Y, Belachew AB, Bennett DA, Biadgo B, Bijani A, Bin Sayeed MS, Brayne C, Carpenter DO, Carvalho F, Catalá-López F, Cerin E, Choi JYJ, Dang AK, Degefa MG, Djalalinia S, Dubey M, Duken EE, Edvardsson D, Endres M, Eskandarieh S, Faro A, Farzadfar F, Fereshtehnejad SM, Fernandes E, Filip I, Fischer F, Gebre AK, Geremew D, Ghasemi-Kasman M, Gnedovskaya EV, Gupta R, Hachinski V, Hagos TB, Hamidi S, Hankey GJ, Haro JM, Hay SI, Irvani SSN, Jha RP, Jonas JB, Kalani R, Karch A, Kasaeian A, Khader YS, Khalil IA, Khan EA, Khanna T, Khoja TAM, Khubchandani J, Kisa A, Kissimova-Skarbek K, Kivimäki M, Koyanagi A, Krohn KJ, Logroscino G, Lorkowski S, Majdan M, Malekzadeh R, März W, Massano J, Mengistu G, Meretoja A, Mohammadi M, Mohammadi-Khanaposhtani M, Mokdad AH, Mondello S, Moradi G, Nagel G, Naghavi M, Naik G, Nguyen LH, Nguyen TH, Nirayo YL, Nixon MR, Ofori-Asenso R, Ogbo FA, Olagunju AT, Owolabi MO, Panda-Jonas S, Passos VMA, Pereira DM, Pinilla-Monsalve GD, Piradov MA, Pond CD, Poustchi H, Qorbani M, Radfar A, Reiner RC Jr, Robinson SR, Roshandel G, Rostami A, Russ TC, Sachdev PS, Safari H, Safiri S, Sahathevan R, Salimi Y, Satpathy M, Sawhney M, Saylan M, Sepanlou SG, Shafieesabet A, Shaikh MA, Sahraian MA, Shigematsu M, Shiri R, Shiue I, Silva JP, Smith M, Sobhani S, Stein DJ, Tabarés-Seisdedos R, Tovani-Palone MR, Tran BX, Tran TT, Tsegay AT, Ullah I, Venketasubramanian N, Vlassov V, Wang YP, Weiss J, Westerman R, Wijeratne T, Wyper GMA, Yano Y, Yimer EM, Yonemoto N, Yousefifard M, Zaidi Z, Zare Z, Vos T, Feigin VL, Murray CJL. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(1):88–106. https://doi.org/10.1016/S1474-4422(18)30403-4.
Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020;16:391.
Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, McDowell I. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol. 2002;156(5):445–53. https://doi.org/10.1093/aje/kwf074.
Belloy ME, Napolioni V, Han SS, Le Guen Y, Greicius MD. Association of Klotho-VS heterozygosity with risk of Alzheimer disease in individuals who carry APOE4. JAMA Neurol. 2020;77(7):849–62. https://doi.org/10.1001/jamaneurol.2020.0414.
Naj AC, Jun G, Reitz C, Kunkle BW, Perry W, Park YS, Beecham GW, Rajbhandary RA, Hamilton-Nelson KL, Wang LS, Kauwe JS, Huentelman MJ, Myers AJ, Bird TD, Boeve BF, Baldwin CT, Jarvik GP, Crane PK, Rogaeva E, Barmada MM, Demirci FY, Cruchaga C, Kramer PL, Ertekin-Taner N, Hardy J, Graff-Radford NR, Green RC, Larson EB, St George-Hyslop PH, Buxbaum JD, Evans DA, Schneider JA, Lunetta KL, Kamboh MI, Saykin AJ, Reiman EM, de Jager PL, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Martin ER, Haines JL, Mayeux RP, Farrer LA, Schellenberg GD, Pericak-Vance MA, Alzheimer Disease Genetics Consortium, Albert MS, Albin RL, Apostolova LG, Arnold SE, Barber R, Barnes LL, Beach TG, Becker JT, Beekly D, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Cantwell LB, Cao C, Carlson CS, Carney RM, Carrasquillo MM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cribbs DH, Crocco EA, DeCarli C, DeKosky S, Dick M, Dickson DW, Duara R, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Kramer JH, LaFerla F, Lah JJ, Leverenz JB, Levey AI, Li G, Lieberman AP, Lin CF, Lopez OL, Lyketsos CG, Mack WJ, Martiniuk F, Mash DC, Masliah E, McCormick W, McCurry S, McDavid A, McKee A, Mesulam M, Miller BL, Miller CA, Miller JW, Murrell JR, Olichney JM, Pankratz VS, Parisi JE, Paulson HL, Peskind E, Petersen RC, Pierce A, Poon WW, Potter H, Quinn JF, Raj A, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosen HJ, Rosenberg RN, Sano M, Schneider LS, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Tanzi RE, Thornton-Wells TA, Trojanowski JQ, Troncoso JC, Valladares O, van Deerlin V, van Eldik L, Vardarajan BN, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Wishnek S, Woltjer RL, Wright CB, Younkin SG, Yu CE, Yu L. Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: a genome-wide association study. JAMA Neurol. 2014;71(11):1394–404. https://doi.org/10.1001/jamaneurol.2014.1491.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, de Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JSK, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, George-Hyslop PS, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–43. https://doi.org/10.1038/ng.801.
Green RC, Cupples LA, Go R, Benke KS, Edeki T, Griffith PA, Williams M, Hipps Y, Graff-Radford N, Bachman D, Farrer LA, MIRAGE Study Group. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA. 2002;287(3):329–36. https://doi.org/10.1001/jama.287.3.329.
Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59(11):1737–46. https://doi.org/10.1001/archneur.59.11.1737.
Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R. Analyses of the National Institute on Aging late-onset Alzheimer’s disease family study: implication of additional loci. Arch Neurol. 2008;65(11):1518–26. https://doi.org/10.1001/archneur.65.11.1518.
Lovestone S, Francis P, Kloszewska I, Mecocci P, Simmons A, Soininen H, Spenger C, Tsolaki M, Vellas B, Wahlund LO, Ward M, on behalf of the AddNeuroMed Consortium. AddNeuroMed—the European collaboration for the discovery of novel biomarkers for Alzheimer’s disease. Ann N Y Acad Sci. 2009;1180(1):36–46. https://doi.org/10.1111/j.1749-6632.2009.05064.x.
Beecham GW, Martin ER, Li Y-J, Slifer MA, Gilbert JR, Haines JL, Pericak-Vance MA. Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet. 2009;84(1):35–43. https://doi.org/10.1016/j.ajhg.2008.12.008.
Weiner MW, Aisen PS, Jack CR, Jagust WJ, Trojanowski JQ, Shaw L, et al. The Alzheimer’s Disease Neuroimaging Initiative: progress report and future plans. Alzheimer’s Dement. 2010;6:202–211.e7.
Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res. 2012;9(6):646–63. https://doi.org/10.2174/156720512801322663.
Weiner MW, Veitch DP, Hayes J, Neylan T, Grafman J, Aisen PS, et al. Effects of traumatic brain injury and posttraumatic stress disorder on Alzheimer’s disease in veterans, using the Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dement. 2014;10:S226–35.
Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS, et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Scientific Data. 2016;3:160089.
Patel D, Mez J, Vardarajan BN, Staley L, Chung J, Zhang X, et al. Association of rare coding mutations with Alzheimer disease and other dementias among adults of European ancestry. JAMA Network Open. 2019;2:e191350.
Wang M, Beckmann ND, Roussos P, Wang E, Zhou X, Wang Q, et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci Data. 2018;5:1–16.
Chen CY, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, Price AL. Improved ancestry inference using weights from external reference panels. Bioinformatics. 2013;29(11):1399–406. https://doi.org/10.1093/bioinformatics/btt144.
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. https://doi.org/10.1038/nature15393.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Naiser T, Ehler O, Kayser J, Mai T, Michel W, Ott A. Impact of point-mutations on the hybridization affinity of surface-bound DNA/DNA and RNA/DNA oligonucleotide-duplexes: comparison of single base mismatches and base bulges. BMC Biotechnol. 2008;8:1–23.
Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, Schlessinger D, Stambolian D, Loh PR, Iacono WG, Swaroop A, Scott LJ, Cucca F, Kronenberg F, Boehnke M, Abecasis GR, Fuchsberger C. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–7. https://doi.org/10.1038/ng.3656.
Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–9. https://doi.org/10.1038/s41586-021-03205-y.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9. https://doi.org/10.1038/ng1847.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaSci. 2015;4(1):7. https://doi.org/10.1186/s13742-015-0047-8.
Rizvi AA, Karaesmen E, Morgan M, Preus L, Wang J, Sovic M, et al. gwasurvivr: an R package for genome-wide survival analysis. Bioinformatics. 2019;35:1968–70.
Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. 2010;36:1–48.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The Ensembl variant effect predictor. Genome Biol. 2016;17(1):122. https://doi.org/10.1186/s13059-016-0974-4.
Waldman YY, Tuller T, Keinan A, Ruppin E. Selection for translation efficiency on synonymous polymorphisms in recent human evolution. Genome Biol Evol. 2011;3:749–61. https://doi.org/10.1093/gbe/evr076.
Sieberts SK, Perumal TM, Carrasquillo MM, Allen M, Reddy JS, Hoffman GE, et al. Large eQTL meta-analysis reveals differing patterns between cerebral cortical and cerebellar brain regions. Sci Data. 2020;7:340.
Desikan RS, Fan CC, Wang Y, Schork AJ, Cabral HJ, Cupples LA, et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. Plos Med. 2017;14:1–17.
Leonenko G, Sims R, Shoai M, Frizzati A, Bossù P, Spalletta G, Fox NC, Williams J, the GERAD consortium, Hardy J, Escott-Price V. Polygenic risk and hazard scores for Alzheimer’s disease prediction. Ann Clin Transl Neurol. 2019;6(3):456–65. https://doi.org/10.1002/acn3.716.
Staley JR, Jones E, Kaptoge S, Butterworth AS, Sweeting MJ, Wood AM, et al. A comparison of Cox and logistic regression for use in genome-wide association studies of cohort and case-cohort design. Eur J Hum Genet. 2017;25(7):854–62. https://doi.org/10.1038/ejhg.2017.78.
Hughey JJ, Rhoades SD, Fu DY, Bastarache L, Denny JC, Chen Q. Cox regression increases power to detect genotype-phenotype associations in genomic studies using the electronic health record. BMC Genomics. 2019;20(1):805. https://doi.org/10.1186/s12864-019-6192-1.
Huang K, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nature Neuroscience. Nature Publishing Group; 2017;20:1052–1061.
Liu L, Caselli RJ. Age stratification corrects bias in estimated hazard of APOE genotype for Alzheimer’s disease. Alzheimer’s Dement. 2018;4:602–8.
Goerdten J, Carrière I, Muniz-Terrera G. Comparison of Cox proportional hazards regression and generalized Cox regression models applied in dementia risk prediction. Alzheimer’s Dement. 2020;6:e12041.
Lawless JF. Two-phase outcome-dependent studies for failure times and testing for effects of expensive covariates. Lifetime Data Anal. 2018;24(1):28–44. https://doi.org/10.1007/s10985-016-9386-8.
Tao R, Zeng D, Lin D-Y. Optimal designs of two-phase studies. J Am Stat Assoc. 2019;0:1–14.
Lin D-Y, Tao R, Kalsbeek WD, Zeng D, Gonzalez F, Fernández-Rhodes L, et al. Genetic association analysis under complex survey sampling: The Hispanic Community Health Study/Study of Latinos. Am J Hum Genet. 2014;95:675–88.
Tao R, Zeng D, Lin D-Y. Efficient semiparametric inference under two-phase sampling, with applications to genetic association studies. J Am Stat Assoc. 2017;112(520):1468–76. https://doi.org/10.1080/01621459.2017.1295864.
Tavana JP, Rosene M, Jensen NO, Ridge PG, Kauwe JS, Karch CM. RAB10: an Alzheimer’s disease resilience locus and potential drug target. Clin Interv Aging. 2019;14:73–9. https://doi.org/10.2147/CIA.S159148.
Ridge PG, Karch CM, Hsu S, Arano I, Teerlink CC, Ebbert MTW, et al. Linkage, whole genome sequence, and biological data implicate variants in RAB10 in Alzheimer’s disease resilience. Genome Med. 2017;9(1):100. https://doi.org/10.1186/s13073-017-0486-1.
Udayar V, Buggia-Prévot V, Guerreiro RL, Siegel G, Rambabu N, Soohoo AL, Ponnusamy M, Siegenthaler B, Bali J, Simons M, Ries J, Puthenveedu MA, Hardy J, Thinakaran G, Rajendran L, Guerreiro R, Brás J, Sassi C, Gibbs JR, Hernandez D, Lupton MK, Brown K, Morgan K, Powell J, Singleton A, Hardy J. A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of β-amyloid production. Cell Rep. 2013;5(6):1536–51. https://doi.org/10.1016/j.celrep.2013.12.005.
Yan T, Wang L, Gao J, Siedlak SL, Huntley ML, Termsarasab P, Perry G, Chen SG, Wang X. Rab10 phosphorylation is a prominent pathological feature in Alzheimer’s disease. J Alzheimers Dis. 2018;63(1):157–65. https://doi.org/10.3233/JAD-180023.
Deng C-Y, Lei W-L, Xu X-H, Ju X-C, Liu Y, Luo Z-G. JIP1 mediates anterograde transport of Rab10 cargos during neuronal polarization. J Neurosci. 2014;34(5):1710–23. https://doi.org/10.1523/JNEUROSCI.4496-13.2014.
Fu M, Holzbaur ELF. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J Cell Biol. 2013;202:495–508.
Shen R, Zhao X, He L, Ding Y, Xu W, Lin S, Fang S, Yang W, Sung K, Spencer B, Rissman RA, Lei M, Ding J, Wu C. Upregulation of RIN3 induces endosomal dysfunction in Alzheimer’s disease. Transl Neurodegeneration. 2020;9(1):26. https://doi.org/10.1186/s40035-020-00206-1.
Giacomini C, Koo C-Y, Yankova N, Tavares IA, Wray S, Noble W, Hanger DP, Morris JDH. A new TAO kinase inhibitor reduces tau phosphorylation at sites associated with neurodegeneration in human tauopathies. Acta Neuropathol Commun. 2018;6(1):37. https://doi.org/10.1186/s40478-018-0539-8.
Kreft KL, van Meurs M, Wierenga-Wolf AF, Melief M-J, van Strien ME, Hol EM, Oostra BA, Laman JD, Hintzen RQ. Abundant kif21b is associated with accelerated progression in neurodegenerative diseases. Acta Neuropathol Commun. 2014;2(1):144. https://doi.org/10.1186/s40478-014-0144-4.
Acknowledgements
Not applicable.
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (https://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Funding
Funding for this study was provided by the Iqbal Farrukh & Asad Jamal Fund, the National Institutes of Health (grants AG066206, AG060747 and AG047366), the Alzheimer’s Association (AARF-20-683984, M.E.B), and the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie (grant agreement No. 890650, Y.L.G.).
The Alzheimer’s Disease Sequencing Project (ADSP) is comprised of two Alzheimer’s Disease (AD) genetics consortia and three National Human Genome Research Institute (NHGRI)-funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer’s Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and non-governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs. Schellenberg, Farrer, Pericak-Vance, Mayeux, and Haines); U01AG049505 to Dr. Seshadri; U01AG049506 to Dr. Boerwinkle; U01AG049507 to Dr. Wijsman; and U01AG049508 to Dr. Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr. Goate, U01AG052410 to Dr. Pericak-Vance, and U01 AG052409 to Drs. Seshadri and Fornage.
The ADGC cohorts include Adult Changes in Thought (ACT) (UO1 AG006781, UO1 HG004610, UO1 HG006375, U01 HG008657), the Alzheimer’s Disease Centers (ADC) (P30 AG019610, P30 AG013846, P50 AG008702, P50 AG025688, P50 AG047266, P30 AG010133, P50 AG005146, P50 AG005134, P50 AG016574, P50 AG005138, P30 AG008051, P30 AG013854, P30 AG008017, P30 AG010161, P50 AG047366, P30 AG010129, P50 AG016573, P50 AG016570, P50 AG005131, P50 AG023501, P30 AG035982, P30 AG028383, P30 AG010124, P50 AG005133, P50 AG005142, P30 AG012300, P50 AG005136, P50 AG033514, P50 AG005681, and P50 AG047270), the Chicago Health and Aging Project (CHAP) (R01 AG11101, RC4 AG039085, K23 AG030944), Indianapolis Ibadan (R01 AG009956, P30 AG010133), the Memory and Aging Project (MAP) (R01 AG17917), Mayo Clinic (MAYO) (R01 AG032990, U01 AG046139, R01 NS080820, RF1 AG051504, P50 AG016574), Mayo Parkinson’s Disease controls (NS039764, NS071674, 5RC2HG005605), University of Miami (R01 AG027944, R01 AG028786, R01 AG019085, IIRG09133827, A2011048), the Multi-Institutional Research in Alzheimer’s Genetic Epidemiology Study (MIRAGE) (R01 AG09029, R01 AG025259), the National Cell Repository for Alzheimer’s Disease (NCRAD) (U24 AG21886), the National Institute on Aging Late Onset Alzheimer’s Disease Family Study (NIA- LOAD) (R01 AG041797), the Religious Orders Study (ROS) (P30 AG10161, R01 AG15819), the Texas Alzheimer’s Research and Care Consortium (TARCC) (funded by the Darrell K Royal Texas Alzheimer’s Initiative), Vanderbilt University/Case Western Reserve University (VAN/CWRU) (R01 AG019757, R01 AG021547, R01 AG027944, R01 AG028786, P01 NS026630, and Alzheimer’s Association), the Washington Heights-Inwood Columbia Aging Project (WHICAP) (RF1 AG054023), the University of Washington Families (VA Research Merit Grant, NIA: P50AG005136, R01AG041797, NINDS: R01NS069719), the Columbia University Hispanic Estudio Familiar de Influencia Genetica de Alzheimer (EFIGA) (RF1 AG015473), the University of Toronto (UT) (funded by Wellcome Trust, Medical Research Council, Canadian Institutes of Health Research), and Genetic Differences (GD) (R01 AG007584). The CHARGE cohorts are supported in part by National Heart, Lung, and Blood Institute (NHLBI) infrastructure grant HL105756 (Psaty), RC2HL102419 (Boerwinkle), and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193.
The CHARGE cohorts participating in the ADSP include the following: Austrian Stroke Prevention Study (ASPS), ASPS-Family study, and the Prospective Dementia Registry-Austria (ASPS/PRODEM-Aus), the Atherosclerosis Risk in Communities (ARIC) Study, the Cardiovascular Health Study (CHS), the Erasmus Rucphen Family Study (ERF), the Framingham Heart Study (FHS), and the Rotterdam Study (RS). ASPS is funded by the Austrian Science Fond (FWF) grant number P20545-P05 and P13180 and the Medical University of Graz. The ASPS-Fam is funded by the Austrian Science Fund (FWF) project I904), the EU Joint Programme - Neurodegenerative Disease Research (JPND) in frame of the BRIDGET project (Austria, Ministry of Science), and the Medical University of Graz and the Steiermärkische Krankenanstalten Gesellschaft. PRODEM-Austria is supported by the Austrian Research Promotion agency (FFG) (Project No. 827462) and by the Austrian National Bank (Anniversary Fund, project 15435). ARIC research is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Neurocognitive data in ARIC is collected by U01 2U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902, 2U01HL096917 from the NIH (NHLBI, NINDS, NIA and NIDCD), and with previous brain MRI examinations funded by R01-HL70825 from the NHLBI. CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants U01HL080295 and U01HL130114 from the NHLBI with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928, and R01AG20098 from the NIA. FHS research is supported by NHLBI contracts N01-HC-25195 and HHSN268201500001I. This study was also supported by additional grants from the NIA (R01s AG054076, AG049607, and AG033040) and NINDS (R01 NS017950). The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007–2013)/grant agreement HEALTH-F4- 2007-201413 by the European Commission under the programme “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2-CT-2002- 01254). High-throughput analysis of the ERF data was supported by a joint grant from the Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam; the Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly (RIDE); the Ministry of Education, Culture and Science; the Ministry for Health, Welfare and Sports; the European Commission (DG XII); and the municipality of Rotterdam. Genetic data sets are also supported by the Netherlands Organization of Scientific Research NWO Investments (175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project 050-060-810. All studies are grateful to their participants, faculty, and staff. The content of these manuscripts is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the US Department of Health and Human Services.
The four LSACs are the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), The American Genome Center at the Uniformed Services University of the Health Sciences (U01AG057659), and the Washington University Genome Institute (U54HG003079).
Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the National Cell Repository for Alzheimer’s Disease (NCRAD, U24AG021886) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA-funded Alzheimer’s Disease Centers (ADCs), and the National Alzheimer’s Coordinating Center (NACC, U01AG016976) and the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private US organizations, or foreign governmental or nongovernmental organizations.
The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P30 AG062428-01 (PI James Leverenz, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P30 AG062421-01 (PI Bradley Hyman, MD, PhD), P30 AG062422-01 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30 AG013854 (PI Robert Vassar, PhD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P30 AG062429-01 (PI James Brewer, MD, PhD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG049638 (PI Suzanne Craft, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P30 AG062715-01 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), and P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
Author information
Authors and Affiliations
Consortia
Contributions
Y.L.G., M.E.B., and Z.H. performed simulation analyses. Y.L.G., M.E.B., V.N., S.J.E, and G.K. performed data processing. Y.L.G. performed whole-exome analyses. Y.L.G., M.E.B., Z.H., and M.D.G. designed study and analyses and obtained funding. Y.L.G., M.E.B., R.T., Z.H., and M.D.G. interpreted statistical analyses and wrote paper. V.N., Z.H., and M.D.G supervised work. Y.L.G. and M.E.B. contributed equally to this work. Z.H. and M.D.G contributed equally to this work. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Participants or their caregivers provided written informed consent in the original studies. The current study protocol was granted an exemption by the Stanford University institutional review board because the analyses were carried out on deidentified, off-the-shelf data; therefore, further informed consent was not required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Le Guen, Y., Belloy, M.E., Napolioni, V. et al. A novel age-informed approach for genetic association analysis in Alzheimer’s disease. Alz Res Therapy 13, 72 (2021). https://doi.org/10.1186/s13195-021-00808-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-021-00808-5