
Abstract
Background
Gastrointestinal malignancies encompass a diverse group of cancers that pose significant challenges to global health. The major histocompatibility complex (MHC) plays a pivotal role in immune surveillance, orchestrating the recognition and elimination of tumor cells by the immune system. However, the intricate regulation of MHC gene expression is susceptible to dynamic epigenetic modification, which can influence functionality and pathological outcomes.
Main body
By understanding the epigenetic alterations that drive MHC downregulation, insights are gained into the molecular mechanisms underlying immune escape, tumor progression, and immunotherapy resistance. This systematic review examines the current literature on epigenetic mechanisms that contribute to MHC deregulation in esophageal, gastric, pancreatic, hepatic and colorectal malignancies. Potential clinical implications are discussed of targeting aberrant epigenetic modifications to restore MHC expression and 0 the effectiveness of immunotherapeutic interventions.
Conclusion
The integration of epigenetic-targeted therapies with immunotherapies holds great potential for improving clinical outcomes in patients with gastrointestinal malignancies and represents a compelling avenue for future research and therapeutic development.
Similar content being viewed by others

Background
Our understanding of epigenetic regulation in physiology and pathophysiology is constantly evolving. Reversible DNA methylation signatures and post-translational modifications to histones, such as methylation, acetylation, phosphorylation, and ubiquitination, play pivotal roles in development and cellular homeostasis. Epigenetic modifications can activate or suppress gene expression in response to both intrinsic and extrinsic stimuli. The counterpoint to this yin/yang arrangement is that inappropriate gene expression can be associated with common pathologies, including cancer etiology. Hypermethylation in gene promoter regions, genome-wide hypomethylation, and histone deacetylation are hallmarks of aberrant gene transcription in various malignancies [1]. A deeper appreciation of tissue-specific epigenetic regulation, with a view to enhanced mechanistic targeting and precision oncology, will open new avenues for cancer interception and novel therapeutic approaches.
Recent advances also have highlighted the crosstalk between epigenetic regulation and the immune system. Major histocompatibility complexes class I and class II (MHC-I and MHC-II) are key components of the adaptive immune response. These human glycoproteins interact with specific pathogenic antigens, resulting in cell surface T cell recognition, and engagement of the host immune system. Epigenetic mechanisms have critical roles in the regulation of MHC-I- and MHC-II-associated genes, the corresponding enhanceosomes, and the master transactivators [2].
Because of their central roles in adaptive immunity, MHC complexes have received interest in the context of immune responses to metastasis and epigenetic regulation. Ideally, antigen-specific CD8+ and CD4+ T cells respond, respectively, to MHC-I and MHC-II cell surface complexes on cancer cells, activating host immune mechanisms that target the tumor for removal [2]. However, altered MHC expression due to malignant transformation and epigenetic deregulation can dampen tumor detection and engagement of the adaptive immune response [2, 3]. The counterpoint, however, is that clinical cancer interception and re-engagement of host immunity are viable strategies arising from the reversibility of epigenetic landscapes in precancer and cancer stages.
We sought to provide an overview of the epigenetic deregulation of MHC-associated factors in the pathogenesis of intestinal, hepatic, and pancreatic malignancies, broadly classified herein as gastrointestinal (GI) cancers. The latter are characterized by epigenetic silencing and concomitant alterations in gene expression that appear in distinct gut pathologies [3]. It is known that GI tumors lack MHC-I and MHC-II expression, which contributes to low CD8+ and CD4+ T cell tumor infiltration and poor prognosis. Thus, new therapeutic approaches that upregulate MHC expression could result in enhanced patient outcomes [4, 5]. Epigenetic landscapes of GI tumors are of current scientific interest with a view to understanding immune evasion in the gut, and to guide future development of novel cancer immunotherapies and immunopreventive approaches. These aspects will be discussed in the following sections.
Epigenetic regulation of MHC-I and MHC-II
Antigen presentation by MHC-I and MHC-II cell surface molecules and the corresponding CD8+ and CD4+ T cell activation have been reviewed extensively [4, 5]. We focused on the epigenetic regulation of MHC components, including human leukocyte antigen (HLA) factors, in the context of specific gut pathologies. Transcription of MHC-I genes is controlled by conserved cis‑acting regulatory elements at the proximal promoters. The promoter generally contains an enhancer A element which contains nuclear factor‑κB (NF‑κB) binding sites, an interferon (IFN)‑stimulated response element (ISRE), and an S-X-Y module. The S-X-Y module consists of four different elements, the W/S, X1, X2 and Y boxes. Specifically, the X1 box binds to the regulatory factor X complex (RFX), which consists of RFX5, RFX‑associated ankyrin‑containing protein (RFXANK) and RFX‑associated protein (RFXAP) [6,7,8,9,10]. The X2 box is bound by cAMP‑responsive element‑binding protein 1 (CREB1) and activating transcription factor 1 (ATF1); whereas the Y box is bound by nuclear transcription factor Y (NFY), which consists of the NFYA, NFYB and NFYC subunits [11,12,13]. Together, the assembly of these proteins to the S-X-Y module is critical for the transcription of MHC-I genes (Fig. 1a) [2, 14].

Transcriptional and epigenetic modulation of MHC class I and class II genes by MHC-I/II enhanceosomes. a MHC-I enhanceosome recruits HATs and HMTs to MHC-I class I gene promoter regions inducing the expression of MHC-I antigen presentation pathway (APP)-associated genes. b MHC-II enhanceosome-induced DNA looping facilitates HAT and AMT recruitment into the proximal promoter of MHC class II genes enabling MHC-II APP-associated gene induction. IFN-γ, interferon-gamma; HAT, histone acetyltransferase; HMT, histone methyltransferase; AMT, arginine methyltransferase; IRF-1, interferon regulatory factor 1; ISRE, interferon-sensitive response element; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; STAT1, signal transducer and activator of transcription 1, CDK7; cyclin-dependent kinase 7; RNA Pol II, RNA polymerase II; TATA, Goldberg–Hogness (TATA) box. Panel (a) adapted from Jongsma MLM et al., Mol Immunol 2019;113:17 and Panel (b) adapted from Masternak K et al., Genes Dev 2000;14(9): 1159. Created with BioRender.com on April 24, 2024
However, MHC-I expression also involves NLR Family CARD Domain Containing 5 (NLRC5), as a ‘master’ MHC-I transactivator [2]. The tripartite domain structure in NLRC5 contains an N-terminal caspase activation and recruitment domain (CARD), a centrally located nucleotide-binding domain (NBD) and carboxy‑terminal leucine‑rich repeats (LRRs) [15,16,17,18]. Moreover, transcription of NLRC5 can be efficiently induced by IFN-γ, via signal transducer and activator of transcription 1 (STAT1) interactions with the corresponding gene promoter region [19, 20]. Genome-wide expression profiles in NLRC5 wild-type or mutant human cell lines revealed that NLRC5 was crucial for the efficient induction of MHC-I associated genes. Notably, NLRC5 not only induced the expression of classical MHC-I genes (HLA‑A, HLA‑B and HLA‑C), but also upregulated accessory components, such as the β2 Microglobulin gene (β2M), Low-molecular mass protein 2 (LMP2, also known as Proteasome 20S subunit beta 9/PSMB9) and Transporter associated with antigen processing 1 (TAP1) (Fig. 1a) [15].
The nucleotide-binding domain (NBD) of NLRC5 is essential for nuclear import and MHC-I gene regulation [21]. Although NLRC5 lacks a DNA‑binding domain, it forms a multi-protein complex with RFX, CREB1-ATF1 and NFY known as the MHC enhanceosome, assembled on the S-X-Y module [22]. Nuclear NLRC5 also acts as a platform for histone-modifying enzymes that regulate chromatin dynamics. For example, NLRC5 has synergistic interactions with histone acetyltransferases (HATs) that activate human MHC class I gene transcription and promote histone H3 lysine 27 methylation (H3K9me) at the proximal promoter of H2-K1, a murine ortholog of classical human MHC-I genes (Fig. 1a) [23, 24].
The polymorphic MHC-I chain-related proteins A and B (MICA and MICB) are induced by cellular stress or neoplastic transformation [25, 26]. Despite homology with classical MHC class I molecules, MICA and MICB are stably expressed on epithelial cells without association with B2-microglobulin, a key subunit of MHC-I complexes [27]. Recognition by CD8+ T cells and NK cells involves the lectin-like NKG2D receptor and plays a role in autoimmunity and tumor recognition [28, 29]. Epigenetic regulation of MIC factors is poorly understood, although recent evidence implicated a role in Merkel cell carcinoma [30].
The discovery of NLRC5 as a key MHC class I transactivator (CITA) paralleled prior research on the corresponding master regulator of MHC-II-dependent genes, known as class II transactivator (CIITA) [31, 32]. Like NLRC5/CITA, CIITA does not bind directly to DNA, but recruits transcription factors and other protein partners that modify chromatin interactions and the local epigenetic landscape [31, 32]. The presence of an S-X-Y module enables interacting proteins, similar to those on MHC-I promoters, to form an MHC-II enhanceosome that acts as a scaffold to recruit CIITA [31, 32]. A regulatory module similar to the X–Y motif is located upstream of some MHC-II genes, such as HLA-DRA. After CIITA binds to the MHC-II enhanceosome, DNA looping facilitates synergistic interactions with chromatin modifiers and HATs, such as CREB binding protein (CBP), P300/CBP-associated factor (PCAF), and steroid receptor coactivator-1 (SRC-1) [33]. Thus, CIITA can modify the chromatin landscape from a repressive ‘closed’ state to an open conformation, allowing for other DNA-binding proteins to interact with MHC-II promoters (Fig. 1b).
Interestingly, CIITA is a direct target for acetylation by CBP and PCAF, a process that can influence CIITA nuclear translocation [34]. Other HATs, such as SRC-1, are recruited to MHC-II promoters only after IFN-γ stimulation and help to dampen estradiol-mediated inhibition of MHC-II expression (Fig. 1b) [35]. Chromatin immunoprecipitation (ChIP) in cell lines that are responsive to IFN-γ established the correlation between CIITA and increased levels of histone H3 and H4 acetylation, synonymous with CIITA promoting an ‘open’ chromatin state [33]. One report [36] proposed that CIITA might possess HAT activity, although this awaits independent corroboration.
As counterpoint to HATs, cellular histone deacetylases 1 and 2 (HDAC1, HDAC2) repress MHC-II gene expression in a broad range of cancers [37]. The HDACs and their corepressors, such as mSin3A, trigger enhanceosome disassembly and override IFN-γ-mediated upregulation of MHC-II genes. In cancer immunotherapy, HDAC inhibitors can increase antitumor immunity by facilitating histone acetylation near MHC-II promoters [33, 37]. Other post-transcriptional modifications, such as mono-ubiquitination, regulate the function of CIITA leading to increased gene activation [38]. Additionally, CIITA can associate with coactivator-associated arginine methyltransferase 1 (CARM1), leading to arginine methylation on histone H3R17 and CBP, enhancing MHC-II promoter interactions and MHC-II gene expression [39]. Moreover, CIITA can promote cyclin-dependent kinase 7-mediated phosphorylation of RNA polymerase II, directly initiating the synthesis of mRNA transcripts from the corresponding genes (Fig. 1b) [33, 40].
Unlike MHC-I complexes which are present in all mammalian cells, MHC-II complexes are expressed mainly in antigen-presenting cells, such as dendric cells, macrophages or B cells. However, as previously noted, IFN-γ induces MHC-II expression in a CIITA-dependent manner in other cell types [41]. Among the four CIITA promoter regions, designated as PI-PIV, IFN-γ acts mainly through CIITA-PIV. Distinct cell types, such as trophoblasts and tumor cells, have mechanisms to diminish the IFN-γ response acting though CIITA-PIV [42]. One such mechanism is DNA hypermethylation at CpG islands around CIITA-PIV, which decreases transcription factor binding [42, 43]. Studies with DNA methyltransferase (DNMT) inhibitors such as 5-azacytidine (5-AZA), plus HDAC inhibitors such as Trichostatin A (TSA), established that epigenetic mechanisms silence CIITA-PIV in tumor cells, and that interference with such mechanisms restores CIITA expression [42, 44, 45]. Thus, downregulation of CIITA and MHC-II-associated gene expression may be a common mechanism of immune escape in cancer cells. Accordingly, studies in uveal melanoma cells observed that treatment with 5-AZA restored the IFN-γ-mediated inducibility of CIITA.
Certain gastric and colorectal cancers do not respond to IFN-γ due to DNA hypermethylation near CIITA-PIV [42, 46]. Histone H3K9 methylation was present as a gene-silencing mark, suggesting that selective inhibition of the corresponding H3K9 methyltransferase might be a therapeutic approach to recover CIITA expression [47]. Collectively, these findings highlight the critical importance of CIITA as a master regulator of MHC-II-associated gene expression, its deregulation in cancer cells, and the contributions of epigenetic mechanisms to pathogenesis.
Immunoepigenetics and the tumor microenvironment
Epigenetic alterations in the tumor microenvironment (TME) affect the dynamic crosstalk between cancer and immune cells exhibiting differential expression of MHC antigen presentation pathway (APP) components. Immune cell subtypes include tumor-associated macrophages (TAMs), neutrophils, natural killer cells, T and B lymphocytes, as well as fibroblasts, stromal cells, and blood vessels [48,49,50]. The TME can either inhibit or promote tumorigenesis and lead to chemotherapy resistance, with implications for cancer development and treatment [51,52,53,54]. For example, a member of the tumor necrosis factor receptor superfamily, Decoy receptor 3 (DcR3), was reported to be overexpressed in pancreatic cancer and other malignancies [49]. DcR3 induced TME dendritic cell apoptosis and TAM differentiation into an M2 phenotype, which was associated with suppression of inflammatory mechanisms and increased angiogenesis, leading to tumor progression [49, 50]. Additionally, DcR3 diminished the expression of genes associated with the MHC-II APP, including classical (HLA-DR, -DP and -DQ) and non-classical (HLA-DM and -DO) MHC-II genes, as well as CD74 and CIITA, in monocyte-derived macrophages (MDMs) [49]. The reduced antigen-presenting capacity in TAMs and T cell anergy diminished the host immune response and promoted cancer immune evasion. Moreover, extracellular signal-regulated kinase/c-Jun N-terminal kinase-associated histone deacetylation has been identified as a mechanism for DcR3-mediated MHC-II suppression [49, 50, 55]. Global histone deacetylation affected all CIITA promoters after MDMs were treated with DcR3, implicating chromatin remodeling and alterations in HDAC/HAT expression, activity, or recruitment; however, the precise mechanisms remain to be elucidated [49, 50]. DcR3 also regulated TAM differentiation in murine models, with increased arginase activity and decreased MHC-II complexes. Downregulation of MHC-II enhanced tumor growth and impaired tumor-associated antigen presentation to T cells, dampening the anti-tumoral adaptive immune response [49, 56]. The HDAC inhibitor sodium valproate/valproic acid (VPA) partially restored MHC-II expression and circumvented the enhanced tumor growth in DcR3-transgenic mice inoculated subcutaneously with murine colon adenocarcinoma cells, indicating that epigenetic modulation of TAM differentiation and MHC-II expression had a crucial role in DcR3-driven tumor progression [56, 57]. The clinical relevance of VPA as an anticancer therapeutic agent is well established, supporting HDAC inhibition for APP enhancement, either as a single agent or by augmenting standard of care in patients with tumors expressing high DcR3 levels, such as pancreatic cancer [56, 58].
Gastric cancer and immunoepigenetic deregulation
Heterogeneous HLA-DRA protein expression was observed in tissue microarrays of human gastric cancer (Fig. 2a), with the Kaplan–Meier survival curves, obtained from The Human Protein Atlas, indicating a non-statistically significant difference for high vs. low HLA-DRA expression of the α subunit of HLA-DR (Fig. 2b P-score = 0.20). In human gastric cancer cell lines, expression of HLA-DR in response to IFN-γ was dampened due to CIITA-PIV hypermethylation [59]. Meazza et al. noted that the upregulation of HLA-DR after introduction of exogenous CIITA led to T cell activation and tumor rejection, suggesting that CIITA could be a valuable target for future therapeutics [60]. Satoh et al. confirmed that 14 of 20 gastric and colon cancer cell lines expressed HLA-DR after IFN-γ stimulation, and that exogenous CIITA in cell lines that lacked HLA-DR prompted re-expression of the target gene, with and without IFN-γ stimulation. Subsequent work noted that CIITA was expressed only when CIITA-PIV was unmethylated [59]. It was confirmed that CIITA-PIV had a CpG island that was methylated close to the transcription start site, and also involved histone deacetylation in the 5’ region of the gene. Supporting the dynamic crosstalk between epigenetic regulatory mechanisms, IFN-γ increased histone acetylation more effectively in gastric cell lines harboring unmethylated CpG sites in the CIIT-PIV region, [59].

MHC-I and MHC-II expression in gastric normal and cancer tissues. a, c Heterogeneous expression of HLA-DRA and HLA-B proteins in human tissue microarrays (TMAs). b, d Kaplan–Meier (KM) curves for HLA-DRA and HLA-B gene expression in gastric cancer. For quantification of percent labeling in TMAs see https://www.proteinatlas.org/
In a similar fashion to HLA-DRA, heterogeneous protein expression was detected for classical MHC-I players, such as HLA-B (Fig. 2c), and survival in patients beyond the 2-year timepoint tended to be improved by high HLA-B levels (Fig. 2d). Downregulation of HLA-A/B/C has been associated with promoter methylation of the corresponding genes in other types of cancer, such as melanoma and neuroblastoma, compared to adjacent non-tumor tissues, suggesting that MHC-I surface complexes could be restored in gastric cancer by DNA methyltransferase inhibitors [61]. These findings re-affirm the integration of reversible epigenetic mechanisms, such as DNA methylation and histone acetylation, and provide avenues for HDAC + DNMT combinatorial therapies in gastric cancer interception.
Pancreatic cancer and immunoepigenetic deregulation
Pancreatic cancer is another malignancy that is influenced by DNA methylation and gene silencing [62,63,64,65,66]. Studies by Cao et al. in a human pancreatic cancer cell line of ductal origin (PANC-1) revealed CIITA promoter hypermethylation and low MHC-II expression, even after stimulation with IFN-γ. Methylation of CIITA was decreased after combined treatment with IFN-γ, 5-AZA and the pan-HDAC inhibitor suberoylanalide hydroxamic acid (SAHA) [67,68,69]. In PANC-1 cells, higher doses of 5-AZA caused complete demethylation of the CIITA promoter, reflected by increased CIITA and MHC-II-associated gene expression [67]. Interestingly, a vaccine was developed using PANC-1 cells with augmented MHC-II expression following epigenetic drug combinations. In murine models, the vaccine enhanced lymphocyte proliferation, CD8+ T cell anti-tumoral activity, and IFN-γ plus IL-2 secretion by Th1-type lymphocytes, while decreasing TGF-β and IL-4 secretion by Th2-type lymphocytes. These findings indicated a shift in the Th1/Th2 ratio, increasing the number of Th1-type cells and resulting in increased activity of cytotoxic T cells [67]. Thus, vaccines produced by a combination of HDAC plus DNMT inhibition could be an approach for pancreatic cancer interception, increasing MHC-II expression and triggering an anti-tumoral immune response. Tissue microarrays showed uniform low levels of CIITA protein in pancreatic cancers, with focal expression (Fig. 3a). Surprisingly, patients with high CIITA levels tended to have worse prognosis, although this was not statistically significant due to the low sample number (Fig. 3b, Pscore = 0.12).

MHC-II-related expression in pancreatic normal and cancer tissues. a Weak-to-moderate CIITA protein expression in human TMAs. b KM survival curves for CIITA gene expression in pancreatic cancer. For quantification of percent labeling in TMAs see https://www.proteinatlas.org/
Colorectal cancer and immunoepigenetic deregulation
In Colon 26 murine colon adenocarcinoma cells, IFN-γ induced transcription from the CIITA-PIV locus, but not from other CIITA promoters, whereas TSA treatment induced CIITA-PIII and not CIITA-PIV [70]. Transfection of Colon 26 cells with a dominant negative CIITA plasmid did not inhibit TSA-mediated MHC-II expression, while IFN-γ-mediated MHC-II expression was impeded [54, 70]. Thus, even in a single murine colon cancer cell line, pathways converge that are CIITA-dependent and CIITA-independent. Furthermore, IFN-γ but not TSA increased protein levels of IRF-1 and phosphorylation of STAT-1, which are essential for CIITA-IV activation [70,71,72]. Combinatorial treatment of IFN-γ with TSA, or the subsequent addition of IFN-γ after TSA treatment, further increased CIITA induction when compared to IFN-γ alone. Interestingly, pretreatment with TSA followed by IFN-γ had the opposite effect, showing repression of total CIITA and CIITA-PIV [37, 70]. This unexpected finding was associated with a decrease in STAT-1α phosphorylation in Colon 26 cells pretreated with TSA. These findings indicate that HDAC inhibition can increase or decrease MHC-I expression in a context-dependent manner [70].
Although TSA increased histone acetylation and induced MHC-II-dependent gene expression, the precise mechanisms are not well understood [37, 70, 73]. As a consequence of TSA-mediated changes in histone acetylation, increased H3K4 methylation and decreased H3K9 methylation was observed, which are hallmarks of enhanced gene expression [70, 74]. The H3K9 methylation mark provided a docking site for heterochromatin protein-1 (HP-1) in the promoter region of MHC-II genes, leading to localized gene silencing; thus, decreased H3K9 methylation after TSA treatment interfered with the repressive actions HP-1 [70, 75]. A TSA-mediated shift between HAT and HDAC activities also affected post-translational modifications on nonhistone proteins, including transcription factors that promote MHC-II expression. For example, Greer et al. observed that CBP/p300 HATs and TSA-mediated HDAC inhibition promoted CIITA monoubiquitylation and enhanced MHC-II expression [70, 76]. However, further studies are needed to determine which mechanism(s) are critical for upregulation of CIITA in Colon 26 cells.
Another approach might be to combine HDAC inhibition with standard of care chemotherapy [77]. For example, the HDAC inhibitor depsipeptide (Dep) enhanced the antitumor efficacy in human colon cancer cells of a first line drug used in the treatment of colorectal cancer, namely 5-fluorouracil (5-FU) [78]. Dep + 5-FU reduced colony formation and cell viability, and increased caspase 3/7 activation in HCT116 cells, compared to 5-FU monotherapy. Microarrays revealed upregulation of genes associated with apoptosis and cell death, as well as MHC-II classical genes such as HLA-DPB1, HLA-DQB1, HLA-DRA and HLA-DRB1 [82]. Other colon cancer cell lines were less responsive to Dep + 5-FU, revealing a modest increase in HLA-DRA, and none of the cell lines exhibited changes in MHC-I genes. In HCT116 cells, increased CIITA, CREB3/5 and PCAF after Dep + 5-FU treatment was observed [78]. As previously established, CREB3 and CREB5 participate in the positive regulation of MHC-II genes, whereas PCAF has HAT activity and when associated with CIITA enhances apoptosis [31, 78, 79].
A colon cancer cell line lacking DNMT1 and DNMT3b exhibited reduced methylation on CIITA-PIV, implicating a role for DNMTs in CIITA regulation [59, 80]. However, CIITA expression occurred only after IFN-γ stimulation, due to transcription factor recruitment to unmethylated CIITA-PIV [59]. Thus, epigenetic modulation of CIITA-PIV by DNMT inhibitors might be a therapeutic approach to enhance immunotherapy. For example, combining the DNTM1/DNMT3a inhibitor hydralazine with VPA increased the expression of MICA and MICB ligands on NK cells and induced NK cell cytotoxicity against tumor cells. This effect was related to the presence of histone H3K4me2 at the MICA and MICB promoters [81]. These studies illustrated how MICA and MICB expression is mostly controlled by DNA methylation, further highlighting the relevance that DNMT inhibitors could have in a subset of colorectal cancer.
On the other hand, metabolomic data from dietary spinach intake in the Apc-mutant polyposis in rat colon (Pirc) model implicated linoleate and butanoate metabolites that targeted HDAC activity and IFN-γ signaling [82]. Mechanistic studies with 13(S)-hydroxyoctadecadienoic acid (13S-HODE) and (S)-2-hydroxybutanoate (S-2HB) revealed elevated β2m protein levels, increased cell surface β2m expression, and induced IL-2 secretion from T cell hybridoma co-cultures, consistent with enhanced MHC-I cell surface presentation on colon cancer cells [82]. Epigenetic and chromatin accessibility marks implicated in the etiology of human colon cancer, obtained from UCSC Genome Browser and ENCODE databases, identified histone acetylation/methylation and DNA methylation, in the regulation of MHC-I and MHC-II gene expression (Fig. 4). For an entire suite of MHC-I and MHC-II genes, low expression in human colorectal cancer was synonymous with worse survival (Fig. 5). In tissue microarrays from colorectal patients, a broad range of MHC-I and MHC-II protein expression was detected, as exemplified for B2M and HLA-DRA, respectively (Fig. 6a, c). Kaplan–Meier curves (Fig. 5, Figure b,d) supported the hypothesis that epigenetic-targeted interventions that re-express MHC factors could enhance survival, and be valuable for immune interception in a subset of colorectal cancer patients.

Epigenetic landscapes and chromatin accessibility of MHC-I and MHC-II genes in human colon cancer cells. Chromatin immunoprecipitation sequencing (ChIP-seq) in HCT116 cells for histone H3K27ac and H3K27me3, or reduced-representation bisulfite sequencing for DNA methylation, with color coding representing methylated CpG island density (fold-change vs. normal). cCREs, cis-regulatory elements (red promoter-like; orange proximal enhancer-like; yellow distal enhancer-like); ATAC-seq, assay for transposase-accessible chromatin with sequencing. Data from the UCSC Genome Browser (https://genome.ucsc.edu) and ENCODE (https://www.encodeproject.org)

KM survival curves for colorectal cancer patients with high vs. low MHC-I (top and middle panels) and MHC-II gene expression (lower panels). Data obtained from the Human Protein Atlas (https://www.proteinatlas.org/)

MHC-I and MHC-II expression in colorectal normal and cancer tissues. a, c Heterogeneous expression of HLA-DRA and B2M proteins in human TMAs. b, d KM survival curves for HLA-DRA and B2M gene expression in colorectal cancer. For quantification of percent labeling in TMAs see https://www.proteinatlas.org/
Esophageal cancer and immunoepigenetic deregulation
Epigenetic modifications also play a critical role in the progression of upper GI malignancies [83]. Kazakh esophageal squamous cell carcinoma (ESCC) exhibited unusually high levels of HLA-DQ, HLA-DR and HLA-DP, whereas later clinical stages had reduced expression of the corresponding genes, indicating that decreased MHC-II molecules facilitated immune escape and tumor progression [84]. Hu et al. demonstrated that HLA-DRB1 methylation levels were lower in Kazakh ESCC when compared to normal tissue, whereas methylation levels of HLA-DQB1 were increased in tumor samples. The methylation status of HLA-DRB1 might promote tumor progression, as evidenced by HLA-DRB1 CpG16 hypermethylation and subsequent gene silencing in later clinical stages and vice versa for HLA-DQB1, where lower methylation levels of CpG16-17 were associated with increased aggressiveness of Kazakh ESCC [84]. These findings suggested that changes in the promoter methylation status of HLA-DRB1 and HLA-DQB1 are tightly associated with Kazakh ESCC progression and could serve as cancer biomarkers. Because HLA genes can harbor differential methylation profiles in Kazakh ESCC, and DNA methyltransferase inhibitors would likely reverse the hypomethylation status of both HLA-DRB1 and HLA-DQB1, it would be important to sub-group ESCC patients before adopting this kind of ‘precision’ epigenetic therapy.
In the case of MHC-I genes, downregulation of HLA-A and β2M has been previously linked to esophageal carcinoma pathogenesis, while overexpression of microRNAs, miR-148a-3p and miR-125a-5p, has been associated with downregulation of MHC-I genes, such as TAP2, in this type of malignancies [85]. Moreover, multiple CpG sites were linked to loss of HLA-B and other associated genes, such as TAP2 and LMP7 [86]. However, human tissue microarrays indicated that patients with head and neck cancer exhibited heterogenous expression of HLA-DRB and LMP7 proteins (Fig. 7a, c), and the corresponding gene expression changes did not predict a significant difference in survival probability [86]. (Fig. 7b, d).

MHC-I and MHC-II expression in head and neck cancer and normal tissues. a, c Heterogeneous expression of HLA-DRB1 and LMP7 proteins in human TMAs. b, d KM survival curves for HLA-DRB1 and LMP7 gene expression in head and neck cancer. For quantification of percent labeling in TMAs see https://www.proteinatlas.org/
Liver cancer and immunoepigenetic deregulation
Vorinostat first exhibited promise as an HDAC inhibitor and anticancer agent in patients with advanced cutaneous T cell lymphoma [87, 88], followed by malignancies such as hepatocellular carcinoma [30, 89, 90]. Vorinostat downregulated microRNA-20a, diminished STAT3 tyrosine phosphorylation, and increased histone acetylation at MICA and MICB promoters, leading to increased expression of these genes and enhanced natural killer (NK) cell-mediated tumor targeting [90]. The HDAC inhibitor MS-275 epigenetically modified exosomes secreted by human liver cancer cells, increasing the expression of MICA, MICB, and heat shock protein 70, and enhancing the cytotoxic effects of NK cells [91]. These data indicated that MICA and MICB expression in liver cancer is regulated by histone acetylation, and that HDAC inhibitors might provide for targeted therapies. Representative examples of MHC-I and MHC-II players in liver cancer, such as B2M and CIITA, showed similar patterns as other cancer types, with heterogenous protein expression in tissue microarrays (Fig. 8a, c) and low gene expression being predictive of poor survival (Fig. 8b, d). Tumor subtype was an additional variable, with hepatocellular carcinoma having high expression and cholangiocarcinoma having low CIITA and B2M protein levels (Fig. 8a, c).

MHC-I and MHC-II expression in hepatic normal and cancer tissues. a, c Heterogeneous expression of CIITA and B2M proteins in human TMAs. b, d KM survival curves for CIITA and B2M gene expression in liver cancer. For quantification of percent labeling in TMAs see https://www.proteinatlas.org/
Conclusions
Demographic and epidemiologic data indicate that the burden of GI cancers and other malignancies remains a major health concern in the US and worldwide [92, 93]. For example, a 50% increase in death from colon cancer and a doubling of rectal cancer mortality is predicted in the US by 2035 [94]. Similarly, new gastric cancer cases diagnosed per year are expected to increase from 1.09 to 1.77 million worldwide from 2020 to 2040, representing a 62% increase in incidence [95]. In this same timeframe, esophageal cancer incidence and mortality are predicted to rise 58.1% and 61.7% worldwide, respectively [96]. Thus, there is an urgent need for new and improved approaches to cancer interception. The reversibility of deregulated epigenetic landscapes in numerous malignances provides an attractive avenue for GI cancer intervention [97]. Understanding the fundamental epigenetic mechanisms that govern the expression of MHC-I, MHC-II, and MHC-associated molecules is an area of growing interest, especially in the field of immunotherapy, via epigenetic monotherapies or as adjuvants to standard of care. Drugs that modify distinct epigenetic processes, such as DNA methylation or histone post-translational modifications, can upregulate MHC-associated molecules in GI malignancies, suggesting the potential to re-engage the host immune system for enhanced cancer immune evasion. It is likely that refinements will be made to the current arsenal of ‘epigenetic’ drugs, to improve efficacy while reducing toxicity. This might involve patient-specific molecular phenotyping combined with the inhibition of selected epigenetic ‘readers’, ‘writers’ and ‘erasers’ [98,99,100]. For example, histone methylation inhibitors targeting the polycomb repressive complex 2 (PRC2) have shown promise in enhancing cancer cell immunogenicity through MHC upregulation [101, 102]. Notably, PRC2 inhibitors are in clinical trials for the treatment of lymphoma and nasopharyngeal carcinoma, and an orally bioavailable PRC2 inhibitor was recently approved for epithelioid sarcoma [103, 104]. Additionally, disrupting the scaffolding capabilities of other histone methyltransferase complexes, through the use of WD repeat-containing protein 5 (WDR5) inhibitors, demonstrated antitumor efficacy in preclinical models of colon cancer [105]. These developments suggest that histone methylation inhibitors will have clinical utility for MHC re-expression, and that drug repurposing could facilitate immunomodulatory targeting of GI malignancies for which altered histone methylation has been implicated [106].
As previously mentioned, MHC upregulation could be a critical mechanism to augment the efficacy of current immunotherapeutic strategies. For example, Birinapant, a mimetic of the second mitochondrial-derived activator of caspases (SMAC), was able to upregulate MHC-I in preclinical melanoma models, which resulted in cancer cell sensitization to CD8+ T cell-dependent cytotoxicity and enhanced immune checkpoint blockade efficacy [107]. Another study demonstrated that MHC-I was upregulated by inhibition of the deubiquitinase USP8, through activation of the NF-κB pathway. USP8 inhibition in combination with PD-1/PD-L1 blockade increased CD8+ T cell infiltration and diminished tumor growth, compared to immune checkpoint blockade monotherapy. This drug combination resulted in improved survival using various preclinical murine tumor models, including mouse colon adenocarcinoma allografts, indicating that findings could be generalized to GI malignancies [108]. Additionally, recent publications examined the role of NLRC5 and MHC-I molecules in cancer immunotherapy sensitivity [109, 110]. These reports shed light on how NLRC5 upregulation and the subsequent increase of MHC-I associated proteins, through the use of epigenetic modifiers such as DNA and histone methylation inhibitors, could reverse unresponsiveness to immunotherapy. Collectively, the findings support the potential use of PRC2 inhibitors and other epigenetic drugs as immunotherapeutic or preventive agents for various GI malignancies. As the field progresses and evolves, new avenues are anticipated for human translation and immunoepigenetic cancer interception via MHC gene upregulation [111,112,113,114]. Pertinent information from the systematic review is summarized in Table 1 and Fig. 9; survival data were obtained from The Human Protein Atlas (Figs. 2, 3, 5, 6, 7, 8) while epigenetic landscape and chromatin accessibility data were attained from the UCSC Genome Browser and the ENCODE database (Fig. 4), as indicated in each figure legend [115,116,117].

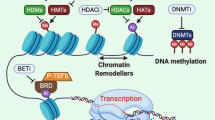
Epigenetic Regulation of MHC-I and MHC-II in gastrointestinal cancer. Epigenetic mechanisms and drugs affecting MHC-I and MHC-II expression in gastrointestinal cancer cells, including histone deacetylase (HDAC) and DNA methyltransferase (DNMT) inhibitors, as well as the possible role of histone methyltransferase (HMT) inhibitors. IFN-γ, interferon-gamma; IL-2, interleukin-2; GrB, Granzyme B; Prf, Perforin; PRC2i, PRC2 inhibitors; WDR5i, WDR5 inhibitors. Created with BioRender.com
Availability of data and materials
The datasets analyzed during the current study are publicly available online as follows: The Human Protein Atlas (https://www.proteinatlas.org/), UCSC Genome Browser (https://genome.ucsc.edu/), and ENCODE (https://www.encodeproject.org/). Datasets were accessed as indicated in the text and figures.
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- MHC:
-
Major histocompatibility complex
- RNA:
-
Ribonucleic acid
- GI:
-
Gastrointestinal
- CD8+ :
-
Cluster of differentiation 8-positive T cells
- CD4+ :
-
Cluster of differentiation 4-positive T cells
- HLA:
-
Human leukocyte antigen
- NF-κB:
-
Nuclear factor-kappa B
- IFN:
-
Interferon
- ISRE:
-
Interferon-stimulated response element
- S-X-Y:
-
Elements in MHC-I gene promoter
- RFX:
-
Regulatory factor X complex
- RFX5:
-
Regulatory factor X 5
- RFXANK:
-
Regulatory factor X-associated ankyrin-containing protein
- RFXAP:
-
Regulatory factor X-associated protein
- CREB1:
-
CAMP-responsive-element-binding protein 1
- ATF1:
-
Activating transcription factor 1
- NFY:
-
Nuclear transcription factor Y
- NLRC5:
-
NLR family CARD domain containing 5
- CARD:
-
Caspase activation and recruitment domain
- NBD:
-
Nucleotide-binding domain
- LRRs:
-
Leucine-rich repeats
- IFN-γ:
-
Interferon-gamma
- STAT1:
-
Signal transducer and activator of transcription 1
- MHC-I:
-
Major histocompatibility complex class I
- β2M:
-
Beta-2 microglobulin
- LMP2:
-
Low-molecular mass protein 2
- PSMB9:
-
Proteasome 20S subunit beta 9
- TAP1/2:
-
Transporter associated with antigen processing 1/2
- HATs:
-
Histone acetyltransferases
- H3K9me:
-
Histone H3 lysine 9 methylation
- MICA:
-
MHC-I chain-related protein A
- MICB:
-
MHC-I chain-related protein B
- NK cells:
-
Natural killer cells
- NKG2D:
-
Natural killer group 2, member D receptor
- CIITA:
-
Class II transactivator
- MHC-II:
-
Major histocompatibility complex class II
- S-X-Y:
-
Elements in MHC-II gene promoter
- CBP:
-
CREB binding protein
- PCAF:
-
P300/CBP-associated factor
- SRC-1:
-
Steroid receptor coactivator-1
- ChIP:
-
Chromatin immunoprecipitation
- HDACs:
-
Histone deacetylases
- mSin3A:
-
Mammalian switch-independent 3A
- PI-PIV:
-
CIITA promoter regions designated as PI-PIV
- CpG:
-
Cytosine-phosphate-guanine
- DNMT:
-
DNA methyltransferase
- TSA:
-
Trichostatin A
- H3K9:
-
Histone H3 lysine 9
- TAMs:
-
Tumor-associated macrophages
- TME:
-
Tumor microenvironment
- T cells:
-
T lymphocytes
- B cells:
-
B lymphocytes
- DcR3:
-
Decoy receptor 3
- TME:
-
Tumor microenvironment
- M2:
-
Alternatively activated macrophages
- APP:
-
Antigen presentation pathway
- HLA-DR:
-
Human leukocyte antigen-DR isotype
- HLA-DP:
-
Human leukocyte antigen-DP isotype
- HLA-DQ:
-
Human leukocyte antigen-DQ isotype
- HLA-DM:
-
Human leukocyte antigen-DM
- HLA-DO:
-
Human leukocyte antigen-DO
- CD74:
-
Cluster of differentiation 74
- MDMs:
-
Monocyte-derived macrophages
- VPA:
-
Valproic acid
- HLA-DRA:
-
Human leukocyte antigen-DR alpha subunit
- HLA-B:
-
Human leukocyte antigen-B isotype
- HDAC + DNMT:
-
Histone deacetylase and DNA methyltransferase
- SAHA:
-
Suberoylanilide hydroxamic acid
- IL-2:
-
Interleukin-2
- Th1/Th2:
-
T-helper 1/T-helper 2 cells
- TGF-β:
-
Transforming growth factor-beta
- IL-4:
-
Interleukin-4
- TSA:
-
Trichostatin A
- IRF-1:
-
Interferon regulatory factor 1
- H3K4:
-
Histone H3 lysine 4
- HP-1:
-
Heterochromatin protein-1
- 5-FU:
-
5-Fluorouracil
- Dep:
-
Depsipeptide
- CREB3/5:
-
CAMP-responsive element-binding protein 3 and 5
- PCAF:
-
P300/CBP-associated factor
- DNMT1:
-
DNA methyltransferase 1
- DNMT3b:
-
DNA methyltransferase 3b
- PRC2:
-
Polycomb repressive complex 2
- ESCC:
-
Esophageal squamous cell carcinoma
- LMP7:
-
Low-molecular mass protein 7
- FDA:
-
Food and drug administration
- STAT3:
-
Signal transducer and activator of transcription 3
- MS-275:
-
Entinostat
- WDR5:
-
WD repeat-containing protein 5
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
References
Zahnow CA, Topper M, Stone M, Murray-Stewart T, Li H, Baylin SB, et al. Inhibitors of DNA methylation, histone deacetylation, and histone demethylation: a perfect combination for cancer therapy. Adv Cancer Res. 2016;130:55–111.
Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol. 2012;12(12):813–20.
Wong CC, Li W, Chan B, Yu J. Epigenomic biomarkers for prognostication and diagnosis of gastrointestinal cancers. Semin Cancer Biol. 2019;55:90–105.
Løvig T, Andersen SN, Thorstensen L, Diep CB, Meling GI, Lothe RA, et al. Strong HLA-DR expression in microsatellite stable carcinomas of the large bowel is associated with good prognosis. Br J Cancer. 2002;87(7):756–62.
Cabrera CM, Jiménez P, Cabrera T, Esparza C, Ruiz-Cabello F, Garrido F. Total loss of MHC class I in colorectal tumors can be explained by two molecular pathways: beta2-microglobulin inactivation in MSI-positive tumors and LMP7/TAP2 downregulation in MSI-negative tumors. Tissue Antigens. 2003;61(3):211–9.
Reith W, Satola S, Sanchez CH, Amaldi I, Lisowska-Grospierre B, Griscelli C, et al. Congenital immunodeficiency with a regulatory defect in MHC class II gene expression lacks a specific HLA-DR promoter binding protein. RF-X Cell. 1988;53(6):897–906.
Jongsma MLM, Guarda G, Spaapen RM. The regulatory network behind MHC class I expression. Mol Immunol. 2019;113:16–21.
Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, et al. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat Genet. 1998;20(3):273–7.
Nagarajan UM, Louis-Plence P, DeSandro A, Nilsen R, Bushey A, Boss JM. RFX-B is the gene responsible for the most common cause of the bare lymphocyte syndrome, an MHC class II immunodeficiency. Immunity. 1999;10(2):153–62.
Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, et al. RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J. 1997;16(5):1045–55.
Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14(9):1156–66.
Moreno CS, Emery P, West JE, Durand B, Reith W, Mach B, et al. Purified X2 binding protein (X2BP) cooperatively binds the class II MHC X box region in the presence of purified RFX, the X box factor deficient in the bare lymphocyte syndrome. J Immunol. 1995;155(9):4313–21.
Jabrane-Ferrat N, Nekrep N, Tosi G, Esserman L, Peterlin BM. MHC class II enhanceosome: how is the class II transactivator recruited to DNA-bound activators? Int Immunol. 2003;15(4):467–75.
van den Elsen PJ. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front Immunol. 2011;2:48.
Ludigs K, Seguín-Estévez Q, Lemeille S, Ferrero I, Rota G, Chelbi S, et al. NLRC5 exclusively transactivates MHC class I and related genes through a distinctive SXY module. PLoS Genet. 2015;11(3):e1005088.
Kuenzel S, Till A, Winkler M, Häsler R, Lipinski S, Jung S, et al. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol. 2010;184(4):1990–2000.
Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hösel M, et al. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem. 2010;285(34):26223–32.
Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J Immunol. 2010;185(3):1681–91.
Staehli F, Ludigs K, Heinz LX, Seguín-Estévez Q, Ferrero I, Braun M, et al. NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. J Immunol. 2012;188(8):3820–8.
Tong Y, Cui J, Li Q, Zou J, Wang HY, Wang RF. Enhanced TLR-induced NF-κB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res. 2012;22(5):822–35.
Meissner TB, Li A, Liu YJ, Gagnon E, Kobayashi KS. The nucleotide-binding domain of NLRC5 is critical for nuclear import and transactivation activity. Biochem Biophys Res Commun. 2012;418(4):786–91.
Gobin SJ, van Zutphen M, Westerheide SD, Boss JM, van den Elsen PJ. The MHC-specific enhanceosome and its role in MHC class I and beta(2)-microglobulin gene transactivation. J Immunol. 2001;167(9):5175–84.
Robbins GR, Truax AD, Davis BK, Zhang L, Brickey WJ, Ting JP. Regulation of class I major histocompatibility complex (MHC) by nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins. J Biol Chem. 2012;287(29):24294–303.
Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, et al. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol. 2012;188(10):4951–8.
Bahram S, Bresnahan M, Geraghty DE, Spies T. A second lineage of mammalian major histocompatibility complex class I genes. Proc Natl Acad Sci USA. 1994;91(14):6259–63.
Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci USA. 1999;96(12):6879–84.
Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci USA. 1996;93(22):12445–50.
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285(5428):727–9.
Cosman D, Müllberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14(2):123–33.
Ritter C, Fan K, Paulson KG, Nghiem P, Schrama D, Becker JC. Reversal of epigenetic silencing of MHC class I chain-related protein A and B improves immune recognition of Merkel cell carcinoma. Sci Rep. 2016;6:21678.
Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109(Suppl):S21-33.
Hake SB, Masternak K, Kammerbauer C, Janzen C, Reith W, Steimle V. CIITA leucine-rich repeats control nuclear localization, in vivo recruitment to the major histocompatibility complex (MHC) class II enhanceosome, and MHC class II gene transactivation. Mol Cell Biol. 2000;20(20):7716–25.
Zika E, Ting JP. Epigenetic control of MHC-II: interplay between CIITA and histone-modifying enzymes. Curr Opin Immunol. 2005;17(1):58–64.
Spilianakis C, Papamatheakis J, Kretsovali A. Acetylation by PCAF enhances CIITA nuclear accumulation and transactivation of major histocompatibility complex class II genes. Mol Cell Biol. 2000;20(22):8489–98.
Tzortzakaki E, Spilianakis C, Zika E, Kretsovali A, Papamatheakis J. Steroid receptor coactivator 1 links the steroid and interferon gamma response pathways. Mol Endocrinol. 2003;17(12):2509–18.
Devaiah BN, Singer DS. CIITA and its dual roles in MHC gene transcription. Front Immunol. 2013;4:476.
Zika E, Greer SF, Zhu XS, Ting JP. Histone deacetylase 1/mSin3A disrupts gamma interferon-induced CIITA function and major histocompatibility complex class II enhanceosome formation. Mol Cell Biol. 2003;23(9):3091–102.
Morgan JE, Shanderson RL, Boyd NH, Cacan E, Greer SF. The class II transactivator (CIITA) is regulated by post-translational modification cross-talk between ERK1/2 phosphorylation, mono-ubiquitination and Lys63 ubiquitination. 2015. Biosci Rep. https://doi.org/10.1042/BSR20150091.
Zika E, Fauquier L, Vandel L, Ting JP. Interplay among coactivator-associated arginine methyltransferase 1, CBP, and CIITA in IFN-gamma-inducible MHC-II gene expression. Proc Natl Acad Sci USA. 2005;102(45):16321–6.
Spilianakis C, Kretsovali A, Agalioti T, Makatounakis T, Thanos D, Papamatheakis J. CIITA regulates transcription onset viaSer5-phosphorylation of RNA Pol II. EMBO J. 2003;22(19):5125–36.
Nanda NK, Birch L, Greenberg NM, Prins GS. MHC class I and class II molecules are expressed in both human and mouse prostate tumor microenvironment. Prostate. 2006;66(12):1275–84.
Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 2006;27(9):405–12.
Holtz R, Choi JC, Petroff MG, Piskurich JF, Murphy SP. Class II transactivator (CIITA) promoter methylation does not correlate with silencing of CIITA transcription in trophoblasts. Biol Reprod. 2003;69(3):915–24.
Liu JH, Bian YM, Xie Y, Lu DP. Epigenetic modification and preliminary investigation of the mechanism of the immune evasion of HL-60 cells. Mol Med Rep. 2015;12(1):1059–65.
Londhe P, Zhu B, Abraham J, Keller C, Davie J. CIITA is silenced by epigenetic mechanisms that prevent the recruitment of transactivating factors in rhabdomyosarcoma cells. Int J Cancer. 2012;131(4):E437–48.
Radosevich M, Jager M, Ono SJ. Inhibition of MHC class II gene expression in uveal melanoma cells is due to methylation of the CIITA gene or an upstream activator. Exp Mol Pathol. 2007;82(1):68–76.
Cornett EM, Ferry L, Defossez PA, Rothbart SB. Lysine methylation regulators moonlighting outside the epigenome. Mol Cell. 2019;75(6):1092–101.
Sun Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016;380(1):205–15.
Chang YC, Chen TC, Lee CT, Yang CY, Wang HW, Wang CC, et al. Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood. 2008;111(10):5054–63.
Hui L, Chen Y. Tumor microenvironment: sanctuary of the devil. Cancer Lett. 2015;368(1):7–13.
Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Mazeedi M, et al. The role of tumor microenvironment in chemoresistance: to survive, keep your enemies closer. Int J Mol Sci. 2017;18(7):1586.
Soysal SD, Tzankov A, Muenst SE. Role of the tumor microenvironment in breast cancer. Pathobiol J Immunopathol Mol Cell Biol. 2015;82(3–4):142–52.
Frankel T, Lanfranca MP, Zou W. The Role of Tumor Microenvironment in Cancer Immunotherapy. Adv Exp Med Biol. 2017;1036:51–64.
Kim M, Park C, Jung J, Yeo SG. The histone deacetylase class I, II inhibitor trichostatin A delays peripheral neurodegeneration. J Mol Histol. 2019;50(2):167–78.
Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. 2019;38(13):2223–40.
Tai SK, Chang HC, Lan KL, Lee CT, Yang CY, Chen NJ, et al. Decoy receptor 3 enhances tumor progression via induction of tumor-associated macrophages. J Immunol. 2012;188(5):2464–71.
Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20(24):6969–78.
Sun Y, Sun Y, Yue S, Wang Y, Lu F. Histone deacetylase inhibitors in cancer therapy. Curr Top Med Chem. 2018;18(28):2420–8.
Satoh A, Toyota M, Ikeda H, Morimoto Y, Akino K, Mita H, et al. Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-gamma-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene. 2004;23(55):8876–86.
Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol. 2003;33(5):1183–92.
Serrano A, Tanzarella S, Lionello I, Mendez R, Traversari C, Ruiz-Cabello F, et al. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2’-deoxycytidine treatment. Int J Cancer. 2001;94(2):243–51.
Natale F, Vivo M, Falco G, Angrisano T. Deciphering DNA methylation signatures of pancreatic cancer and pancreatitis. Clin Epigenetics. 2019;11(1):132.
Mishra NK, Guda C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget. 2017;8(17):28990–9012.
Liu B, Pilarsky C. Analysis of DNA hypermethylation in pancreatic cancer using methylation-specific PCR and bisulfite sequencing. Methods Mol Biol. 2018;1856:269–82.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52.
Cao W, Zhou G, Qiu J, Xu L, Ding X, Zhang H, et al. Research on the epigenetic modification of pancreatic cancer vaccine. Hepatogastroenterology. 2014;61(130):272–7.
Tao Y, Lin F, Li T, Xie J, Shen C, Zhu Z. Epigenetically modified pancreatic carcinoma PANC-1 cells can act as cancer vaccine to enhance antitumor immune response in mice. Oncol Res. 2013;21(6):307–16.
Maslov AY, Lee M, Gundry M, Gravina S, Strogonova N, Tazearslan C, et al. 5-aza-2’-deoxycytidine-induced genome rearrangements are mediated by DNMT1. Oncogene. 2012;31(50):5172–9.
Bubna AK. Vorinostat-an overview. Indian J Dermatol. 2015;60(4):419.
Chou SD, Khan AN, Magner WJ, Tomasi TB. Histone acetylation regulates the cell type specific CIITA promoters, MHC class II expression and antigen presentation in tumor cells. Int Immunol. 2005;17(11):1483–94.
Zenke K, Muroi M, Tanamoto KI. IRF1 supports DNA binding of STAT1 by promoting its phosphorylation. Immunol Cell Biol. 2018;96(10):1095–103.
Abou El Hassan M, Huang K, Eswara MB, Xu Z, Yu T, Aubry A, et al. Properties of STAT1 and IRF1 enhancers and the influence of SNPs. BMC Mol Biol. 2017;18(1):6.
Beresford GW, Boss JM. CIITA coordinates multiple histone acetylation modifications at the HLA-DRA promoter. Nat Immunol. 2001;2(7):652–7.
Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301(5634):798–802.
Richards EJ, Elgin SC. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002;108(4):489–500.
Greer SF, Zika E, Conti B, Zhu XS, Ting JP. Enhancement of CIITA transcriptional function by ubiquitin. Nat Immunol. 2003;4(11):1074–82.
Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18(7):1414.
Okada K, Hakata S, Terashima J, Gamou T, Habano W, Ozawa S. Combination of the histone deacetylase inhibitor depsipeptide and 5-fluorouracil upregulates major histocompatibility complex class II and p21 genes and activates caspase-3/7 in human colon cancer HCT-116 cells. Oncol Rep. 2016;36(4):1875–85.
Moreno CS, Beresford GW, Louis-Plence P, Morris AC, Boss JM. CREB regulates MHC class II expression in a CIITA-dependent manner. Immunity. 1999;10(2):143–51.
Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416(6880):552–6.
Chávez-Blanco A, De la Cruz-Hernández E, Domínguez GI, Rodríguez-Cortez O, Alatorre B, Pérez-Cárdenas E, et al. Upregulation of NKG2D ligands and enhanced natural killer cell cytotoxicity by hydralazine and valproate. Int J Oncol. 2011;39(6):1491–9.
Chen YS, Li J, Neja S, Kapoor S, Tovar Perez JE, Tripathi C, et al. Metabolomics of acute vs. chronic spinach intake in an apc-mutant genetic background: linoleate and butanoate metabolites targeting HDAC activity and IFN-γ signaling. Cells. 2022;11(3):573.
Kailasam A, Mittal SK, Agrawal DK. Epigenetics in the pathogenesis of esophageal adenocarcinoma. Clin Transl Sci. 2015;8(4):394–402.
Hu JM, Li L, Chen YZ, Liu C, Cui X, Yin L, et al. HLA-DRB1 and HLA-DQB1 methylation changes promote the occurrence and progression of Kazakh ESCC. Epigenetics. 2014;9(10):1366–73.
Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. 2021;12:636568.
Sheyhidin I, Hasim A, Zheng F, Ma H. Epigenetic changes within the promoter regions of antigen processing machinery family genes in Kazakh primary esophageal squamous cell carcinoma. Asian Pacific J Cancer Prevent APJCP. 2014;15(23):10299–306.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27.
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109(1):31–9.
Sun T, Li Y, Yang W, Wu H, Li X, Huang Y, et al. Histone deacetylase inhibition up-regulates MHC class I to facilitate cytotoxic T lymphocyte-mediated tumor cell killing in glioma cells. J Cancer. 2019;10(23):5638–45.
Yang H, Lan P, Hou Z, Guan Y, Zhang J, Xu W, et al. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br J Cancer. 2015;112(1):112–21.
Xiao W, Dong W, Zhang C, Saren G, Geng P, Zhao H, et al. Effects of the epigenetic drug MS-275 on the release and function of exosome-related immune molecules in hepatocellular carcinoma cells. Eur J Med Res. 2013;18(1):61.
Zhang Y, Wu Q, Xu L, Wang H, Liu X, Li S, et al. Sensitive detection of colorectal cancer in peripheral blood by a novel methylation assay. Clin Epigenetics. 2021;13(1):90.
Han YD, Oh TJ, Chung TH, Jang HW, Kim YN, An S, et al. Early detection of colorectal cancer based on presence of methylated syndecan-2 (SDC2) in stool DNA. Clin Epigenetics. 2019;11(1):51.
Araghi M, Soerjomataram I, Jenkins M, Brierley J, Morris E, Bray F, et al. Global trends in colorectal cancer mortality: projections to the year 2035. Int J Cancer. 2019;144(12):2992–3000.
Morgan E, Arnold M, Camargo MC, Gini A, Kunzmann AT, Matsuda T, et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020–40: a population-based modelling study. EClinicalMedicine. 2022;47:101404.
Morgan E, Soerjomataram I, Rumgay H, Coleman HG, Thrift AP, Vignat J, et al. The global landscape of esophageal squamous cell carcinoma and esophageal adenocarcinoma incidence and mortality in 2020 and projections to 2040: new estimates from GLOBOCAN 2020. Gastroenterology. 2022;163(3):649-58.e2.
Ge T, Gu X, Jia R, Ge S, Chai P, Zhuang A, et al. Crosstalk between metabolic reprogramming and epigenetics in cancer: updates on mechanisms and therapeutic opportunities. Cancer Commun. 2022;42(11):1049–82.
Kapoor S, Gustafson T, Zhang M, Chen YS, Li J, Nguyen N, et al. Deacetylase plus bromodomain inhibition downregulates ERCC2 and suppresses the growth of metastatic colon cancer cells. Cancers. 2021;13(6):1438.
Rajendran P, Johnson G, Li L, Chen YS, Dashwood M, Nguyen N, et al. Acetylation of CCAR2 Establishes a BET/BRD9 acetyl switch in response to combined deacetylase and bromodomain inhibition. Cancer Res. 2019;79(5):918–27.
Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sánchez-Rivera FJ, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21(10):1163–71.
Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An evolutionarily conserved function of polycomb silences the MHC Class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell. 2019;36(4):385-401.e8.
Monterroza L, Parrilla MM, Samaranayake SG, Rivera-Rodriguez DE, Yoon SB, Bommireddy R, et al. Tumor-intrinsic enhancer of zeste homolog 2 controls immune cell infiltration, tumor growth, and lung metastasis in a triple-negative breast cancer model. Int J Mol Sci. 2024;25(10):5392.
Straining R, Eighmy W. Tazemetostat: EZH2 Inhibitor. J Adv Pract Oncol. 2022;13(2):158–63.
Chu L, Qu Y, An Y, Hou L, Li J, Li W, et al. Induction of senescence-associated secretory phenotype underlies the therapeutic efficacy of PRC2 inhibition in cancer. Cell Death Dis. 2022;13(2):155.
Barghout SH, Machado RAC, Barsyte-Lovejoy D. Chemical biology and pharmacology of histone lysine methylation inhibitors. Biochim Biophys Acta Gene Regul Mech. 2022;1865(6):194840.
Zha L, Cao Q, Cui X, Li F, Liang H, Xue B, et al. Epigenetic regulation of E-cadherin expression by the histone demethylase UTX in colon cancer cells. Med Oncol. 2016;33(3):21.
Gu SS, Zhang W, Wang X, Jiang P, Traugh N, Li Z, et al. Therapeutically Increasing MHC-I expression potentiates immune checkpoint blockade. Cancer Discov. 2021;11(6):1524–41.
Xiong W, Gao X, Zhang T, Jiang B, Hu MM, Bu X, et al. USP8 inhibition reshapes an inflamed tumor microenvironment that potentiates the immunotherapy. Nat Commun. 2022;13(1):1700.
Shukla A, Cloutier M, Appiya Santharam M, Ramanathan S, Ilangumaran S. The MHC Class-I transactivator NLRC5: implications to cancer immunology and potential applications to cancer immunotherapy. Int J Mol Sci. 2021;22(4):1964.
Li L, Song Q, Zhou J, Ji Q. Controllers of histone methylation-modifying enzymes in gastrointestinal cancers. Biomed Pharmacother. 2024;174:116488.
Dashwood RH, Ho E. Dietary histone deacetylase inhibitors: from cells to mice to man. Semin Cancer Biol. 2007;17(5):363–9.
Kawazu M, Ueno T, Saeki K, Sax N, Togashi Y, Kanaseki T, et al. HLA Class I analysis provides insight into the genetic and epigenetic background of immune evasion in colorectal cancer with high microsatellite instability. Gastroenterology. 2022;162(3):799–812.
Anderson P, Aptsiauri N, Ruiz-Cabello F, Garrido F. HLA class I loss in colorectal cancer: implications for immune escape and immunotherapy. Cell Mol Immunol. 2021;18(3):556–65.
West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology. 2014;3(1):e27414.
The Human Protein Atlas. https://www.proteinatlas.org/. Accessed 10 October 2023.
UCSC Genome Browser. https://genome.ucsc.edu/. Accessed 10 October 2023.
ENCODE. https://www.encodeproject.org/. Accessed 10 October 2023.
Acknowledgements
We thank members of the Dashwood and Rajendran laboratories for valuable discussion of topics related to the review article, including Wan Mohaiza Dashwood, Nivedhitha Mohan, Sultan Abda Neja, and Ahmed Muhsin.
Funding
Research reviewed herein was supported in part by NIH grants CA122959 and CA257559, by the John S. Dunn Foundation, and by a Chancellor’s Research Initiative from Texas A&M University.
Author information
Authors and Affiliations
Contributions
Manuscript design and revisions were performed by JETP, PR and RD. Manuscript composition and figure creation were undertaken by JETP, SZ, WH, and RD. SK and KK enhanced the manuscript design and provided intellectual insights. The final manuscript was reviewed and endorsed by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tovar Perez, J.E., Zhang, S., Hodgeman, W. et al. Epigenetic regulation of major histocompatibility complexes in gastrointestinal malignancies and the potential for clinical interception. Clin Epigenet 16, 83 (2024). https://doi.org/10.1186/s13148-024-01698-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01698-8