
Abstract
Background
The epigenetic status of patients 6-month post-COVID-19 infection remains largely unexplored. The existence of long-COVID, or post-acute sequelae of SARS-CoV-2 infection (PASC), suggests potential long-term changes. Long-COVID includes symptoms like fatigue, neurological issues, and organ-related problems, regardless of initial infection severity. The mechanisms behind long-COVID are unclear, but virus-induced epigenetic changes could play a role.
Methods and results
Our study explores the lasting epigenetic impacts of SARS-CoV-2 infection. We analyzed genome-wide DNA methylation patterns in an Italian cohort of 96 patients 6 months after COVID-19 exposure, comparing them to 191 healthy controls. We identified 42 CpG sites with significant methylation differences (FDR < 0.05), primarily within CpG islands and gene promoters. Dysregulated genes highlighted potential links to glutamate/glutamine metabolism, which may be relevant to PASC symptoms. Key genes with potential significance to COVID-19 infection and long-term effects include GLUD1, ATP1A3, and ARRB2. Furthermore, Horvath's epigenetic clock showed a slight but significant age acceleration in post-COVID-19 patients. We also observed a substantial increase in stochastic epigenetic mutations (SEMs) in the post-COVID-19 group, implying potential epigenetic drift. SEM analysis identified 790 affected genes, indicating dysregulation in pathways related to insulin resistance, VEGF signaling, apoptosis, hypoxia response, T-cell activation, and endothelin signaling.
Conclusions
Our study provides valuable insights into the epigenetic consequences of COVID-19. Results suggest possible associations with accelerated aging, epigenetic drift, and the disruption of critical biological pathways linked to insulin resistance, immune response, and vascular health. Understanding these epigenetic changes could be crucial for elucidating the complex mechanisms behind long-COVID and developing targeted therapeutic interventions.
Similar content being viewed by others


Background
In the wake of the COVID-19 pandemic, a remarkably high number of individuals worldwide have been affected. While the pathophysiology and immune response to SARS-CoV-2 (the virus causing COVID-19) infection have been extensively studied to predict acute disease progression and prognosis [1,2,3,4,5,6,7], there is limited knowledge about the long-term effects caused to the host by the infection. The condition of persistent clinical symptoms after the acute phase of infection is common to many viruses—e.g., Epstein–Barr virus (EBV) [8, 9]; cytomegalovirus (CMV) [10, 11], and human herpesvirus 6 (HHV-6) [12]—but in the case of COVID-19, this condition takes on great significance as it appears to affect a large percentage of individuals. The persistence of COVID-19 clinical symptoms for at least 12 weeks or more (several months) is referred to as post-acute sequelae of SARS-CoV-2 infection (PASC) or long-COVID [13] affecting individuals with both severe acute symptoms and those with mild or asymptomatic disease progression [14]. These symptoms encompass organ (e.g., fatigue, post-exertional malaise, headache, insomnia, tachycardia) and neurological (e.g., brain fog, memory/speech/language issues, sleeping problems) manifestations [15, 16]. Estimating the accurate prevalence of this condition proves to be highly challenging, primarily because inherent studies are shaped by a wide array of variables, including age, gender, ethnicity, the severity of acute symptoms, the type of SARS-CoV-2 variant, follow-up durations, viral load, the presence of concurrent medical conditions, vaccination history, preexisting social, economic, and medical factors, among others [17]. Among the most updated meta-analyses, the prevalence is reported between 30 and 60% [18,19,20,21,22]. Another meta-analysis revealed that COVID-19 long-term symptoms are slightly associated with older age and strongly associated with female sex and preexisting comorbidities (e.g., diabetes and obesity) [23].
The exact causes of long-COVID are still being investigated, but some hypotheses have emerged over time. SARS-Cov-2 can reach (via hematogenous spreading) and infect cells of the central nervous system (CNS), producing neuroinflammation [24]. It has also been hypothesized that the SARS-CoV-2 virus may persist in specific tissues long after the acute phase [13], leading to potential long-term health complications [25]. Disease risk factors include cell death and immune dysfunction after SARS-Cov-2 infection [26, 27], uncontrolled and persistent release of cytokines [28, 29], multiple cell fusion in infected organ (syncytia) [30, 31], autoantibodies causing immunodeficiency (against type I IFN) [32, 33] or microclots [33, 34], and persistent viral infection [35, 36].
The possibility that these dysfunctions are mediated over time by epigenetic changes has been explored through EWAS approaches in a limited number of studies capable of identifying specific epigenetic signatures obtained by analyzing small cohorts of post-/long-COVID-19 [37,38,39]. Lee and colleagues [38] investigated DNA methylation changes in immune response-associated genes in post-COVID-19 patients (after 3 months from the acute phase), identifying the gene IFI44L (interferon-induced protein 44 like) as the primary target. IFI44L plays a critical role in antiviral and antibacterial activity. The study of [37] compared methylation changes in the acute phase, and after one year, highlighting the persistence of pathways related to viral response and inflammation. The study performed by Nikesjo and colleagues [39] found a unique DNAm signature in PACS patients involving modified pathways related to angiotensin II and muscarinic receptor signaling and mitochondrial function.
Here, we present a genome-wide study using the Illumina 850 K EPIC BeadChips of a large cohort of ninety-six individuals whose blood samples were collected 6 months after COVID-19 infection. The follow-up examination has highlighted the presence of suggestive long-term clinical features in twenty-eight patients. This study aims to assess potential epigenetic changes 6 months after COVID-19 exposure.
Results
Characteristics of the sample population
Demographic and clinical features, including age, sex, and cellular components of subjects sampled 6 months after the initial SARS-CoV-2 infection (cases) and healthy subjects with no history of SARS-CoV-2 infection (references), are summarized in Table 1. The age and sex distributions of the sample population were analyzed to understand the demographic profile of the participants: The two cohorts differ statistically in terms of age distribution (Mann–Whitney test, p < 0.05) and sex ratio (Fisher's exact test, p < 0.05). The estimation of cellular components of peripheral blood obtained from the EpiDISH package [40] was evaluated to explore any variations in the composition of blood samples between the reference and the post-COVID-19 groups. The analysis showed no significant differences in immune cell composition.
Exploratory
We conducted an exploratory approach to investigate the epigenetic differences between the two groups (post-COVID-19 vs. reference) through a principal component analysis (PCA) both at the CpG site and region levels (genes, CpG islands, and promoter). The analysis (Fig. 1) did not reveal distinct patterns of epigenetic variation between the two groups since the patterns observed were almost overlapping, indicating a lack of solid differences in DNA methylation at CpG sites and regions between the two groups.

Scatter plots of principal component analysis (PCA). Scatter plot distribution of samples and the first two principal components at a sites, b genes, c promoters, and d CpG islands
Differential methylation and over-representation (ORA) analysis
The differential methylation analysis between the two groups (post-COVID-19 vs reference) was performed using linear models for microarray data (LIMMA) [41]. Confounding factors (sex, chronological age, and cellular component estimations) [40, 42] were considered by adjusting for their effects in the analysis (see Methods for details). Limma results are available as Supplementary File 1. At the site level, the study revealed a set of 42 CpG sites overlapping 53 genes exhibiting significant differential methylation between the two groups (FDR < 0.05): 24 hypo-methylated (log2FC < 0; ∆β < 0) and 18 hyper-methylated (log2FC > 0; ∆β > 0) (Fig. 2).

Circos plot visualizes the genomic distribution of differentially methylated sites throughout the human genome. The blue (hyper-methylated) and red (hypo-methylated) dots represent the genomic position of sites that have exceeded the statistical significance threshold (FDR < 0.05) and are spatially arranged according to the -log10 (unadjusted p-value). The solid red line indicates the FDR significance threshold, while the dashed red line represents the Bonferroni significance threshold. X and Y chromosomes are omitted from the analysis.
Genomic localization of the 42 deregulated CpG sites showed a significant enrichment at the level of CpG islands (OR = 3.426, p-value = 1.58 × 10–4). In comparison, 18 out of 42 sites (43%) are functionally related to promoter regions (TSS200, TSS1500, 5'UTR, and 1stExon) (data not shown).
Differentially methylated cytosines were annotated with their corresponding genes for functional annotation and prioritization (Supplementary File 1).
Over-representation analysis (methylGSA) revealed KEGG pathways associated with amino acid metabolism, including "Alanine, aspartate, and glutamate metabolism" (hsa00250) and "D-glutamine and D-glutamate metabolism" (hsa00471), albeit with nominal significance (Supplementary File 2).
Next, we focused on differently methylated genes to determine their potential relevance to COVID infection and symptoms associated with post-acute sequelae of SARS-CoV-2 infection (PASC) by using the relevant keywords (e.g., COVID-19, long-COVID, post-acute sequelae of SARS-CoV-2 infection; neuronal; inflammation, virus infection); we ranked genes based on their most suitable phenotype associations (VarElect) (Supplementary File 3). Among the top-rated genes to note: GLUD1 (glutamate dehydrogenase 1) (cg00167275; log2FC = 1.5, ∆β = 0.31, FDR = 1.66e-35), ATP1A3 (alpha 3 subunit of the Na + /K + ATPase) (cg13628106; log2FC = -0.32, ∆β = -0.16, FDR = 3.2e-4), RNASEH2C (C subunit of ribonuclease H) (cg25294185; log2FC = -0.82, ∆β = -0.04, FDR = 1.96e-12), SMAD2 (SMAD family member 2) (cg05100634; log2FC = 0.51, ∆β = 0.02, FDR = 9.4e-7), TNIP1 (TNFAIP3 interacting protein 1) (cg22178392; log2FC = -0.18, ∆β = -0.03, FDR = 1.5e-2), PRKCI (protein kinase C iota) (cg18139307; log2FC = 0.18, ∆β = 0.31, FDR = 3.4e-6), and ARRB2 (arrestin beta 2) (cg10047026; log2FC = 0.24, ∆β = 0.025, FDR = 6e-3).
Considering the top 200 nominally significant genes, we did not significantly enrich KEGG/PANTHER pathways (Supplementary File 4). However, at the gene ontology level, we identified a significant enrichment at the level of cellular components (CC), primarily involving GO terms related to the proper function and structure of the Golgi apparatus (GO:0000139, "Golgi membrane"; GO:0098791, "Golgi subcompartment"; GO:0044431, "Golgi apparatus part"; GO:0005794"Golgi apparatus"; GO:0031984," organelle subcompartment").
At the regional level, the differential analysis revealed significant epigenetic changes limited to a specific CpG island region on chromosome 6 (chr6:41,068,476–41069343) (adjusted p-value = 0.006, diff.meth = + 1%). This region encompasses the 3' terminal portion of NFYA (nuclear transcription factor Y subunit alpha) and the first exon of the pseudogene ADCY10P1 (ADCY10 pseudogene 1).
Meta-analysis
To enhance the robustness and validation of our findings, we conducted a comprehensive literature search for analogous studies. After identifying the sole survey with a similar design, we integrated our nominal p-values derived from the differential analysis of CpG sites with those obtained from Lee and colleagues [38], who conducted a comparison between COVID-19 positive and negative cases, using a meta-analytical method. The meta-analysis (Supplementary File 5) detected 13 significant (FDR < 0.05) CpG sites with identical directions of effect/deregulation across the two datasets. This analysis was conducted considering that there is indeed a discrepancy between the two studies regarding the timing of sample collection post-infection (6 months vs 3 months). The thirteen genes associated to the 13 CpG sites are (in order of significance): DYRK2 (dual-specificity tyrosine phosphorylation-regulated kinase 2), ATP5PF (ATP synthase peripheral stalk subunit F6), B4GAT1 (beta-1,4-glucuronyltransferase 1), PRKXP1 (PRKX Pseudogene 1), SLC25A21-AS1 (SLC25A21 antisense RNA 1), NRDC (nardilysin convertase), SMAD2 (SMAD Family Member 2), OSBPL6 (oxysterol-binding protein like 6), IFI44L (interferon-induced protein 44 like), PATJ(PATJ Crumbs cell polarity complex component), PRH1-PRR4 (PRH1-PRR4 readthrough), and UBAC2 (UBA domain containing 2). The thirteenth probe is located in an intergenic region and aligns simultaneously with two genes, ACAN (Aggrecan) and ISG20 (interferon-stimulated exonuclease gene 20).
Age acceleration
Methylation profiles were evaluated through Horvath's epigenetic clock tool to assess whether exposure to the COVID-19 virus impacted biological age estimates. We observed a slight but significant epigenetic age acceleration (EAA) in post-COVID-19 patients across all available clocks [42,43,44,45], (Horvath, p-value = 3 × 10–3; Hannum, p-value = < < 0.05; PhenoAge, p-value = 8.8 × 10–4; SkinBlood, p-value = 2.7 × 10–3) (Fig. 3 panels A, C, D, E). On the contrary, despite an increasing trend, no significant differences were found when evaluating the DNAm GrimAge predictor of lifespan (linear regression adjusted for covariates, p-value = 0.42) (Fig. 3B).

Boxplots showing the distribution of age acceleration differences for different epigenetic clocks: A Horvath, B GrimAge, C Hannum, D PhenoAge, and E SkinBlood. The thick horizontal line in each box represents the median of the distribution, while the box itself represents the interquartile range (IQR). In the "ggplot" boxplot function, the whiskers extend to the data points located within 1.5 times the IQR from the box by default. Dots represent outliers (single values exceeding 1.5 IQRs). (F) Telomere length evaluation (DNAmTLadjAge)
Moreover, we also assessed DNAmTLadjAge, a DNA methylation-based estimator of telomere length [46]. DNAmTLadjAge corresponds to the age-adjusted parameter that relates DNAmTL to chronological age. Negative values of DNAmTLadjAge indicate DNAmTL that is shorter than expected based on age, while positive values indicate the opposite. Our analysis revealed that DNAmTLadjAge was significantly lower in post-COVID patients compared to controls. Results of linear regression adjusted for covariates: telomere length (DNAmTL) (p-value < < 0.05).
Epigenetic drift
The assessment of epigenetic drift was carried out through the identification of stochastic epigenetic mutations (SEMs) as described in [3, 47,48,49,50,51]. To detect SEMs, we first examined the distribution and variability of methylation levels in the control population for all the probes: A reference methylation range for each probe was generated using the formula Q1-(3 × IQR) = lower limit and Q3 + (3 × IQR) = upper limit. Methylation levels of cases falling outside this extreme interval were identified as SEMs. Differences between cases and controls were then investigated using two distinct metrics to evaluate the epigenetic drift: one examining the broader influence of SEMs (Global-Epi Mutation Load (EML)), while the other focused on assessing the burden of SEMs at the gene level (Gene-EML).
In the case cohort (Global-EML), the median value of SEMs stood at 488.5, IQR (373–922), whereas in the reference group, it was 369, IQR (314–520.5). The multiple regression model that accounted for sex and principal component covariates, which included age and cellular components, confirmed a significant increase of SEMs in the post-COVID-19 group compared to the control/reference group (with an estimated incremental log10 transformed value of 0.1667 and a p-value of 3.74 × 10–06) as shown in Fig. 4. The increase is confirmed even when SEMs are hyper-methylated and hypo-methylated categories (data not shown).

Boxplot showing the distribution of SEMs in Reference and post-COVID-19 groups. The thick horizontal line in the box represents the median of the distribution, while the box represents the interquartile range. By default, in the "ggplot" boxplot function, the whiskers extend to the data points located within 1.5 times the interquartile range (IQR) from the box. Dots represent outliers (single values exceeding 1.5 IQRs)
Differences in Gene-EML between cases and controls were investigated using a sequence kernel association test (SKAT) [52]. After correction for appropriate covariates (sex and principal components, as described for the differential methylation step), the analysis identified 790 SEMs-enriched genes with statistically significant associations with sample group variable (Perm P value < 0.05) (Supplementary File 6). The roster of genes exhibiting increased drift in cases underwent an over-representation analysis (Fig. 5) (Supplementary File 7).

Bar chart showing enrichment ratio of a KEGG and b PANTHER pathways
ORA identified several enriched pathways, some remaining significant even after multiple testing corrections. Notably, the "Insulin resistance" pathway (hsa04931) remained significant (FDR = 0.04), alongside the "VEGF signaling" pathway (hsa04370) and the "Apoptosis signaling pathway" (P00006) (FDR = 0.005). Other pathways, while not significant after correction, are noteworthy, including "Hypoxia response via HIF activation," "Axon guidance mediated by netrin," "Relaxin signaling pathway," "T-cell activation," and the "Endothelin signaling pathway."
Discussion
This study aimed to investigate potential epigenetic changes 6 months after COVID-19 exposure. Currently, within this timeframe of exposure, it stands out as one of the epigenetic studies with robust statistical power, analyzing the methylation profile of nearly a hundred individuals.
The role of DNA methylation in developing long-term COVID-19 symptoms has been examined in three previously published studies [37,38,39]. However, these studies utilized diverse approaches, primarily focusing on post-acute sequelae of SARS-CoV-2 infection (PASC), and employed study designs with reduced sample sizes. We examined peripheral blood samples collected from individuals 6 months post-infection, regardless of persistent COVID-19 symptoms, with a primary focus on investigating whether the virus-induced significant epigenetic remodeling or reprogramming in the host organism.
The initial suggestive discovery reveals no discernible variance in the immune system landscape between the two groups, as evidenced by comparable estimates of blood cellular composition (CD8T, CD4T, NK, Bcell, Mono, Gran) (Supplementary File 8).
Although not directly comparing the same populations at the same time point (3 months post-infection), [38] confirmed the absence of an inflammatory state, as they observed no significant differences in the various cell types under consideration.
The principal component analysis (PCA) showed no distinct patterns of epigenetic variation between the two groups, indicating a lack of solid differences in DNA methylation at CpG sites and regions (genes, promoters, CpG islands) level. However, the differential methylation analysis between the two groups revealed 42 CpG sites exhibiting significant differential methylation. The over-representation analysis highlighted a pathway related to glutamate/glutamine metabolism. The dysregulation of this pathway has been reported as significant in COVID-19 since studies have shown that glutamine and glutamate metabolism play a crucial role in COVID-19 severity, with elevated glutamate levels associated with an increased risk of infection and severe disease. In contrast, elevated glutamine levels are linked to a decreased risk of infection and severe COVID-19 [53]. Moreover, the functional prioritization analysis enabled the identification of genes, including GLUD1, ATP1A3, RNASEH2C, SMAD2, TNIP1, PRKCI, and ARRB2, which are particularly intriguing for their potential involvement in the neurological and immunological processes associated with post-COVID symptoms.
Among the genes with the highest prioritization scores, we identified GLUD1 (glutamate dehydrogenase 1), which is interesting because it plays a role in maintaining glutamate levels. The hyper-methylation of GLUD1 observed in this study aligns with the elevated levels of glutamate resulting from systemic inflammation caused by SARS-CoV-2 infection. This finding might contribute to explaining a number of neurotoxic effects, contributing to neuronal dysfunctions such as altered learning, memory, and neuroplasticity highlighted in post-COVID patients [54, 55].
Additionally, after prioritization analysis, ATP1A3 (alpha three subunits of the Na + /K + ATPase) emerged as another intriguing gene. Although not directly linked to COVID-19, it has been associated with various neurological disorders [56] and cardiac abnormalities [57], suggesting a potential indirect role in the neurological manifestations and cardiovascular complications observed in some patients.
Emerging findings indicate that also the other genes, RNASEH2C, SMAD2, TNP1, PRKCI, and ARRB2, may play a role in the pathophysiology of long-COVID by influencing various aspects of the immune response, inflammation, or cellular processes contributing to prolonged symptoms following COVID-19 infection. Mutations in the C subunit of ribonuclease H (RNASEH2C), for example, have been reported to affect the immune response and potentially result in severe COVID-19 outcomes [58, 59]. SMAD2 (SMAD family member 2) is a protein that plays a role in the TGF-β signaling pathway, which is involved in the regulation of cell growth, differentiation, and immune response [60]. Research has shown that SMAD2, along with other genes like SMAD1 and SMAD3, plays a role in modulating T-cell immunity and viral infection responses, contributing to symptoms such as chronic inflammation and immune dysregulation observed in long-COVID [61].
TNFAIP3 interacting protein 1 (TNIP1) is a hub protein associated with autoimmune diseases [62] and plays a role in COVID-19. Research suggests that TNIP1 is involved in the immune response and inflammation regulation, making it a potential target for therapeutic interventions in COVID-19 patients [63].
Among the latest interesting genes, we found PRKCI and ARRB2. The PRKCI gene encodes a protein kinase C iota (PKCi), which regulates cellular functions such as cell proliferation, division, differentiation, survival, migration, and polarization [64, 65]. On the other hand, ARRB2, a member of the arrestin/beta-arrestin protein family, is involved in desensitizing G-protein-coupled receptors and regulating signaling pathways related to cell proliferation, migration, and inflammation. Mutations in ARRB2 have been linked to neurodegenerative diseases [66, 67], cardiovascular alterations [68, 69], and cancer [70]. Additionally, β-arrestin 2 promotes the production of IFN-β and virus clearance in macrophages, although some viruses may degrade it to evade the immune response [71]. Both ARRB2 and PRKCI regulate toll-like receptor (TLR) signaling, which is critical for inducing inflammation in response to microbes and host molecules.
An additional analysis aimed to boost the over-representation analysis (ORA) sensitivity by incorporating the first 200 nominally significant genes into the gene list. This effort was made to capture potentially relevant genetic factors that may enhance ORA accuracy. The results revealed considerable enrichment in gene ontology (GO) terms associated with "Golgi apparatus functionality" under "cellular components." This finding is noteworthy as it aligns with previous research indicating that SARS-CoV-2 infection induces Golgi fragmentation, which aids in viral trafficking and release [72]. Additionally, Golgi fragmentation is commonly observed in brain samples from individuals with Alzheimer's disease and can be triggered by excessive neuronal activation [73].
After concluding the differential methylation analysis, the results were compared with those of a previous study using a meta-analytical approach to produce a more reliable list of genes. This comparative analysis identified 13 genes showing notably consistent epigenetic differences among studies. Despite the differing study designs, this gene list allows for a focus on more robust results.
Furthermore, the study explored additional facets of epigenetic regulation and investigated whether exposure to the SARS-CoV-2 virus affected biological age and epigenetic drift.
We observed a slight but significant age acceleration (AgeAccelerationDiff) and telomere shortening in post-COVID-19 patients, suggesting that SARS-CoV-2 virus exposure might accelerate aging. The effects of viral infections, specifically COVID-19, as well as immune responses on the process of biological aging are currently a topic of debate: several studies have explored this aspect among individuals with acute phase COVID-19, examining both the comparison between healthy controls and COVID-19 cases and the assessment of varying severity levels within the COVID-19 patient group, comparing mild and severe cases. While specific studies have shown no distinctions between chronological and biological age [3, 74], others have observed an acceleration [75]. In partial contrast to our findings, it is worth noting the absence of this acceleration in Lee et al., 2022 [38], where no significant differences are observed; however, it is essential to consider that the time points between our study (after 6 months) and Lee's [38] (after 3 months) are pretty different. Telomere shortening is a widely observed and confirmed aspect in the context of COVID-19, as demonstrated in other studies involving subjects with severe forms of the disease [75,76,77] and in post-COVID-19 survivors [78].
Another critical issue in understanding the biological effects of COVID-19 virus infection after 6 months is the results obtained by evaluating epigenetic drift. Epigenetic drift refers to the changes in DNA methylation patterns that occur over time, contributing to aging. It can be influenced by genetics and environmental exposure, including viral infections, exerting an influence on individual health by increasing genomic instability and promoting abnormal gene expression [79]. Stochastic epigenetic mutations (SEMs) can be considered a reliable measure of epigenetic drift [3, 50, 80]. For example, the burden of SEMs was recently found to be associated with Parkinson's disease (referred to as epigenetic mutation load) [81] or with amyotrophic lateral sclerosis [80]. Two different metrics have been used to assess epigenetic drift: one that analyzed the broader impact of SEMs (Global-EML) and another that concentrated on evaluating the burden of SEMs at the gene level (Gene-EML). Interestingly, our analyses showed a significantly increased Global-EML in the post-COVID-19 group compared to controls (Fig. 4).
Moreover, to assess epigenetic drift at the gene level (Gene-EML), we employed a sequence kernel association test (SKAT). Designed initially for rare variant studies, this method has recently found applications in other areas like copy number variations (CNVs) and epigenetic modifications. It has been widely used in numerous studies aiming to identify genetic associations with diseases such as Alzheimer's disease, schizophrenia, and autism spectrum disorder, amyotrophic lateral sclerosis [80, 82,83,84]. This analysis has identified a list of genes that exhibit significantly different epigenetic drift between the two study groups. The ORA analysis considering this list of genes revealed several significantly enriched biochemical pathways, most of which may directly or indirectly relate to COVID-19.
The "VEGF signaling pathway" and "Hypoxia response via HIF activation" pathways have been associated with COVID-19 due to their involvement in vascular dysfunction and inflammation observed during disease progression [85, 86]. Vascular endothelial growth factor (VEGF) plays a crucial role in angiogenesis and regulates various activities such as vascular permeability, cell migration, proliferation, and survival [87]. Hypoxia, or low oxygen conditions, can activate the hypoxia-inducible factor (HIF), a key regulator in the response to hypoxia. The concurrent activation of HIF and proinflammatory signaling leads to the upregulation of VEGF, which is elevated in COVID-19 patients compared to healthy controls [88, 89] and significantly higher in patients with severe outcomes compared to survivors [86, 90], suggesting that extensive activation of endothelial cells significantly contributes to disease progression. Unfortunately, we cannot speculate on whether these pathways are up- or down-regulated in our analysis; as for the SKAT analysis, we considered overall epigenetic dysregulation by aggregating hyper- and hypo-methylated SEMs.
Moreover, pathways like "Insulin Resistance" and the "Insulin/IGF pathway-protein kinase B signaling cascade" could become relevant due to COVID-19's association with various metabolic alterations, such as impacts on insulin sensitivity and glucose metabolism. Studies have shown that COVID-19 patients, even those with mild cases, may experience increased insulin resistance, which can persist long after the acute phase of the infection [91].
Another attractive deregulated pathway is represented by the "Apoptosis signaling pathway"; this pathway plays a central role in the pathogenesis of COVID-19, and its dysregulation may also contribute to disease severity. However, the role of apoptosis is very complex, and both the induction and inhibition of apoptosis have been suggested as potential therapeutic targets at different stages of the disease [92].
The "T-cell receptor signaling pathway" and "T-cell activation" pathways are highly suggestive concerning COVID-19. These pathways are relevant because the SARS-CoV-2 virus can directly impact the immune response of T-cells. During COVID-19 infection, there has been significant observed impact on T-cell populations and their activation, as T-cells play a crucial role in the immune response against the virus: Longitudinal studies [93, 94] show that immune abnormalities may persist after a severe COVID-19 progression, with sustained activation of myeloid cells, the presence of proinflammatory cytokines, and consistently activated T-cells still detectable between 8 to 12 months after the initial COVID-19 infection.
In conclusion, these results provide comprehensive insights into the epigenetic consequences of SARS-CoV-2 exposure after 6 months, emphasizing potential associations with aging, SEM accumulation, and dysregulation in critical pathways linked to insulin resistance, immune response, and vascular function.
We emphasize, for completeness of information, that an additional epigenetic analysis was conducted considering the subcohort of 28 samples that exhibited long-COVID symptoms. However, although these results confirm the findings already highlighted regarding the SARS-CoV-2 exposure factor, they should be taken and interpreted with caution due to the low sample size. The results of this additional analysis are reported in Supplementary File 9.
The study has some limitations Due to experimental constraints, cases and controls were not perfectly balanced on each BeadChip, necessitating careful evaluation and correction for any potential batch effects. To validate the results obtained from an EWAS (epigenome-wide association study), a biological replicate of the experiment with a different validation cohort would be necessary. Furthermore, although the sample size may be considered large, it may still be insufficient to detect very subtle differences between cohorts that could prove significant in a genomic framework or large genetic association study. However, it is essential to note that the analysis framework developed in this study aims to apply not only the most commonly adopted methods in the field of DNA methylation, such as the study of age predictors and the analysis of differential methylation, but also extends to less common analytical approaches by integrating the study of epigenetic drift and pathways enriched in genes significantly enriched in stochastic epigenetic mutations.
Materials and methods
Patient recruitment and study design
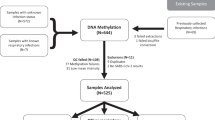
This study, conducted across multiple centers, intended to examine the long-term effects of COVID-19 by assessing ninety-six individuals 6 months after they contracted the virus (years 2020 and 2021). Inclusion criteria encompassed hospitalization during the infection period, with a subgroup of patients requiring intensive care unit admission and the use of forced ventilation. As a reference methylation profile, a control group of 191 selected individuals from an unrelated study with no history of COVID-19 (confirmed through serological testing) was included. At the time of sampling, control individuals did not exhibit any symptoms suggestive of COVID-19. The ethics committees of the participating hospital centers approved the study protocol. Informed consent was obtained from all participants before their inclusion in the study. All data collected were treated with strict confidentiality and adhered to relevant data protection regulations.
DNA extraction
DNA isolation from peripheral blood from all patients was performed using automatic equipment and a commercial kit based on magnetic beads separation. Total genomic DNA quantification was carried out using an N60 Implen Nanophotometer. Samples showing aberrant protein (260/280) and organic compounds (230/260) ratios were discarded or purified.
Methylation assay
Following the manufacturer's instructions, 900 ng of high-quality genomic DNA was bisulfite converted using the EZ DNA methylation kit (D5001, Zymo Research Corporation). Illumina incubation conditions were used to increase the efficiency and reproducibility of the bisulfite conversion. Quality control/quantification of bisulfite-converted DNA (bsDNA) was performed using an N60 Implen Nanophotometer. Approximately 200 ng/ul of bisulfite-converted DNA was hybridized on Illumina Infinium Methylation EPIC BeadChips. Fluorescent signals were detected using the (two-color laser—532 nm/660 nm) Illumina iScan scanner and saved as intensity data files (*.idat). The methylation level for each CpG site is represented as β-values based on the fluorescent intensity ratio between methylated and unmethylated probes. β-values may range between 0 (non-methylated) and 1 (completely methylated).
Quality control and differential methylation analysis
The generation of the β-value dataset of all samples was carried out using the ChAMP package [95]: After the quality control/preprocessing step, 12,777 probes with a detection p-value above 0.01, 5179 probes with a bead count < 3 in at least 5% of samples, 2875 NoCG probes, 94,318 probes with potential SNPs [96], 11 probes that align to multiple locations, and 16,424 probes located on X and Y chromosomes were filtered out. 734,334 probes and 287 samples were retained. After BMIQ normalization, the variability due to the batch effect was corrected by using the ComBat function [97], which performs parametric empirical Bayesian adjustments. To infer the proportions of a priori known cell types (e.g., CD8+T and CD4+T-cells, natural killer (NK) cells, B cells, monocytes, granulocytes (basophils, eosinophils, and neutrophils)) present in blood samples, we used EpiDISH package [40] in R environment.
Differential methylation analysis at the CpG sites level was conducted at the group level by computing p-values using the “Limma” package [41]. At the region level (Genes, promoters, CpG island, tiling), the “RnBeads” differential methylation (Limma) module was alternatively used [98].
To adjust for potential confounding factors, a principal component analysis (PCA) was performed to evaluate the association of age and blood cell estimations with both dependent (disease groups) and independent (methylation values) variables: The principal components summarizing 80% of the variability were used as covariates in the differential methylation step. Principal components (PCs) were used to avoid collinearity of covariates. Estimation of biological age (Horvath, Hannum, PhenoAge, DNAm skin, and blood) [43,44,45] and telomere length (surrogate marker DNAmTL) [46] were assessed using the DNA methylation age calculator analysis tool [42] (https://dnamage.genetics.ucla.edu/). Enrichment analysis on differentially methylated sites was performed using the "methylGSA" package [99] (methylglm function) in the R environment.
Individual sample analyses were carried out by identifying stochastic epigenetic mutations (SEMs) as described in [3, 47,48,49,50,51]. SEMs represent extreme aberrant methylation data points and were identified for each CpG site by comparing the methylation profile of each case to a reference methylation range, calculated from a control population as follows: upper value = Q3 + (k * IQR), lower value = Q1—(k * IQR), where Q1 represents the first quartile, Q3 corresponds to the third quartile, IQR (Interquartile Range) equals Q3—Q1, and k is set at 3. Outlier values were then classified as hyper-methylated or hypo-methylated with respect to the median values of the controls' corresponding probes. Gene annotation of SEMs was obtained using the web tool wANNOVAR [100]. For testing associations between cases and SEM enriched genes, a method for rare variants analysis was applied using the SKAT‐O method implemented in the RVTESTS package [52]. Organization/investigation of results was conducted according to the clinical characteristics/keywords by using the available prioritization tools, including WEB-based gene set analysis toolkit (WebGestalt) [101] which uses, among others, gene ontology, KEGG (Kyoto Encyclopedia of Genes and Genomes) and PANTHER (protein analysis through evolutionary relationships) databases, and VarElect (the next-generation sequencing phenotyper) [102]. Data/results were visualized using the "ggplot2" package for PCA charts and boxplots, the "Pheatmap" package for the heatmaps, and the "CMplot" for the Manhattan plots. Linear regressions (after checking assumptions) or the Mann–Whitney function were used to evaluate statistical differences in age, cell-type composition, and burden of SEMs between cases and controls. Unless otherwise stated, the statistical significance threshold was set to 0.05.
Meta-analysis The meta-analysis was performed using the computational tool METAL [103], specifically designed for (epi) genome-wide analysis. Only the overlapping site-specific p-values (n = 723,656) derived from the differential methylation analyses were integrated into the meta-analysis.
Availability of data and materials
Raw data related to our study are available on the GEO repository under accession number GSE247869 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE247869). Public access to the GEO repository is open, and thus administrative permission to access and use the data is not needed.
Abbreviations
- COVID-19:
-
Coronavirus disease 2019
- PACS:
-
Post-acute sequelae of SARS-CoV-2
- IQR:
-
Interquartile range
- PCA:
-
Principal component analysis
- SEMs:
-
Stochastic epigenetic mutations
- WEBGESTALT:
-
Web-based gene set analysis toolkit
- CNS:
-
Central nervous system
- FDR:
-
False discovery rate
- LOG2FC:
-
Logarithm of fold change
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PANTHER:
-
Protein analysis through evolutionary relationships
- LIMMA:
-
Linear models for microarray data
- RVTEST:
-
Rare variant tests
- SKAT:
-
Sequence kernel association test
- ∆β :
-
Delta beta
References
Balnis J, Madrid A, Hogan KJ, Drake LA, Chieng HC, Tiwari A, et al. Blood DNA methylation and COVID-19 outcomes. Clin Epigenetics. 2021;13(1):118.
Bradic M, Taleb S, Thomas B, Chidiac O, Robay A, Hassan N, et al. DNA methylation predicts the outcome of COVID-19 patients with acute respiratory distress syndrome. J Transl Med. 2022;20(1):526.
Calzari L, Zanotti L, Inglese E, Scaglione F, Cavagnola R, Ranucci F, et al. Role of epigenetics in the clinical evolution of COVID-19 disease. Epigenome-wide association study identifies markers of severe outcome. Eur J Med Res. 2023;28(1):81.
Castro de Moura M, Davalos V, Planas-Serra L, Alvarez-Errico D, Arribas C, Ruiz M, et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine. 2021;66:103339.
Corley MJ, Pang APS, Dody K, Mudd PA, Patterson BK, Seethamraju H, et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J Leukoc Biol. 2021;110(1):21–6.
Konigsberg IR, Barnes B, Campbell M, Davidson E, Zhen Y, Pallisard O, et al. Host methylation predicts SARS-CoV-2 infection and clinical outcome. Commun Med (London). 2021;1(1):42.
Zhou S, Zhang J, Xu J, Zhang F, Li P, He Y, et al. An epigenome-wide DNA methylation study of patients with COVID-19. Ann Hum Genet. 2021;85(6):221–34.
Kimura H, Cohen JI. Chronic active Epstein–Barr virus disease. Front Immunol. 2017;8:1867.
Kristiansen MS, Stabursvik J, O’Leary EC, Pedersen M, Asprusten TT, Leegaard T, et al. Clinical symptoms and markers of disease mechanisms in adolescent chronic fatigue following Epstein–Barr virus infection: an exploratory cross-sectional study. Brain Behav Immun. 2019;80:551–63.
Britt W. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol. 2008;325:417–70.
Goodrum F, Caviness K, Zagallo P. Human cytomegalovirus persistence. Cell Microbiol. 2012;14(5):644–55.
Straus SE. Studies of herpesvirus infection in chronic fatigue syndrome. Ciba Found Symp. 1993;173:132–9; discussion 9–45.
Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol. 2021;12: 698169.
Tabacof L, Tosto-Mancuso J, Wood J, Cortes M, Kontorovich A, McCarthy D, et al. Post-acute COVID-19 syndrome negatively impacts physical function, cognitive function, health-related quality of life, and participation. Am J Phys Med Rehabil. 2022;101(1):48–52.
Monje M, Iwasaki A. The neurobiology of long COVID. Neuron. 2022;110(21):3484–96.
Raman B, Bluemke DA, Luscher TF, Neubauer S. Long COVID: post-acute sequelae of COVID-19 with a cardiovascular focus. Eur Heart J. 2022;43(11):1157–72.
Lippi G, Sanchis-Gomar F, Henry BM. COVID-19 and its long-term sequelae: what do we know in 2023? Pol Arch Intern Med. 2023;133(4):16402.
Chen C, Haupert SR, Zimmermann L, Shi X, Fritsche LG, Mukherjee B. Global prevalence of post-coronavirus disease 2019 (COVID-19) condition or long COVID: a meta-analysis and systematic review. J Infect Dis. 2022;226(9):1593–607.
Fernandez-de-Las-Penas C, Palacios-Cena D, Gomez-Mayordomo V, Florencio LL, Cuadrado ML, Plaza-Manzano G, et al. Prevalence of post-COVID-19 symptoms in hospitalized and non-hospitalized COVID-19 survivors: a systematic review and meta-analysis. Eur J Intern Med. 2021;92:55–70.
Han Q, Zheng B, Daines L, Sheikh A. Long-term sequelae of COVID-19: a systematic review and meta-analysis of one-year follow-up studies on post-COVID symptoms. Pathogens. 2022;11(2):269.
Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med. 2021;27(4):601–15.
O’Mahoney LL, Routen A, Gillies C, Ekezie W, Welford A, Zhang A, et al. The prevalence and long-term health effects of long Covid among hospitalised and non-hospitalised populations: a systematic review and meta-analysis. eClinicalMedicine. 2023;55: 101762.
Notarte KI, de Oliveira MHS, Peligro PJ, Velasco JV, Macaranas I, Ver AT, et al. Age, sex and previous comorbidities as risk factors not associated with SARS-CoV-2 infection for long COVID-19: a systematic review and meta-analysis. J Clin Med. 2022;11(24):7314.
Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, Skriabine S, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med. 2021;218(3):e20202135.
Vibholm LK, Nielsen SSF, Pahus MH, Frattari GS, Olesen R, Andersen R, et al. SARS-CoV-2 persistence is associated with antigen-specific CD8 T-cell responses. EBioMedicine. 2021;64: 103230.
Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. 2023;21(3):133–46.
Halpin SJ, McIvor C, Whyatt G, Adams A, Harvey O, McLean L, et al. Postdischarge symptoms and rehabilitation needs in survivors of COVID-19 infection: a cross-sectional evaluation. J Med Virol. 2021;93(2):1013–22.
Diamond MS, Kanneganti TD. Innate immunity: the first line of defense against SARS-CoV-2. Nat Immunol. 2022;23(2):165–76.
Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184(1):149-68 e17.
Braga L, Ali H, Secco I, Chiavacci E, Neves G, Goldhill D, et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature. 2021;594(7861):88–93.
Lin L, Li Q, Wang Y, Shi Y. Syncytia formation during SARS-CoV-2 lung infection: a disastrous unity to eliminate lymphocytes. Cell Death Differ. 2021;28(6):2019–21.
Damoiseaux J, Dotan A, Fritzler MJ, Bogdanos DP, Meroni PL, Roggenbuck D, et al. Autoantibodies and SARS-CoV2 infection: the spectrum from association to clinical implication: report of the 15th Dresden Symposium on Autoantibodies. Autoimmun Rev. 2022;21(3): 103012.
Kruger A, Vlok M, Turner S, Venter C, Laubscher GJ, Kell DB, et al. Proteomics of fibrin amyloid microclots in long COVID/post-acute sequelae of COVID-19 (PASC) shows many entrapped pro-inflammatory molecules that may also contribute to a failed fibrinolytic system. Cardiovasc Diabetol. 2022;21(1):190.
Turner S, Naidoo CA, Usher TJ, Kruger A, Venter C, Laubscher GJ, et al. Increased levels of inflammatory and endothelial biomarkers in blood of long COVID patients point to thrombotic endothelialitis. Semin Thromb Hemost. 2023;50:288–94.
Frank MG, Nguyen KH, Ball JB, Hopkins S, Kelley T, Baratta MV, et al. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: evidence of PAMP-like properties. Brain Behav Immun. 2022;100:267–77.
Yang B, Fan J, Huang J, Guo E, Fu Y, Liu S, et al. Clinical and molecular characteristics of COVID-19 patients with persistent SARS-CoV-2 infection. Nat Commun. 2021;12(1):3501.
Balnis J, Madrid A, Hogan KJ, Drake LA, Adhikari A, Vancavage R, et al. Persistent blood DNA methylation changes one year after SARS-CoV-2 infection. Clin Epigenetics. 2022;14(1):94.
Lee Y, Riskedal E, Kalleberg KT, Istre M, Lind A, Lund-Johansen F, et al. EWAS of post-COVID-19 patients shows methylation differences in the immune-response associated gene, IFI44L, three months after COVID-19 infection. Sci Rep. 2022;12(1):11478.
Nikesjo F, Sayyab S, Karlsson L, Apostolou E, Rosen A, Hedman K, et al. Defining post-acute COVID-19 syndrome (PACS) by an epigenetic biosignature in peripheral blood mononuclear cells. Clin Epigenetics. 2022;14(1):172.
Teschendorff AE, Breeze CE, Zheng SC, Beck S. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinf. 2017;18(1):105.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7): e47.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–67.
Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY). 2018;10(7):1758–75.
Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10(4):573–91.
Lu AT, Seeboth A, Tsai PC, Sun D, Quach A, Reiner AP, et al. DNA methylation-based estimator of telomere length. Aging (Albany NY). 2019;11(16):5895–923.
Gentilini D, Garagnani P, Pisoni S, Bacalini MG, Calzari L, Mari D, et al. Stochastic epigenetic mutations (DNA methylation) increase exponentially in human aging and correlate with X chromosome inactivation skewing in females. Aging (Albany NY). 2015;7(8):568–78.
Gentilini D, Scala S, Gaudenzi G, Garagnani P, Capri M, Cescon M, et al. Epigenome-wide association study in hepatocellular carcinoma: Identification of stochastic epigenetic mutations through an innovative statistical approach. Oncotarget. 2017;8(26):41890–902.
Gentilini D, Somigliana E, Pagliardini L, Rabellotti E, Garagnani P, Bernardinelli L, et al. Multifactorial analysis of the stochastic epigenetic variability in cord blood confirmed an impact of common behavioral and environmental factors but not of in vitro conception. Clin Epigenetics. 2018;10:77.
Guida V, Calzari L, Fadda MT, Piceci-Sparascio F, Digilio MC, Bernardini L, et al. Genome-wide DNA methylation analysis of a cohort of 41 patients affected by Oculo-auriculo-vertebral spectrum (OAVS). Int J Mol Sci. 2021;22(3):1190.
Spada E, Calzari L, Corsaro L, Fazia T, Mencarelli M, Di Blasio AM, et al. Epigenome wide association and stochastic epigenetic mutation analysis on cord blood of preterm birth. Int J Mol Sci. 2020;21(14):5044.
Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics. 2016;32(9):1423–6.
Li XK, Tu B, Zhang XA, Xu W, Chen JH, Zhao GY, et al. Dysregulation of glutamine/glutamate metabolism in COVID-19 patients: a metabolism study in African population and mini meta-analysis. J Med Virol. 2023;95(1): e28150.
Boldrini M, Canoll PD, Klein RS. How COVID-19 Affects the Brain. JAMA Psychiat. 2021;78(6):682–3.
Yesilkaya UH, Sen M, Balcioglu YH. COVID-19-related cognitive dysfunction may be associated with transient disruption in the DLPFC glutamatergic pathway. J Clin Neurosci. 2021;87:153–5.
Vezyroglou A, Akilapa R, Barwick K, Koene S, Brownstein CA, Holder-Espinasse M, et al. The phenotypic continuum of ATP1A3-related disorders. Neurology. 2022;99(14):e1511–26.
Balestrini S, Mikati MA, Alvarez-Garcia-Roves R, Carboni M, Hunanyan AS, Kherallah B, et al. Cardiac phenotype in ATP1A3-related syndromes: a multicenter cohort study. Neurology. 2020;95(21):e2866–79.
Kuang SY, Li Y, Yang SL, Han X. Child neurology: aicardi-goutieres syndrome presenting as recurrent ischemic stroke. Neurology. 2022;99(9):393–8.
Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of Aicardi–Goutieres syndrome. Am J Hum Genet. 2007;81(4):713–25.
Wang Q, Xiong F, Wu G, Wang D, Liu W, Chen J, et al. SMAD proteins in TGF-beta signalling pathway in cancer: regulatory mechanisms and clinical applications. Diagnostics (Basel). 2023;13(17):2769.
Lai LYS, Gracie NP, Gowripalan A, Howell LM, Newsome TP. SMAD proteins: mediators of diverse outcomes during infection. Eur J Cell Biol. 2022;101(2): 151204.
Shamilov R, Aneskievich BJ. TNIP1 in autoimmune diseases: regulation of toll-like receptor signaling. J Immunol Res. 2018;2018:3491269.
Islam T, Rahman MR, Aydin B, Beklen H, Arga KY, Shahjaman M. Integrative transcriptomics analysis of lung epithelial cells and identification of repurposable drug candidates for COVID-19. Eur J Pharmacol. 2020;887: 173594.
Murray NR, Kalari KR, Fields AP. Protein kinase Ciota expression and oncogenic signaling mechanisms in cancer. J Cell Physiol. 2011;226(4):879–87.
Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010;11(2):103–12.
Fang Y, Jiang Q, Li S, Zhu H, Xu R, Song N, et al. Opposing functions of beta-arrestin 1 and 2 in Parkinson’s disease via microglia inflammation and Nprl3. Cell Death Differ. 2021;28(6):1822–36.
Jiang T, Yu JT, Wang YL, Wang HF, Zhang W, Hu N, et al. The genetic variation of ARRB2 is associated with late-onset Alzheimer’s disease in Han Chinese. Curr Alzheimer Res. 2014;11(4):408–12.
Mathieu NM, Nakagawa P, Grobe CC, Reho JJ, Brozoski DT, Lu KT, et al. ARRB2 (beta-Arrestin-2) deficiency alters fluid homeostasis and blood pressure regulation. Hypertension. 2022;79(11):2480–92.
Ren H, Liu Y, Tan Z, Luo G, Zhang M, Li S, et al. A common variant of ARRB2 promoter region associated with the prognosis of heart failure. Hum Hered. 2023;88(1):68–78.
Zhou B, Song H, Xu W, Zhang Y, Liu Y, Qi W. The comprehensive analysis of hub gene ARRB2 in prostate cancer. Dis Markers. 2022;2022:8518378.
Zhang Y, Li M, Li L, Qian G, Wang Y, Chen Z, et al. beta-arrestin 2 as an activator of cGAS-STING signaling and target of viral immune evasion. Nat Commun. 2020;11(1):6000.
Wang Y, Gandy S. The Golgi apparatus: Site for convergence of COVID-19 brain fog and Alzheimer’s disease? Mol Neurodegener. 2022;17(1):67.
Thayer DA, Jan YN, Jan LY. Increased neuronal activity fragments the Golgi complex. Proc Natl Acad Sci U S A. 2013;110(4):1482–7.
Franzen J, Nuchtern S, Tharmapalan V, Vieri M, Nikolic M, Han Y, et al. Epigenetic clocks are not accelerated in COVID-19 patients. Int J Mol Sci. 2021;22(17):9306.
Cao X, Li W, Wang T, Ran D, Davalos V, Planas-Serra L, et al. Accelerated biological aging in COVID-19 patients. Nat Commun. 2022;13(1):2135.
Bejaoui Y, Humaira Amanullah F, Saad M, Taleb S, Bradic M, Megarbane A, et al. Epigenetic age acceleration in surviving versus deceased COVID-19 patients with acute respiratory distress syndrome following hospitalization. Clin Epigenetics. 2023;15(1):186.
Sanchez-Vazquez R, Guio-Carrion A, Zapatero-Gaviria A, Martinez P, Blasco MA. Shorter telomere lengths in patients with severe COVID-19 disease. Aging (Albany NY). 2021;13(1):1–15.
Mongelli A, Barbi V, Gottardi Zamperla M, Atlante S, Forleo L, Nesta M, et al. Evidence for biological age acceleration and telomere shortening in COVID-19 survivors. Int J Mol Sci. 2021;22(11):6151.
Li Y, Tollefsbol TO. Age-related epigenetic drift and phenotypic plasticity loss: implications in prevention of age-related human diseases. Epigenomics. 2016;8(12):1637–51.
Brusati A, Peverelli S, Calzari L, Tiloca C, Casiraghi V, Sorce MN, et al. Exploring epigenetic drift and rare epivariations in amyotrophic lateral sclerosis by epigenome-wide association study. Front Aging Neurosci. 2023;15:1272135.
Chen GK, Yan Q, Paul KC, Kusters CDJ, Folle AD, Furlong M, et al. Stochastic epigenetic mutations influence Parkinson’s disease risk, progression, and mortality. J Parkinsons Dis. 2022;12(2):545–56.
Fernandez MV, Kim JH, Budde JP, Black K, Medvedeva A, Saef B, et al. Analysis of neurodegenerative Mendelian genes in clinically diagnosed Alzheimer disease. PLoS Genet. 2017;13(11): e1007045.
Vojinovic D, Brison N, Ahmad S, Noens I, Pappa I, Karssen LC, et al. Variants in TTC25 affect autistic trait in patients with autism spectrum disorder and general population. Eur J Human Genet EJHG. 2017;25(8):982–7.
Zhao M, Ma J, Li M, Zhu W, Zhou W, Shen L, et al. Different responses to risperidone treatment in Schizophrenia: a multicenter genome-wide association and whole exome sequencing joint study. Transl Psychiatry. 2022;12(1):173.
Jahani M, Dokaneheifard S, Mansouri K. Hypoxia: a key feature of COVID-19 launching activation of HIF-1 and cytokine storm. J Inflamm (Lond). 2020;17:33.
Josuttis D, Schwedler C, Heymann G, Gumbel D, Schmittner MD, Kruse M, et al. Vascular endothelial growth factor as potential biomarker for COVID-19 severity. J Intensive Care Med. 2023;38(12):1165–73.
Melincovici CS, Bosca AB, Susman S, Marginean M, Mihu C, Istrate M, et al. Vascular endothelial growth factor (VEGF): key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. 2018;59(2):455–67.
Pine AB, Meizlish ML, Goshua G, Chang CH, Zhang H, Bishai J, et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm Circ. 2020;10(4):2045894020966547.
Smadja DM, Philippe A, Bory O, Gendron N, Beauvais A, Gruest M, et al. Placental growth factor level in plasma predicts COVID-19 severity and in-hospital mortality. J Thromb Haemost. 2021;19(7):1823–30.
Rovas A, Osiaevi I, Buscher K, Sackarnd J, Tepasse PR, Fobker M, et al. Microvascular dysfunction in COVID-19: the MYSTIC study. Angiogenesis. 2021;24(1):145–57.
He X, Liu C, Peng J, Li Z, Li F, Wang J, et al. COVID-19 induces new-onset insulin resistance and lipid metabolic dysregulation via regulation of secreted metabolic factors. Signal Transduct Target Ther. 2021;6(1):427.
Bader SM, Cooney JP, Pellegrini M, Doerflinger M. Programmed cell death: The pathways to severe COVID-19? Biochem J. 2022;479(5):609–28.
Phetsouphanh C, Darley DR, Wilson DB, Howe A, Munier CML, Patel SK, et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat Immunol. 2022;23(2):210–6.
Taeschler P, Adamo S, Deng Y, Cervia C, Zurbuchen Y, Chevrier S, et al. T-cell recovery and evidence of persistent immune activation 12 months after severe COVID-19. Allergy. 2022;77(8):2468–81.
Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, et al. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics. 2014;30(3):428–30.
Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017;45(4): e22.
Wang M, Huang J, Liu Y, Ma L, Potash JB, Han S. COMBAT: a combined association test for genes using summary statistics. Genetics. 2017;207(3):883–91.
Muller F, Scherer M, Assenov Y, Lutsik P, Walter J, Lengauer T, et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 2019;20(1):55.
Ren X, Kuan PF. methylGSA: a Bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics. 2019;35(11):1958–9.
Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. 2012;49(7):433–6.
Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):W199–205.
Stelzer G, Plaschkes I, Oz-Levi D, Alkelai A, Olender T, Zimmerman S, et al. VarElect: the phenotype-based variation prioritizer of the GeneCards Suite. BMC Genom. 2016;17(Suppl 2):444.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–1.
Acknowledgements
We would like to acknowledge the patients and study participants who made this study possible. We also express our gratitude to the staff of the institutions involved in the collection of blood and clinical data.
Funding
This work was supported by two grants from the Italian Ministry of Health (Ricerca Corrente Reti 2020-RCR-2020–23670065 and Ricerca Corrente Reti 2021-RCR-2021–23671212). CG is supported by Fondi 5xMille and Ricerca Corrente Rete Aging. AF is supported by the Italian National Research Council (CNR)—(SAC.AD002.173.027) Progetti@CNR—IMMUNAGE.
Author information
Authors and Affiliations
Contributions
DG and GP conceived the study, contributed to the analysis, and co-wrote the first draft of the manuscript. The recruitment of patients (and clinical information) was managed by LP, IC, EI, RD, AMDB, LZ, SDM, AF, VB, MGZ, and CG and carried out by EI and IC. LC and DFD performed the DNA methylation Illumina microarray assay. DG, LC, DFD, RC, and AB developed the informatics pipeline for this study. LC, DG, DFD, AB, RC, FR, and GNB analyzed the data. GC contributed by aiding in interpreting the results. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Approval for this study was granted by the local Istituto Auxologico Italiano Ethics Committee (Approval number: 2020_03_26_02), ASST Grande Ospedale Metropolitano Niguarda Ethics Committee (Approval number: 232–22,042,021) and by the Ethical Committee of Istituti Clinici Scientifici Maugeri Spa SB (Pavia) (Document: METH-COVID-19—Studio epigenetico sulle conseguenze dell’infezione da SarsCoV-2 (ID: 2543 CE)).
Consent for publication
All study participants gave informed written consent including specific consent to genetic testing and permission to publish the results.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luciano, C., Fernando, D.D., Lucia, Z. et al. Epigenetic patterns, accelerated biological aging, and enhanced epigenetic drift detected 6 months following COVID-19 infection: insights from a genome-wide DNA methylation study. Clin Epigenet 16, 112 (2024). https://doi.org/10.1186/s13148-024-01724-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01724-9