
Abstract
Background
E-cadherin, a major actor of cell adhesion in the intestinal barrier, is encoded by the CDH1 gene associated with susceptibility to Crohn Disease (CD) and colorectal cancer. Since epigenetic mechanisms are suspected to contribute to the multifactorial pathogenesis of CD, we studied CpG methylation at the CDH1 locus. The methylation of the CpG island (CGI) and of the 1st enhancer, two critical regulatory positions, was quantified in surgical specimens of inflamed ileal mucosa and in peripheral blood mononuclear cells (PBMC) of 21 CD patients. Sixteen patients operated on for a non-inflammatory bowel disease, although not normal controls, provided a macroscopically normal ileal mucosa and PBMC for comparison.
Results
In ileal mucosa, 19/21 (90%) CD patients vs 8/16 control patients (50%) (p < 0.01) had a methylated CDH1 promoter CGI. In PBMC, CD patients with methylated CGI were 11/21 (52%) vs 7/16 controls (44%), respectively. Methylation in the 1st enhancer of CDH1 was also higher in the CD group for each of the studied CpGs and for their average value (45 ± 17% in CD patients vs 36 ± 17% in controls; p < 0.001). Again, methylation was comparable in PBMC. Methylation of CGI and 1st enhancer were not correlated in mucosa or PBMC.
Conclusions
Methylation of several CpGs at the CDH1 locus was increased in the inflamed ileal mucosa, not in the PBMC, of CD patients, suggesting the association of CDH1 methylation with ileal inflammation. Longitudinal studies will explore if this increased methylation is a risk marker for colorectal cancer.
Similar content being viewed by others

Introduction
While its exact pathogenesis remains unknown, CD is considered to result from a dysregulated immune response to altered microbiota or other various environmental factors in genetically predisposed patients [1]. Since CD incidence has increased in newly industrialized countries [2], the westernized environment, including diet [3,4,5] and changes in microbiota [6], is suspected. While environmental factors still remain hypothetical, as in most multifactorial gene-environment diseases, our knowledge of genetic susceptibility has greatly increased [7]. Indeed, genome-wide association studies (GWAS) have identified as many as 71 CD susceptibility loci [8, 9]. However, most loci contribute to CD risk with low odds ratios (< 1.15), suggesting a limited genetic contribution to CD causality [10] already demonstrated by the high rates of discordance in monozygotic twins [11].
A growing body of evidence supports the role of epigenetic mechanisms in multifactorial diseases [12] due to the dynamic interaction of epigenetics with environment [13]. A widely studied epigenetic mechanisms is CpG methylation, a mitotically heritable process able to modulate the transcription of many genes [14, 15] in blood cells and solid human tissues [16]. The earliest CpG methylation marks are programmed in two waves during embryogenesis [17, 18], then, during later fetal and postnatal life, environmental cues may modify CpG methylation marks at certain loci of our genome [19,20,21,22]. On the other side, genetic variation in cis or trans plays a major role in the establishment of methylation marks, either directly or in interaction with environmental exposures [23]. CpG methylation can therefore mediate the effects of gene–environment interactions on gene expression [24]. Indeed, the level of methylation of CpGs in promoter or enhancer regions can modulate the transcription of certain genes [15], thus can play a causal role in health and diseases [25], including inflammatory bowel disease (IBD) [26, 27]. It is important to stress that, while variation in methylation marks can contribute to disease causation it can as well be a consequence of disease or disease related changes in lifestyle or treatments.
Several studies have reported differentially methylated CpG sites in the blood cells of patients with CD ileitis [28,29,30,31,32,33,34,35]. In contrast, only three studies have examined CpG methylation in intestinal tissues of adult patients with long standing CD [36]. One of them studied samples of diseased ileal mucosa collected during surgery in 5 patients with CD [37], another studied rectal biopsies of 16 CD patients (8 with and 8 without rectal inflammation) [38], the third one studied diseased ileal mucosa from 7 CD patients with perforation or fistula [39]. In addition to these studies, distinctive CpG methylation marks associated with CD were also found in purified epithelial cells from mucosal biopsies collected from terminal ileum or ascending or sigmoid colons of 43 children newly diagnosed with CD [40]. None of the four studies cited above found that the CDH1 locus was a differentially methylated region (DMR) in CD patients.
Several reasons, however, prompted us to study methylation specifically at the CDH1 locus in the intestinal mucosa of CD patients. First, the CDH1 gene encodes E-cadherin, a transmembrane glycoprotein expressed in epithelial cells and a major actor in cell adhesion, intestinal barrier, and dynamic balance of epithelial tissues [41]. E-cadherin contributes to epithelial-to-mesenchymal transition (EMT) a phenomenon that allows the conversion of adherent epithelial cells to a mesenchymal cell phenotype, which enhances migratory capacity and invasiveness [42], physiological functions that are at the forefront of the pathogenic phenomena leading to CD. In addition, fine-mapping studies of the 16q22.1 region found that rs16260, a single-nucleotide variant located in the CDH1 promoter, is associated with ileal CD [9]. Functionality of this SNP has been demonstrated, suggesting that it could itself modulate E-cadherin expression in vivo [43,44,45]. Another reason for exploring methylation at the CDH1 locus is that methylation changes at this locus have already been reported in ulcerative colitis (UC), the other major IBD [38, 46,47,48] and were said to contribute to the prediction of UC severity [42, 49]. If the methylation marks observed at the CDH1 locus are triggered by intestinal UC-associated inflammation, comparable marks might also occur in response to CD-associated inflammation. In addition, CDH1 is a tumor-suppressor gene involved in the predisposition to several cancers, notably gastric and colorectal cancer. Predisposition to colorectal cancer (CRC) occurs through both genetic predisposition [50, 51] and changes in CpG methylation [52, 53]. Since patients with CD are at increased risk of CRC diagnosis and death [54], we had an additional reason to focus on the CDH1 locus.
The current study explored CpG methylation at the CDH1 locus in patients with ileal CD. To find epigenetic signatures that might reflect local disease mechanisms, we measured CDH1 methylation in inflamed mucosa. We chose a candidate gene approach focused on the CDH1 locus instead of attempting agnostic epigenome-wide association studies (EWAS) [55, 56]. The reason was that we could not collect ileal samples in large enough numbers of patients with CD or non-IBD gut pathology to feed an EWAS, which requires analyzing several hundreds of patients and controls and is for this reason always carried out in blood cells [56, 57], rather than in the diseased tissues.
Materials and methods
Study population
From September 2021 to September 2022, twenty-one consecutive patients who underwent ileal or ileocolic resection in the Bicêtre Department of Digestive Surgery were prospectively included into the studied CD group. The diagnosis of CD was based on biopsies of lesions located on the colon or ileum performed during colonoscopy and enteroscopy. Crohn disease activity index (CDAI) was > 150 in all patients. Average duration of CD since diagnosis was 11.7 years (1–34 years). Imaging and endoscopic examination confirmed the presence of fistula for 12/21 and/or stricture for 14/21, leading to the surgical indication. At the time of surgery, 12/21 CD patients were treated with anti-TNFα drugs and 6/21 were receiving Azathioprine.
Sixteen patients who had non-inflammatory intestinal disease underwent ileal or ileocolic resection; they were used as a “control” group for comparison with CD patients, also they cannot be considered healthy control. This group was composed of: 4 stoma closures, 6 right sporadic colonic adenocarcinomas, 3 colonic polyps with low-grade dysplasia, 1 ileal non-inflamed stricture of undetermined etiology and 2 right colonic diverticulosis without diverticulitis. All of them had an erythrocyte sedimentation rate < 10 mm/h, normal hemoglobin and platelets, and normal C reactive protein.
The research protocol in the Department of Surgery was agreed by Paris Sud University Institutional Review Board. During the pre-operative consultation, patients signed informed consent for the current study and for genetic analysis, according to the French rules of bioethics.
Table 1 presents the main characteristics of the studied patients collected from the electronic medical record system of Bicêtre Hospital. Additional file 1: Table S1 describes the detailed results of the methylation levels for each sample analysed.
Procedural methods
Venous blood samples were collected and peripheral blood mononuclear cells (PBMC) were immediately purified from fresh blood (10 mL) [58]. All CD patients had inflammatory lesions complicated by fistula (associated with intra-abdominal sepsis) and/or stricture (associated with small bowel obstruction). A surgical specimen was taken under sterile conditions in the operating room and stored for further analysis. A 1 × 1 cm sample of superficial ileal mucosa without muscularis was latter isolated from a macroscopically inflamed area and placed in a dry cryotube. The sample was frozen at − 80 °C in liquid nitrogen. For the control patients, a macroscopically healthy non-inflamed ileal specimen was taken at least 10 cm away from the surgical lesion and processed similarly.
Choice of CpGs at CDH1 locus
We studied the CpG island (CGI) made of 104 CpGs located in CDH1 promoter, exon 1, intron 1 and exon 2. We also selected 4 CpGs for study, named according to their position towards the transcription start site (TSS) of CDH1 and located near 4724 bp upstream the TSS, recently identified as the 1st enhancer [59]. The FANTOM5 consortium has identified several enhancers in the CDH1 gene using the CAGE (short for Cap Analysis of Gene Expression) technology where a bidirectional transcription CAGE pattern was detected. Particularly, the 1st active enhancer of CDH1 chr16: 68732016-68732406 (hg38 version), was detected to interact with the promoter regions (also a CGI region) of CDH1 https://pressto.binf.ku.dk/about.php#h_enhancers_description_long. In addition, Activity By Contact (ABC) enhancer study [60] further identified that most of them are “pleiotropic” enhancers where enhancers were detected to be contacted with multiple genes in different cell-line or tissues [61]. Additional file 1: Fig. S1 summarizes the genomic map of the CDH1 locus with the CpG sites of interest (CGI and 4 CpGs of the 1st enhancer) and the SNP rs16260.
DNA isolation and bisulfite conversion
Total DNA was extracted from samples and purified on spin-column with the "DNeasy Blood & Tissue kit" (Qiagen, Hilden, Germany), then stored at − 20 °C in 1.5 mL tubes. The concentrations of extracted DNA were assayed with NanoDrop spectroscopy (Nyxor Biotech, Paris, France). An optimal degree of purity and quality of the extracted DNA was assessed by 260 nm/230 nm and 260 nm/280 nm ratios. 400 ng of DNA were converted to sodium bisulfite using the EZ DNA Methylation-Gold Kit (Zymo Research Corporation, CA, USA).
Methylation specific PCR-based bisulfite analysis
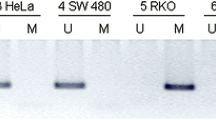
Methylation-specific PCR (MSP) requires only small quantities of DNA, is sensitive to 0.1% methylated alleles of a given CGI. Analysis of CDH1 CGI was performed by MSP as described [62].
To amplify methylated CGI, primers were: The PCR was performed using the Taq'Ozyme HS (Ozyme, France) with a melting temperature of 52 °C. Briefly, bisulfite converted DNA were amplified using Taq'Ozyme HS polymerase (Ozyme, France) with specific primers for unmethylated or methylated DNA [respectively 5′-TTAGGTTAGAGGGTTATCGCGT-3′ (forward) and 5′-TAACTAAAAATTCACCTAC CGAC-3′ (reverse) and 5′-TAATTTTAGGTTAGAGGGTTATTGT-3′ (forward) and 5′-CACAACCA ATCAACAACACA-3′ (reverse)]. PCR mix contained 5X Buffer, 0.6 µM of each primer, 3% DMSO and 1 unit Taq DNA polymerase. Amplifications were performed in X cycles with an annealing temperature of 52 °C (methylated CGI primers) or 53 °C (unmethylated CGI primers). PCR products were assessed by electrophoretic migration on the QIAxcel Advanced automate (Qiagen, Hilden, Germany).
Pyrosequencing-based bisulfite PCR analysis
First, a PCR amplification of the genomic sequence containing the 4 CpGs of interest was performed using unbiased primers: 5′-TTGTTATAAGGAAATTTGGAG-3′ (forward) and 5′-CCTAAAACTATACACAAACCTATC-3′ (reverse with a biotin linked in 5′ position). The PCR was performed using the Epimark Hot Start Taq DNA polymerase (New England BioLabs) with a melting temperature of 52 °C and reagents in the following proportions to give a total volume per sample of 50μL (Buffer 5X, 0.2 mM DNTP, 0.6 µM each primer, 1.25 mM MgCl2, DMSO 3% and 1.25 units / 50 µl PCR mix of Epimark polymerase).
Biotin-labeled single stranded amplicon was isolated according to protocol using the PyroMark Q96 ID Pyrosequencing instrument (Qiagen, Hilden, Germany) and underwent pyrosequencing with 0.5 μM primer. The primers used were the following according to CpG position: 5′-TGGAGTTTGTGATTTTATTA-3′, 5′-GATAGGGTTTTTTATTTAT-3′, 5′-GATGTTTGAA ATTTTATTGT-3′ and 5′-GTAATGGGTTTTATTATTT-3′. Primers (including for PCR) were generated using MethPrimer (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi) [63]. The methylation percentage for each CpG was calculated using PyroQ CpG Software (Qiagen, Hilden, Germany).
Genotyping of SNP rs16260
The rs16260 (C > A) SNP is adjacent to the CpG Island of CDH1. The rs16260 SNP was genotyped by TaqMan® pre-designed SNP genotyping assay technology (LifeTech, Assay ID C_11934298_10, ThermoFisher Scientific) with TaqPath™ ProAmp Master Mix under conditions recommended by the manufacturer. PCR cycling conditions were 60 °C for 30 s, 95 °C for 5 min, 40 cycles at 95 °C for 15 s and 60 °C for 45 s. Results were generated by LightCycler® Software (Detection format: dual color hydrolysis/UPL Probe and Analysis by Endpoint Genotyping).
Statistical analysis
Continuous data were reported as mean ± SD according to the normality of the distribution, checked graphically on a histogram and by the Shapiro–Wilk test at the 20% threshold. Intergroup comparisons of continuous variables were performed using Student t-test or the Mann–Whitney U test, depending on the distribution of the variables. Intergroup comparisons of classified variables were performed using the chi-square test or Fisher’s exact test depending on sample size. Classified variables were reported in absolute terms and percentages. The correlation between methylation of the studied enhancer CpGs was both analyzed by Pearson correlation analysis. Multivariate regressions were performed to assess the association between methylation level and Crohn disease according to the main demographic variables (sex, age, BMI). P-values < 0.05 were considered statistically different. All P-values were two-sided. The analyses were performed using R 4.1.2 for MacOS.
Results
Methylation of CDH1 CpG island (CGI) in mucosa and PBMC.
In the ileal mucosa, 19/21 (90%) of the CD patients had a methylated CGI of the CDH1 promoter compared to 8/16 (50%) of the control patients (p < 0.01). In PBMC, 11/21 (52%) of the CD patients and 7/16 (44%) of the controls had a methylated CGI of CDH1. CGI methylation was more frequent in mucosa than in PBMC of CD patients (90% vs 52% in controls, p < 0.01), while no difference was observed between mucosa and PBMC in controls (50% vs 44% in controls, p = 0.7). Only 11/21 CD patients had a methylated CGI in both tissues, while 7/8 of the controls showed a methylated CGI in both mucosa and PBMC.
In the CD group, we found no association of CGI methylation in ileal mucosa or PBMC with age, sex, BMI, smoker status, treatment with anti-TNFα or azathioprine, or CDAI score. CDH1 CGI methylation was not associated with the duration of CD. The presence of fistula or stricture was not associated with CGI methylation. In the controls, no association was found between age, sex, BMI, or smoker status and CDH1 CGI methylation in the ileal mucosa. Three out of 6 patients with colorectal cancer had a methylated CGI in the ileal mucosa, a proportion similar to that of the entire control group.
Methylation of CDH1 enhancer CpGs in mucosa and PBMC
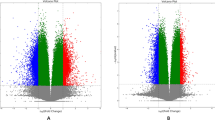
Figure 1 describes the methylation of the 4 CpGs located in the CDH1 enhancer sequence in control and CD patients in the ileal mucosa and in circulating PBMC. Detailed results are given in Table 2. In the ileal mucosa, the degree of methylation of the 4 CpGs was significantly higher in the CD group, for each individual CpG and for the average of the 4 CpGs. A ROC curve assessing the performance of average methylation of the 4 CpGs in predicting CD yielded an AUC value of 0.85 in mucosa samples (Additional file 1: Fig S2). In contrast, CDH1 enhancer methylation in PBMC was not different in CD and in controls. In patients with CD, the level of methylation of the 4 CpGs in mucosa was higher (45 ± 17%) than in PBMC (37 ± 9%, p = 0.002). In controls, no difference was observed between mucosa and PBMC.

Methylation of the 4 CpGs in the 1st enhancer of CDH1 gene A in mucosa; and B in PBMC B patients with CD or non-inflammatory disease controls. * p 0.03; ** p < 0.02; ***p 0.01
There was a significant inter-correlation of methylation across enhancer CpGs in ileal mucosa (Additional file 1: Fig S3) or in PBMC (Additional file 1: Fig S4). The methylation of the 4 CpGs in ileal mucosa was not correlated with their methylation in PBMC (r = 0.3, p = NS). No association was found between methylation of the CGI and enhancer CpGs as shown in Additional file 1: Fig S5.
No significant association was demonstrated between methylation of enhancer CpGs and age in the control and/or the CD groups, nor with sex, BMI, or smoker status. In the CD group, no association was found with duration of clinical disease, CDAI score, medical treatment or the presence of fistula or stricture.
Lack of association of rs16260 SNP with methylation
Additional file 1: Table S2 shows rs16260 (C > A) genotypes (no patient had an AA genotype). The rs16260 SNP was not associated with CD or with methylation of CDH1 CGI or enhancer CpGs.
Discussion
The main finding of the current study is that both the CDH1 promoter CGI and a group of CpGs located within the 1st enhancer of CDH1 showed increased methylation in the inflamed ileal mucosa of CD patients. Notably CDH1 CGI was found to be methylated in the ileal mucosa of 90% of CD patients, versus only 50% of our « control» group. Also, the increased average methylation of the 4 CpGs in CDH1 enhancer was relatively large (45 ± 17% in CD patients vs 35 ± 17% in controls). The intestinal epithelium is constantly renewed by intestinal stem cells throughout life. It is interesting to note that the CDH1 locus was also hypermethylated in the inflamed mucosa of patients with UC [38, 46,47,48] and that this methylation of the CDH1 locus could contribute to predicting the severity of UC [33, 42, 43]. This observation supports that intestinal inflammation or its local consequences could increase methylation at the CDH1 locus in both CD and UC. While the intestinal epithelium responds to changes in diet, age, microbiome, and immune activation, it is unknown whether these responses occur in mature or progenitor cells and whether they involve epigenetic reprogramming. Indeed, epigenetic mechanisms have recently been recognized as operating at the interface between the microbiome and the intestinal epithelial cell genome [64,65,66].
In PBMC, percentages of methylated CDH1 CGI in our control group matched those observed in a study of 1036 healthy controls [67]. Consistent with previous EWAS performed in blood cells [28,29,30,31,32,33,34,35], we found no difference of CGI methylation between CD and control patients. Methylation levels of CDH1 CGI and the 4 enhancer CpGs in PBMC correlated with those found in intestinal mucosa in our controls, as reported in normal people [68]. This correlation suggests that progenitors of PBMC and intestinal epithelium may share a common early programming of CDH1 methylation in the endoderm germ layer, with derived cell types retaining these patterns decades later as a stable lineage mark [69]. In contrast, the absence of correlation between intestinal and PBMC methylation in CD patients supports that increased methylation has more recently occurred in progenitors or mature mucosal cells of CD patients. As discussed by Heijmans et al. [70], high inter-tissue concordance may be present for DNA methylation changes induced early in development (and potentially propagated soma-wide). In contrast, changes occurring during aging are more likely to remain tissue-specific.
Mucosal tissue from surgical specimens is composed of heterogeneous cell populations that are different in non-inflamed « control» mucosa and inflamed ileal areas of CD. There is a clear concern that our analyses, as those of others [38, 39], were conducted using whole mucosal samples containing mixed epithelial and non-epithelial cell populations, known to have different methylation [38]. Given that methylation signatures are cell-type-specific, the question arises as to whether the epigenetic patterns we observed in CD mucosa arise from the epithelial or non-epithelial cells, and whether they might be confounded by the different cell populations present in inflamed CD versus control mucosa [43]. Among non-epithelial cells, intra-epithelial lymphocytes show lineages diversity and functional states in the intestinal mucosa under both healthy and CD conditions, as well as altered spatial distribution that potentially correlates with transmural inflammation [71]. Since we did not perform mucosal cell purification [71], CDH1 methylation could not be analyzed in infiltrated intra-mucosal lymphocytes. The only indirect information regarding mucosal lymphocytes in our CD patients comes from circulating PBMC, in which CDH1 methylation was comparable to non-inflamed control mucosa or control PBMC. Since enterocyte number largely exceeds non-epithelial cells in inflamed ileal lesions, it is unlikely that the increased methylation levels observed in inflamed CD mucosa could be explained by infiltrated lymphocytes. Indeed, if local inflammation were able to increase CGI methylation in resident lymphocytes or non-epithelial cells, this would not result in the 90% proportion of methylated CGI that we observed in our mucosal samples.
Our findings suggest that mucosal cells of UC and CD may share epigenetic mechanisms at the CDH1 locus despite heterogeneity in location, severity of inflammation and phenotypes [72].
The current observation may also be relevant to the risk of colorectal cancer (CRC) in CD patients. Numerous genes, notably CDH1 have been reported to be hypermethylated and silenced in sporadic CRC [73, 74]. In addition, hypermethylation of CDH1 CGI characterizes UC-associated CRC [53] and was supposed to serve as a useful biomarker for detecting UC patients at high risk of developing CRC [75]. Since CD increases the risk of CRC [54], the question arises whether the increased methylation that we found in CD at the CDH1 locus could help predict disease course and associated CRC.
Clearly, methylation marks at the CDH1 locus were not influenced by genomic sequence variation in cis and thus contribute to the epigenetic signature of the inflamed mucosa independently from genetics.
Many questions remain, since the current study is only observational and exploratory and carries several weaknesses. Although the ileal mucosa studied in the control group was macroscopically normal and taken out of pathological lesions, one cannot consider our control population as truly normal, i.e., non-pathological. Also, our study does not provide extensive information about all CpG residues at the CDH1 locus. Also, it does not provide information about non-inflamed CD mucosa, as obtained from intestinal biopsies [38] in a study of CD that did not provide a comparison between non-penetrating and normal mucosa [38]. Last but not least, a major weakness of our work is the lack of information about CDH1 expression in the ileal samples. This was due to the fact that most surgical specimens were collected and stored in conditions that would not have allowed a reliable measurement of CDH1 mRNA.
More globally, our observation does not elucidate whether increased methylation contributes to the causality of inflammatory lesions of CD, or is instead a secondary consequence of the local inflammation induced by CD. The first hypothesis is that preexisting increased methylation predisposes some regions of the ileal mucosa of future CD patients to inflammation and possibly CRC, as it is postulated for UC. Following this hypothesis, methylation marks preexisting in the intestinal mucosa would result from epigenetic programming and environmental exposures occurring during the pre-disease life of patients. The increased methylation observed here in mucosa was not present in PBMC, and is thus posterior to the embryonic differentiation of patients’ endodermal (gut) and mesodermal (PBMC) cells. Another possibility is that the methylation changes that we observed in CD mucosa occur during post-disease life, and are induced by the local inflammation or other environmental exposures of intestinal cells, such as changes in microbiome [64,65,66]. In fact, the two hypotheses are not mutually exclusive. They could combine their effects to increase both the preexisting and secondary methylation at the CDH1 regulatory locus, which might perpetuate local inflammation and trigger CRC risk in the intestinal epithelium of CD patients.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Change history
18 March 2024
A Correction to this paper has been published: https://doi.org/10.1186/s13148-024-01654-6
References
De Souza HSP, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13(1):13–27.
Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–78.
Lee D, Albenberg L, Compher C, Baldassano R, Piccoli D, Lewis JD, et al. Diet in the pathogenesis and treatment of inflammatory bowel diseases. Gastroenterology. 2015;148(6):1087–106.
Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M. Role of “Western Diet” in inflammatory autoimmune diseases. Curr Allergy Asthma Rep. 2014;14(1):404.
Lo CH, Lochhead P, Khalili H, Song M, Tabung FK, Burke KE, et al. Dietary inflammatory potential and risk of Crohn’s disease and ulcerative colitis. Gastroenterology. 2020;159(3):873-883.e1.
Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. 2012;9(10):599–608.
Roda G, Chien Ng S, Kotze PG, Argollo M, Panaccione R, Spinelli A, et al. Crohn’s disease. Nat Rev Dis Primers. 2020;6(1):22.
Franke A, McGovern DPB, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118–25.
Elding H, Lau W, Swallow DM, Maniatis N. Dissecting the genetics of complex inheritance: linkage disequilibrium mapping provides insight into Crohn disease. Am J Hum Genet. 2011;89(6):798–805.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–53.
Gordon H, Trier Moller F, Andersen V, Harbord M. Heritability in inflammatory bowel disease: from the first twin study to genome-wide association studies. Inflamm Bowel Dis. 2015;21:1428–34.
Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456(7218):18–21.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13(2):97–109.
Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21.
Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–22.
Oliva M, Demanelis K, Lu Y, Chernoff M, Jasmine F, Ahsan H, et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat Genet. 2023;55(1):112–22.
Horsthemke B. A critical view on transgenerational epigenetic inheritance in humans. Nat Commun. 2018;9(1):2973.
Slieker RC, Roost MS, Van Iperen L, Suchiman HED, Tobi EW, Carlotti F, et al. DNA methylation landscapes of human fetal development. Reik W, éditeur. PLOS Genet. 2015;11(10):e1005583.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(S3):245–54.
Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LTY, Kohlbacher O, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500(7463):477–81.
Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG Island context. PLoS Genet. 2009;5(8):e1000602.
Martin EM, Fry RC. Environmental influences on the epigenome: exposure-associated DNA methylation in human populations. Annu Rev Public Health. 2018;39(1):309–33.
Villicaña S, Bell JT. Genetic impacts on DNA methylation: research findings and future perspectives. Genome Biol. 2021;22(1):127.
Schübeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321–6.
Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018;392(10149):777–86.
Ventham NT, Kennedy NA, Nimmo ER, Satsangi J. Beyond Gene discovery in inflammatory bowel disease: the emerging role of epigenetics. Gastroenterology. 2013;145(2):293–308.
Hornschuh M, Wirthgen E, Wolfien M, Singh KP, Wolkenhauer O, Däbritz J. The role of epigenetic modifications for the pathogenesis of Crohn’s disease. Clin Epigenetics. 2021;13(1):108.
Lin Z, Hegarty JP, Yu W, Cappel JA, Chen X, Faber PW, et al. Identification of disease-associated DNA methylation in B cells from Crohn’s disease and ulcerative colitis patients. Dig Dis Sci. 2012;57(12):3145–53.
Harris AR, Nagy-Szakal D, Pedersen N, Opekun A, Bronsky J, Munkholm P, et al. Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18(12):2334–41.
Nimmo ER, Prendergast JG, Aldhous MC, Kennedy NA, Henderson P, Drummond HE, et al. Genome-wide methylation profiling in Crohnʼs disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis. 2012;18(5):889–99.
Adams AT, Kennedy NA, Hansen R, Ventham NT, O’Leary KR, Drummond HE, et al. Two-stage genome-wide methylation profiling in childhood-onset Crohnʼs disease implicates epigenetic alterations at the VMP1/MIR21 and HLA loci. Inflamm Bowel Dis. 2014;20(10):1784–93.
Moret-Tatay I, Cerrillo E, Sáez-González E, Hervás D, Iborra M, Sandoval J, et al. Identification of epigenetic methylation signatures with clinical value in Crohnʼs disease. Clin Transl Gastroenterol. 2019;10(10): e00083.
Somineni HK, Venkateswaran S, Kilaru V, Marigorta UM, Mo A, Okou DT, et al. Blood-derived DNA methylation signatures of Crohn’s disease and severity of intestinal inflammation. Gastroenterology. 2019;156(8):2254-2265.e3.
Gasparetto M, Payne F, Nayak K, Kraiczy J, Glemas C, Philip-McKenzie Y, et al. Transcription and DNA methylation patterns of blood-derived CD8+ T cells are associated with age and inflammatory bowel disease but do not predict prognosis. Gastroenterology. 2021;160(1):232-244.e7.
Joustra V, Hageman IL, Satsangi J, Adams A, Ventham NT, De Jonge WJ, et al. Systematic review and meta-analysis of peripheral blood DNA methylation studies in inflammatory bowel disease. J Crohns Colitis. 2023;17(2):185–98.
Agliata I, Fernandez-Jimenez N, Goldsmith C, Marie JC, Bilbao JR, Dante R, et al. The DNA methylome of inflammatory bowel disease (IBD) reflects intrinsic and extrinsic factors in intestinal mucosal cells. Epigenetics. 2020;15(10):1068–82.
Lin Z, Hegarty J, Cappel J, Yu W, Chen X, Faber P, et al. Identification of disease-associated DNA methylation in intestinal tissues from patients with inflammatory bowel disease. Clin Genet. 2011;80(1):59–67.
Cooke J, Zhang H, Greger L, Silva AL, Massey D, Dawson C, et al. Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18(11):2128–37.
Li Y, Wang Z, Wu X, Wang G, Gu G, Ren H, et al. Intestinal mucosa-derived DNA methylation signatures in the penetrating intestinal mucosal lesions of Crohn’s disease. Sci Rep. 2021;11(1):9771.
Howell KJ, Kraiczy J, Nayak KM, Gasparetto M, Ross A, Lee C, et al. DNA methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology. 2018;154(3):585–98.
Biswas KH. Molecular mobility-mediated regulation of E-cadherin adhesion. Trends Biochem Sci. 2020;45(2):163–73.
Tahara T, Shibata T, Okubo M, Ishizuka T, Nakamura M, Nagasaka M, et al. DNA methylation status of epithelial-mesenchymal transition (EMT)—related genes is associated with severe clinical phenotypes in ulcerative colitis (UC). PLoS ONE. 2014;9(10):e107947.
Li LC, Chui RM, Sasaki M, Nakajima K, Perinchery G, Au HC, et al. A single nucleotide polymorphism in the E-cadherin gene promoter alters transcriptional activities. Cancer Res. 2000;60(4):873–6.
Nakamura A, Shimazaki T, Kaneko K, Shibata M, Matsumura T, Nagai M, et al. Characterization of DNA polymorphisms in the E-cadherin gene (CDH1) promoter region. Mutat Res Mol Mech Mutagen. 2002;502(1–2):19–24.
Cattaneo F, Venesio T, Molatore S, Russo A, Fiocca R, Frattini M, et al. Functional analysis and case-control study of -160C/A polymorphism in the E-cadherin gene promoter: association with cancer risk. Anticancer Res. 2006;26(6B):4627–32.
Saito S, Kato J, Hiraoka S, Horii J, Suzuki H, Higashi R, et al. DNA methylation of colon mucosa in ulcerative colitis patients: correlation with inflammatory status. Inflamm Bowel Dis. 2011;17(9):1955–65.
Taman H, Fenton CG, Hensel IV, Anderssen E, Florholmen J, Paulssen RH. Genome-wide DNA methylation in treatment-naïve ulcerative colitis. J Crohns Colitis. 2018;12(11):1338–47.
Barnicle A, Seoighe C, Greally JM, Golden A, Egan LJ. Inflammation-associated DNA methylation patterns in epithelium of ulcerative colitis. Epigenetics. 2017;12(8):591–606.
Venkateswaran S, Somineni HK, Matthews JD, Kilaru V, Hyams JS, Denson LA, et al. Longitudinal DNA methylation profiling of the rectal mucosa identifies cell-specific signatures of disease status, severity and clinical outcomes in ulcerative colitis cell-specific DNA methylation signatures of UC. Clin Epigenetics. 2023;15(1):50.
Lennerz JK, Van Der Sloot KWJ, Le LP, Batten JM, Han JY, Fan KC, et al. Colorectal cancer in Crohn’s colitis is comparable to sporadic colorectal cancer. Int J Colorectal Dis. 2016;31(5):973–82.
Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, et al. Genomic alterations observed in colitis-associated cancers are distinct from those found in sporadic colorectal cancers and vary by type of inflammatory bowel disease. Gastroenterology. 2016;151(2):278-287.e6.
Kim YH, Lee HC, Kim SY, Yeom YI, Ryu KJ, Min BH, et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann Surg Oncol. 2011;18(8):2338–47.
Li YX, Lu Y, Li CY, Yuan P, Lin SS. Role of CDH1 promoter methylation in colorectal carcinogenesis: a meta-analysis. DNA Cell Biol. 2014;33(7):455–62.
Olén O, Erichsen R, Sachs MC, Pedersen L, Halfvarson J, Askling J, et al. Colorectal cancer in Crohn’s disease: a Scandinavian population-based cohort study. Lancet Gastroenterol Hepatol. 2020;5(5):475–84.
Birney E, Smith GD, Greally JM. Epigenome-wide association studies and the interpretation of disease-omics. PLOS Genet. 2016;12(6):e1006105.
Campagna MP, Xavier A, Lechner-Scott J, Maltby V, Scott RJ, Butzkueven H, et al. Epigenome-wide association studies: current knowledge, strategies and recommendations. Clin Epigenetics. 2021;13(1):214.
Heijmans BT, Mill J. Commentary: the seven plagues of epigenetic epidemiology. Int J Epidemiol. 2012;41(1):74–8.
Belot MP, Castell AL, Le Fur S, Bougnères P. Dynamic demethylation of the IL2RA promoter during in vitro CD4+ T cell activation in association with IL2RA expression. Epigenetics. 2018;13(5):459–72.
Tedaldi G, Molinari C, São José C, Barbosa-Matos R, André A, Danesi R, et al. Genetic and epigenetic alterations of CDH1 regulatory regions in hereditary and sporadic gastric cancer. Pharmaceuticals. 2021;14(5):457.
Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593(7858):238–43.
Laiker I, Frankel N. Pleiotropic enhancers are ubiquitous regulatory elements in the human genome. Genome Biol Evol. 2022;14(6):0evac71.
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–6.
Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–31.
Ansari I, Raddatz G, Gutekunst J, Ridnik M, Cohen D, Abu-Remaileh M, et al. The microbiota programs DNA methylation to control intestinal homeostasis and inflammation. Nat Microbiol. 2020;5(4):610–9.
Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CGK, Mullican SE, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature. 2013;504(7478):153–7.
Yu DH, Gadkari M, Zhou Q, Yu S, Gao N, Guan Y, et al. Postnatal epigenetic regulation of intestinal stem cells requires DNA methylation and is guided by the microbiome. Genome Biol. 2015;16(1):211.
Cho YH, McCullough LE, Gammon MD, Wu HC, Zhang YJ, Wang Q, et al. Promoter hypermethylation in white blood cell DNA and breast cancer risk. J Cancer. 2015;6(9):819–24.
Karatzas PS, Mantzaris GJ, Safioleas M, Gazouli M. DNA Methylation profile of genes involved in inflammation and autoimmunity in inflammatory bowel disease. Medicine (Baltimore). 2014;93(28): e309.
Loyfer N, Magenheim J, Peretz A, Cann G, Bredno J, Klochendler A, et al. A DNA methylation atlas of normal human cell types. Nature. 2023;613(7943):355–64.
Heijmans BT, Tobi EW, Lumey LH, Slagboom PE. The epigenome: Archive of the prenatal environment. Epigenetics. 2009;4(8):526–31.
Jaeger N, Gamini R, Cella M, Schettini JL, Bugatti M, Zhao S, et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat Commun. 2021;12(1):1921.
Furey TS, Sethupathy P, Sheikh SZ. Redefining the IBDs using genome-scale molecular phenotyping. Nat Rev Gastroenterol Hepatol. 2019;16(5):296–311.
Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–54.
Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Dong Chen W, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36(4):417–22.
Yi JM. DNA methylation change profiling of colorectal disease: screening towards clinical use. Life. 2021;11(5):412.
Funding
INSERM 1169 and GETDOC Association 1901 provided the funding.
Author information
Authors and Affiliations
Contributions
PB supervised the project and wrote the manuscript with support from CdP and SA. CdP carried out methylation analyses, data preprocessing, quality control, and statistical analyses. SA carried out methylation analyses. CdP, SA, PB contributed to the interpretation of the results. MPB provided technical support to methylation analyses. XS analyzed data bases. CdP, SA, AB and CP recruited the patients and contributed to the interpretation of the results. All authors provided critical feedback and helped shape the research, analysis, and manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
The research protocol in the Dept of Surgery was reviewed and approved by Paris Sud University IRB. in accordance with the French regulations. During the pre-operative consultation, participants provided written informed consent to participate. Additional consent to review medical records and for genetic analysis was obtained through signed written consent.
Consent for publication
Not applicable.
Competing interests
P.Bougnères is the scientific founder of Adrenas Therapeutics, which had no role in this research. MPB is an employee of Therapy Design Consulting who gave technical advice to CdP at initiation of the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Fig. S1. Scheme of the genomic map (5’) proximal from the CDH1 gene. Studied CpGs are figured as CGI, or as lollipops for the 1st enhancer. The rs16260 A/C variant is shown. Fig. S2. ROC curve testing the association between CD and the methylation at the CDH1 locus in ileal mucosa. A logistic regression explored the association between CD and the enhancer CpGs according to the methylation status of CGI. The coefficient of the latter adjustment variable was used to weigh the enhancer CpGs value based on the CGI methylation status. Fig. S3. Correlation of the methylation of the 1st enhancer studied CpGs in the mucosa of pooled CD and control patients. R (p value). Fig. S4: Correlation of the methylation of the 1st enhancer studied CpGs in PBMC of pooled CD and control patients. R (p-value). Fig. S5. Methylation of the 1st enhancer CpGs in (A) mucosa; (B) PBMC of patients with CD or controls, according to the methylation status of the CGI. No significant differences were detected. Table S1. Complete individual methylation data in the studied CD patients and controls. Table S2. Methylation in mucosa does not depend on rs16260 (C>A) genotype. However, we observed non-significant trend for increased CGI methylation in patients with CC genotype. No had AA genotype was present. Mean (±SD); decimal values rounded to the nearest integer.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
de Ponthaud, C., Abdalla, S., Belot, MP. et al. Increased CpG methylation at the CDH1 locus in inflamed ileal mucosa of patients with Crohn disease. Clin Epigenet 16, 28 (2024). https://doi.org/10.1186/s13148-024-01631-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01631-z