
Abstract
Background
Acute myeloid leukaemia (AML) is a deadly disease characterised by the uncontrolled proliferation of immature myeloid cells within the bone marrow. Altered regulation of DNA methylation is an important epigenetic driver of AML, where the hypoxic bone marrow microenvironment can help facilitate leukaemogenesis. Thus, interactions between epigenetic regulation and hypoxia signalling will have important implications for AML development and treatment.
Main body
This review summarises the importance of DNA methylation and the hypoxic bone marrow microenvironment in the development, progression, and treatment of AML. Here, we focus on the role hypoxia plays on signalling and the subsequent regulation of DNA methylation. Hypoxia is likely to influence DNA methylation through altered metabolic pathways, transcriptional control of epigenetic regulators, and direct effects on the enzymatic activity of epigenetic modifiers. DNA methylation may also prevent activation of hypoxia-responsive genes, demonstrating bidirectional crosstalk between epigenetic regulation and the hypoxic microenvironment. Finally, we consider the clinical implications of these interactions, suggesting that reduced cell cycling within the hypoxic bone marrow may decrease the efficacy of hypomethylating agents.
Conclusion
Hypoxia is likely to influence AML progression through complex interactions with DNA methylation, where the therapeutic efficacy of hypomethylating agents may be limited within the hypoxic bone marrow. To achieve optimal outcomes for AML patients, future studies should therefore consider co-treatments that can promote cycling of AML cells within the bone marrow or encourage their dissociation from the bone marrow.
Similar content being viewed by others

Background
Characterised by the uncontrolled proliferation and diminished differentiation of immature myeloid cells in the bone marrow (BM), acute myeloid leukaemia (AML) is among the deadliest blood cancers [1]. Affecting ~ 5 in 100,000 individuals and carrying a dismal 5-year survival rate of ~ 25%, AML predominantly occurs in adults over the age of 60 [2, 3]. AML patients are typically treated with a standard combination of chemotherapies, such as daunorubicin and cytarabine [4]. While most patients respond well to these treatments and achieve remission, relapse often occurs within 3 years of diagnosis [5]. Unfortunately, many relapsed patients are typically non-responsive to further treatment, causing overall survival to be as low as 6 months from recurrence [6].
In comparison with most solid cancers, AML carries a relatively low mutational burden with an average of 13 mutations per patient [7, 8]. Among the most frequently mutated genes are several regulators of DNA methylation (DNMT3A, TET2, IDH1/2), showing that epigenetic dysregulation is an important driver of AML pathogenesis [9]. The BM microenvironment (BMME), in which AML develops, also influences disease progression through altered cell–cell interactions and extracellular factors [10, 11]. Since epigenetic mechanisms are known to respond to environmental stimuli [12], the crosstalk between the BMME and regulators of DNA methylation has clear relevance for AML. In this review, we consider how the low oxygen availability in the BMME may influence DNA methylation in AML. First, the importance of DNA methylation in AML development and treatment is described. Second, the effects of hypoxia on AML cells are summarised. Finally, we discuss likely modes of crosstalk between DNA methylation and hypoxia signalling, as well as implications for AML treatment.
DNA methylation and AML
Regulation of DNA methylation
DNA methylation is a critical component of the epigenome, with indispensable roles in regulating gene expression during development [13]. Methylation of the 5’ carbon in cytosine residues (5’-methylcytosine, 5mC) occurs in CpG dinucleotides in mammalian genomes. While majority of CpGs in the genome are methylated, short CpG-rich sequences termed CpG islands (CGIs) are generally hypomethylated. Most CGIs are located in gene promoters, and methylation of these loci can lead to transcriptional repression [14].
Regulation of DNA methylation is achieved through the combined actions of several enzyme families. De novo DNA methyltransferase (DNMT) enzymes, DNMT3A and DNMT3B, establish new DNA methylation marks within the genome [15, 16], while DNMT1 ensures that methylation profiles are stably inherited during cell division. In contrast, ten–eleven translocase (TET) enzymes initiate active DNA demethylation by oxidising 5mC to 5’-hydroxymethylcytosine (5hmC), and other modified bases that are then excised and replaced by DNA repair enzymes [17, 18]. Importantly, TET activity depends on several co-factors including oxygen and α-ketoglutarate (α-KG), which is produced by isocitrate dehydrogenase (IDH) enzymes [19, 20]. In AML, mutations have been detected in genes encoding DNMT3A, TET2, and IDH enzymes. These mutations disrupt DNA methylation patterns leading to dysregulated gene expression and altered differentiation (as outlined below).
Dysregulation of DNA methylation in AML
DNMT3A, TET2, and IDH enzymes are all required for appropriate haematopoiesis [20,21,22,23,24], with loss of enzyme activity causing proliferation of immature cells, impaired differentiation, and lineage skewing [25,26,27,28,29]. Mutations in these epigenetic regulators are insufficient to trigger overt leukaemia, and are detected in healthy individuals [30, 31], clonal haematopoiesis [32], and pre-leukaemic myelodysplasias [33,34,35,36], with the frequency of mutations increasing with age [37]. Upon acquisition of an AML driver mutation (e.g. FLT3, NPM1), the expanded progenitor population transforms to leukaemia [9, 38]. Thus, mutations in epigenetic regulators reshape the genetic landscape that is permissive for AML development.
DNMT3A mutations are observed in 12–22% of AML patients and are associated with reduced survival [39,40,41,42]. Missense mutations of arginine 882 (R882) are the most common abnormality [43, 44], causing reduced enzyme activity [45, 46] and altered patterns of DNA methylation [47]. While the global level of DNA methylation is not markedly affected [42], hypomethylation has been observed at many loci and in many genomic contexts [45, 48] (Fig. 1). The transcriptional consequences of these changes have been challenging to discern, since hypomethylated genes are not always upregulated in DNMT3A-mutant AML [42, 45, 47, 48]. Recently, single-cell analysis has been applied to resolve this issue. In clonal haematopoiesis patients with mosaic DNMT3A mutations, loss of DNA methylation at MYC binding sites in DNMT3A-mutant cells was accompanied by increased expression of MYC target genes [49]. Thus, changes in DNA methylation caused by DNMT3A mutations perturb key transcriptional programmes critical for myeloid differentiation.

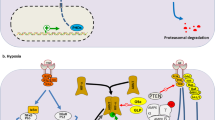
Common mutations in epigenetic regulators and their influence on DNA methylation in AML. In wild-type cells (black; top, left), the equilibrium between methylated (5mC) and unmodified (C) cytosines is governed by balanced DNMT3A and TET2 activity. IDH1/2 activity produces α-KG (orange), which is required for TET function. Loss of function DNMT3A mutations (green; bottom, left) lead to impaired DNMT3A activity and hypomethylation (purple). Conversely, loss of function TET2 (pink; top, right) or IDH1/2 (blue; bottom right) mutations result in hypermethylation (red). Mutations in IDH1/2 lead to the production of the oncometabolite 2-HG, which acts as a competitive inhibitor for TET2, resulting in decreased TET2 activity and hypermethylation (blue; bottom, right)
TET2 is mutated in ~ 20% of AML patients [38, 48, 50, 51] and is associated with unfavourable outcomes [38, 50, 52, 53]. Frameshift and nonsense mutations are spread across the whole TET2 coding sequence, and missense mutations occur in two conserved domains that are important for enzyme function [38, 50]. Mutations in TET2 prevent the conversion of 5mC to 5hmC [53, 54] and result in a hypermethylated phenotype that can encourage myeloid proliferation [55, 56] (Fig. 1). For example, in patients with clonal haematopoiesis and clonal cytopenia, TET2 mutations were associated with increased methylation of enhancers linked to myeloid differentiation [57].
Mutations in IDH1 or IDH2 are found in ~ 10% of AML patients, with missense mutations common at IDH1 R132 and IDH2 R140 or R172 residues [58, 59]. IDH enzymes support the catalytic activity of TET and other oxoglutarate enzymes by producing their essential co-factor, α-KG [60]. However, mutant IDH enzymes produce an oncometabolite, 2-hydroxyglutarate (2-HG), which competes with α-KG [61,62,63]. In turn, TET activity is reduced by 2-HG, leading to a global reduction in 5hmC levels and subsequent increases in 5mC [53] (Fig. 1). Given that IDH mutations result in loss of TET function, they seldom co-occur with TET2 mutations and are associated with hypermethylation of MYC target sites, as seen in TET2-mutant cells [20, 64]. While there are many similarities between TET2 and IDH-mutant AMLs, it is important to note that TETs are not the only enzymes dependent on α-KG. Other members of the α-KG-dependent dioxygenase family include: an N6-methyladenosine RNA demethylase (e.g. FTO) [65], histone demethylases (e.g. KDM7A/2B/5C) [60], and prolyl hydroxylase domain (PHD) enzymes, which are negative regulators of hypoxia-inducible factors (HIFs) [66]. Thus, IDH mutations may impact multiple aspects of epigenetic regulation, as well as responses to hypoxia.
Intriguingly, common mutations in AML have contrary effects on DNA methylation: DNMT3A mutations are associated with hypomethylation, while TET2 and IDH1/2 mutations lead to increases in DNA methylation (Fig. 1). These mutations can also co-occur, with around 40% of DNMT3A-mutant cases carrying a mutation in either TET2, IDH1 or IDH2 [48]. This suggests that AML is not driven by specific patterns of DNA methylation, but rather by disruption of the epigenetic equilibrium within blast cells. As such, DNA methylation can be dysregulated and clinically relevant in AML, even when the mutations described above are absent.
One interesting source of dysregulated DNA methylation in AML is the high levels of reactive oxygen species (ROS) observed in patients [67,68,69]. While ROS and oxidative stress can deplete the DNMT co-factor S-adenosyl methionine (SAM) resulting in reduced enzyme activity [70, 71], ROS can also act directly on DNA to convert 5mC to 5hmC [71]. If production remains unregulated, ROS can convert guanine bases to 8-hydroxydeoxyguanosine (8-OHdG), inhibiting the maintenance of methylation at nearby cytosines [72, 73]. Oxidative DNA damage also influences the formation of epigenetic complexes including DNMT1, DNMT3B, and polycomb repressive complex 4, as well as promote tighter binding of DNMT1 to chromatin, trapping the enzyme and causing transcriptional repression [74].
Several studies have profiled DNA methylation in large patient cohorts revealing epigenetic differences between AML subtypes [75, 76]. New molecular subtypes with differences in patient survival were identified on the basis of DNA methylation alone [76]. High levels of 5hmC can also predict inferior survival [53], suggesting that active remodelling of the methylome may promote AML growth. More recent work has used computational approaches to estimate intra-tumoural DNA methylation heterogeneity in patient samples [77, 78]. These analyses have demonstrated that high DNA methylation heterogeneity is associated with specific genetic abnormalities (e.g. IDH1/2 and CEBPA mutations) and reduced time to relapse. Information from 26 loci was sufficient to divide patients into low- and high-risk groups with significantly different relapse-free survival [77]. Dysregulated DNA methylation was associated with variable expression of neighbouring genes, suggesting that epigenetic heterogeneity allows diversification of transcriptional states within a tumour [77]. This may in turn promote AML progression by increasing the collective fitness of a population of cancer cells.
AML therapies targeting DNA methylation
Given the important role of DNA methylation in the initiation and progression of AML, it is not surprising that therapeutics targeting DNA methylation are being used to treat AML patients.
Hypomethylating agents (HMAs)
DNA hypomethylating agents (HMAs) are used as alternatives to standard chemotherapies for older AML patients, due to their low toxicity. Decitabine (DAC, 2’-deoxy-5-azacytidine) and azacytidine (AZA, 5-azacytidine) are two HMAs approved for the treatment of AML and a pre-leukaemic dysplasia known as myelodysplastic syndrome (MDS). Early clinical trials showed that higher-risk MDS patients treated with AZA had significantly increased overall survival (OS) compared to the conventional care group (24.5 vs. 15 months) [79]. Similar results have been observed for DAC, and in various other patient groups [80,81,82,83,84,85].
Despite these benefits, the use of HMAs is limited by variable patient responses, with only 20–30% of patients benefitting from therapy [86]. Many studies have investigated genetic, epigenetic, and other determinants of patient response. Some have noted improved responses in patients with DNMT3A [87], TET2 [88] or IDH1/2 [89] mutations, while others have yielded contrary results [90]. Studies have also suggested that DNA methylation levels before treatment may be a better predictor of HMA response rather than changes induced by therapy [91]. Changes to the BMME may also influence HMA response, as indicated in a study of AZA treatment in MDS patients. Non-responding patients were found to have higher proportions of quiescent progenitor cells in the BM. These cells expressed high levels of integrin alpha 5, a cell-surface protein important for cell–extracellular matrix adhesion within the BMME [92]. Thus, interactions between malignant blasts and the BMME may influence HMA efficacy.
DAC and AZA are cytidine analogues that are incorporated into DNA during replication [93,94,95,96]. This leads to degradation of DNMT enzymes, loss of DNA methylation, decreased growth, and increased immunogenicity in AML cells [96,97,98,99,100,101,102]. HMA-induced promoter demethylation has been associated with re-expression of tumour suppressor genes; however, many studies also show wide-spread increases in gene expression that are independent of promoter demethylation [96, 99, 102, 103]. This suggests that not all transcriptional changes induced by HMA treatment are dependent on methylation changes at cis-regulatory elements. DAC and AZA can also trigger a ‘viral mimicry’ response by upregulating the expression of endogenous retroviral (ERV) elements scattered across the genome. These transcripts are then recognised by the viral defence pathway, promoting apoptosis via an interferon response [104,105,106]. AZA-responsive patients have also shown a greater upregulation of many transposable elements compared to non-responding patients, demonstrating the clinical relevance of viral mimicry [107].
IDH1/2 therapies
Therapies have recently been developed to target mutant IDH enzymes. Ivosidenib (AG-120) and enasidenib (AG-221) are now approved for treatment of IDH1- and IDH2-mutant cancers, respectively. By reducing 2-HG production, IDH inhibitors can trigger epigenetic reprogramming and restoration of myeloid differentiation in AML [108, 109]. However, these agents are currently only used in relapsed or refractory (r/r) AML cases, where standard treatments are no longer beneficial.
In patients with advanced IDH1-mutant AML, ivosidenib induced remission in 30.4% of patients, with an associated median OS of 14.5 months [110]. Similar benefits were observed in newly diagnosed IDH1-mutant AML patients ineligible for standard chemotherapy [111], and in clinical trials of enasidenib treatment in IDH2-mutant AML [112, 113]. Despite these promising results, patient responses remain variable, and relapse is common. As such, further studies are required to improve the clinical utility of IDH inhibitors.
Combination therapies
To sustain long-term treatment responses and improve patient survival, combinatorial treatment strategies are being designed to enhance the efficacy of epigenetic therapies.
IDH inhibitors are currently being tested in combination with HMAs to treat IDH-mutant AML patients. By reducing DNA methylation through DNMT inhibition, and simultaneously blocking 2-HG production to restore TET2 activity, this combination strategy may improve efficacy in patients. In a phase IB trial, newly diagnosed IDH1-mutant AML patients treated with ivosidenib and AZA showed deep and durable treatment responses (overall response rate (ORR) 78.3%; complete remission (CR) 60.9%; and 12-month survival estimate 82%) [114]. Promising results have also been obtained from a recent phase III clinical trial [115], studies combining enasidenib with AZA [116, 117], and pre-clinical studies of newly developed IDH inhibitors [118].
Venetoclax is a selective BCL-2 inhibitor that is widely used to promote apoptosis in haematological malignancies [119]. The benefits of combining venetoclax and HMA treatment have been shown in several studies of AML, including treatment-naïve and r/r cases [120,121,122,123]. For example, one study reported superior responses in r/r patients (64% ORR vs. 19% AZA alone), as well as patients with IDH1/2 or TP53 mutations (67% ORR) [120]. Combined HMA and venetoclax therapy has also enhanced survival in elderly treatment-naïve AML patients [123, 124], and improved outcomes in patients with MDS [125, 126], as well as those undergoing stem cell transplantation [127, 128].
E-selectin is an endothelial cell adhesion molecule typically found in the BM, which regulates haematopoietic stem cell (HSC) self-renewal, homing and engraftment potential [129]. In AML, leukaemic blasts can bind to E-selectin on endothelial cells, holding them within the BM to avoid the effects of certain chemotherapies. Currently, an E-selectin inhibitor known as uproleselan (GMI-1271) is in phase III clinical trials in combination with chemotherapy for r/r AML [130]. Uproleselan has been shown to release quiescent AML blasts into the cell cycle, blocking pro-survival pathways, and reducing the retention of blasts in the BM [129]. In turn, uproleselan may also enhance the efficacy of other therapies, like HMAs, that are particularly dependent on replication. Encouragingly, the combination of uproleselan with venetoclax and HMAs has demonstrated improved survival in pre-clinical models of AML [131], and in AML patients [132]. In a phase I clinical trial of elderly or treatment-naïve AML patients, 75% of patients achieved remission [132]. Overall, E-selectin inhibition is a promising therapeutic avenue that highlights the need to target AML cells in the context of the BM.
Hypoxia and AML
Hypoxia signalling
While we know that AML develops within the BM, the direct role of the microenvironment on leukemogenesis is only beginning to be elucidated. The BM contains two distinct hypoxic niches with differing capillary types: the endosteal region with thick-walled, low-permeable vessels that enforce low oxygen tensions (1% O2), and the more oxygen tense sinusoidal-vascular region (5% O2) that contains more fenestrated, permeable capillaries [133, 134].
For cells to sense, coordinate, and adapt to these low oxygen conditions, oxygen-sensing transcription factors known as hypoxia-inducible factors (HIFs) are required to activate hypoxia-responsive genes. In low oxygen conditions, oxygen-sensitive alpha (α) subunits (HIF1α, HIF2α, HIF3α) and constitutively expressed beta (β) subunits (HIF1β, HIF-2β) dimerise to form a HIF complex. These HIF complexes can then translocate into the nucleus, where they bind to hypoxia-responsive elements (HREs) in promoter or enhancer regions of hypoxia-responsive genes [135, 136]. HIF-induced transcription of these genes can modulate oxygen consumption (e.g. pyruvate dehydrogenase kinase 1—PDK1) [137], erythrocyte production (e.g. erythropoietin—EPO) [138], angiogenesis (e.g. vascular endothelial growth factor—VEGF) [139], mitochondrial metabolism (e.g. IDH1, cytochrome c oxidase subunit 4—COX4) [140, 141], and cellular quiescence (e.g. early growth response 1—EGR1, signal transducer and activator of transcription 5—STAT5) [142, 143]. Conversely, in regions with higher oxygen tension, HIFα subunits are modified by prolyl hydroxylase domains, von Hippel–Lindau proteins, or factor inhibiting HIF1 enzymes to trigger degradation or inhibition of HIFα complexes [144,145,146].
Hypoxia also triggers metabolic reprogramming. While oxidative phosphorylation (OXPHOS) is the predominant source of energy for cells in oxygen-rich tissues, anaerobic glycolysis is used in hypoxic regions. HIF1α transcription factors upregulate the expression of glucose transporter 1 (GLUT1 encoded by SLC2A1) [147] and lactate dehydrogenase A (LDHA) [148] to increase glucose intake and the downstream conversion of pyruvate to lactate, respectively. HIF1α further supports anaerobic glycolysis by upregulating PDK1 expression, which prevents mitochondrial respiration and OXPHOS by inhibiting pyruvate dehydrogenase (PDH) [149]. Compared to OXPHOS, the energy generated by anaerobic glycolysis is very low (~ 2 ATP molecules vs. ~ 32 molecules, respectively) [150], meaning that suppressed cell growth is crucial for homeostasis in hypoxic conditions. OXPHOS also has the ability to generate ROS in the form of superoxide radicals (O2−) whereby the reduction of oxygen produces electrons that can leak from complexes in the electron transport chain. Superoxides are then converted to hydrogen peroxide (and O2) by mitochondrial superoxide dismutase’s (SODs) [151, 152].
Interestingly, ROS production within HPSCs is related to their proliferative state. Quiescent HSCs with low-cycling rates are associated with low levels of ROS production, while HSPCs with short-term repopulating capacity and increased cycling have higher ROS production [153, 154]. Thus, HSCs can use the endosteal niche to enter a quiescent state that protects them from oxidative DNA damage [154].
Effects of hypoxia in AML
The effects of hypoxia on AML cells and patients are complex, with in vitro and in vivo analyses yielding contradictory results [155,156,157,158,159,160,161,162,163]. Some evidence suggests that AML cells are capable of proliferating rapidly within the hypoxic BM, while other data suggest that they can avoid the effects of chemotherapy by entering quiescence within hypoxic microenvironments.
Several studies have demonstrated that increased HIF1A expression in AML is associated with upregulation of genes such as VEGF, GLUT1, and heme oxygenase-1 (HO-1); encouraging disease progression through increased angiogenic, metabolic, and apoptotic processes, respectively [164,165,166,167]. These effects are reversed with HIF1α inhibition, suppressing the growth of AML cell lines, and encouraging various apoptotic pathways [166]. The glycolytic switch induced by hypoxia can also promote AML cell growth, viability, and survival [160, 168,169,170,171,172]. A distinct glycolytic profile is observed in AML patients, with high levels of glycolytic metabolites predicting poor survival [173]. Furthermore, markers of anaerobic glycolysis are increased in patients who do not achieve remission [165]. These studies suggest that AML cell growth is encouraged within the hypoxic BM, and accordingly, pre-clinical studies show that AML blasts can outcompete healthy myeloid cells [158]. Importantly, this over-proliferation of AML cells generates an increasingly hypoxic environment that forces healthy HSCs to enter quiescence [159].
In contrast to the studies described above, hypoxia can also suppress AML cell growth in certain circumstances. Most in vitro studies of AML cell lines have reported increased HIF1A expression, together with reduced cell growth, in the context of low oxygen environments [155,156,157]. HIF1α can encourage entry of AML blasts into the G0/G1 phase of the cell cycle, while upregulating an S-phase inhibitory protein, known as p27 [157], which can enhance resistance to replication-dependent drugs such as cytarabine (Ara-C) [174, 175]. Similarly, HIF1α-induced GLUT1 activity has been associated with poor therapeutic response in AML [165, 176]. Together, these studies suggest that leukaemic stem cells (LSCs) may localise to the hypoxic BM environment following chemotherapy [177], where a glycolytic shift and resulting quiescence can protect them from treatment. To circumvent this possibility, pre-clinical studies are exploring the benefits of therapeutically targeting hypoxia-induced signalling and metabolic reprogramming in haematological tumours [69, 178, 179].
A role for HIF2α in AML is also becoming more evident, where blast cells in the BM of leukaemic mice exhibit higher HIF2A expression compared to healthy mice. AML cell lines support this, demonstrating that HIF2A deletion can decrease cell proliferation and prolong survival in xenograft experiments [180]. Knock-out in primary AML cells also lowered the engraftment and survival capacity of those cells [181].
In summary, the capacity of AML cells to survive and thrive in hypoxic environments is a contributor to poor prognosis [159, 182]. Within the BM, AML blasts can proliferate rapidly during disease progression, while LSCs lie quiescent to avoid chemotherapy. Optimised AML treatments will depend on a thorough understanding of hypoxia signalling in AML cells, as well as strategies to prevent the rapid adaptation of AML cells to changes in their environment.
Crosstalk between hypoxia and DNA methylation
As described above, both epigenetic regulation and the hypoxic BM play important roles in AML development and progression. Here, we explore the interactions between hypoxia and DNA methylation in AML, as well as any implications for therapeutic efficacy.
Hypoxia and TETs
During active demethylation, TET enzymes require oxygen to convert 5mC to 5hmC (Fig. 2a), implying that hypoxia may limit TET activity to create a hypermethylated state. In a study conducted by Thienpont et al., 5hmC levels were significantly decreased across 11 cancer cell lines in hypoxia (0.5% O2), with TET expression showing a positive correlation with 5hmC levels [54]. Further investigations using MCF7 breast cancer cells revealed that reductions of 5hmC in hypoxia were accompanied by increases in 5mC levels, with changes especially pronounced at gene enhancers, promoters, and actively transcribed regions.

Complex interactions between hypoxia and DNA methylation. Hypoxia may influence DNA methylation via direct effects on TET activity, altered transcription, or metabolic reprogramming. DNA methylation may also influence hypoxia responses. A Oxygen is an essential co-factor for TET enzymes, and hypoxia reduces TET-mediated DNA hydroxymethylation in some cancers. B In certain cell types, hypoxia-inducible factors (HIFα, HIFβ) bind to hypoxia-responsive elements (HREs) in TET and DNMT promoters to induce their expression. C In hypoxia, cancer cells can induce a metabolic switch from oxidative phosphorylation to glutamine metabolism. In IDH wild-type cells, upregulated glutamine metabolism has been associated with production of 2-HG which can inhibit TET enzymes. D DNA methylation can prevent the binding of HIF complexes to HREs, altering transcriptional responses induced by hypoxia
HIF signalling can also influence TET expression and activity depending on the cancer type (Fig. 2b). For example, in the hypoxic microenvironment of metastatic melanoma and glioblastoma, knockdown of HIF1α was associated with increased TET2 expression and 5hmC levels [183], while in neuroblastoma [184, 185] and hepatocellular carcinoma [186] studies, TET expression and 5hmC levels were increased in hypoxia. The above study by Thienpont also performed ChIP-seq to demonstrate HIF binding within TET promoter regions [54].
Until recently, the relationship between hypoxia and TET activity in AML had not been explored. In 2021, an analysis of KG-1 AML cells exposed to hypoxia (1–3% O2) demonstrated a positive correlation between HIF1A and TET2 expression [170]. Enhanced TET2 transcription was also associated with increased binding of HIF1α to the TET2 promoter, which subsequently increased 5hmC and decreased 5mC. As such, HIF1α-induced TET expression may override any reduction in TET activity caused by low oxygen availability in the context of AML.
TET3 may also be regulated by hypoxia in haematological malignancies. In a chronic myeloid leukaemia cell line (K562), the enhancer for TET3 was identified as a strong target for HIF1α binding, with TET3 expressed at higher levels than TET1 and TET2 in hypoxia [187]. Deletion of HIF1α binding sites in the TET3 enhancer resulted in decreased TET3 expression that reduced cell viability and impaired erythroid differentiation.
Overall, these studies demonstrate that the effects of hypoxia on TET expression and activity may be tissue dependent. In some cases, like leukaemia, hypoxia can enhance TET2 or TET3 expression to induce hypomethylation, while in others like breast cancer, it can reduce TET activity and promote hypermethylation across the genome.
Hypoxia and IDH1/2
Hypoxia can also influence the availability of the TET co-factor, α-KG. IDH enzymes, particularly IDH2, function in the mitochondria to convert isocitrate metabolites of the tricarboxylic acid (TCA) cycle into α-KG. However, when cells are deprived of oxygen, IDH metabolism and α-KG production are altered, influencing downstream TET activity and hence epigenetic regulation.
While hypoxia is well known to induce a glycolytic switch, some studies have found that hypoxia can also prompt a shift from OXPHOS to the glutamine-derived metabolic cycle (Fig. 2c). In IDH wild-type glioblastomas, hypoxia not only decreased TCA cycle activity, but also concomitantly increased glutamine-derived α-KG and the oncometabolite, 2-HG [188,189,190]. While α-KG can be produced by catabolism of glutamine, a non-reductive form of carboxylation can generate 2-HG without any IDH mutation [191]. Thus, hypoxia can mimic the effects of mutant IDH enzymes by generating 2-HG through alternate metabolic pathways.
In IDH-mutant cancers, the production of 2-HG can indirectly mimic a hypoxic state by promoting HIF1α activity [192]. PHDs are α-KG-dependent enzymes that typically ubiquitinate HIF1α in normoxic conditions. As a result, the production of 2-HG can reduce PHD activity, stabilising HIF1α to transcribe hypoxia-responsive genes. Thus, mutations in IDH have the potential to increase hypoxia signalling via loss of negative regulation [192, 193].
The production of 2-HG in primary IDH-mutant AML cells also inhibits the activity of cytochrome C oxidase (COX; Complex IV) enzymes that break down oxygen in the mitochondria for aerobic energy generation [194]. By decreasing the activity of Complex IV, oxygen consumption is reduced, such that the cell’s metabolic processes mimic a hypoxic state. This can create a glycolytic or glutaminergic metabolic shift that increases the anti-apoptotic effects of BCL-2 [195, 196], encouraging disease survival.
While studies in leukaemia are limited, a relationship between hypoxia and IDH enzymes is beginning to emerge. On the one side, hypoxia itself can cause a metabolic switch in IDH wild-type cells that indirectly produces 2-HG and inhibits α-KG-dependent enzymes, like TETs. On the other hand, production of 2-HG in cells with IDH mutations can promote a hypoxic-like state by either: promoting HIF signalling through PHD inhibition or decreasing OXPHOS via inhibition of Complex IV enzymes in the mitochondria.
Hypoxia and DNMTs
Hypoxia also influences the expression of DNMT enzymes in cancers such as liver, prostate, and breast cancer. Increased activity of HIF1α in hypoxia is associated with enhanced binding to the DNMT1 and DNMT3B promoters (Fig. 2b), promoting methylation and repression of tumour suppressor gene expression, such as protein sprouty homolog 2 (SPRY2) [197, 198]. Furthermore, DNMTs can act in a negative feedback loop to suppress HIF expression. In foetal lung fibroblasts, HIF2α-induced DNMT1 expression was followed by methylation of the HIF2A promoter, which in turn dampened the hypoxic response (Fig. 2d) [199]. Renal cell carcinomas and glioblastoma cell lines support this finding, where reduced DNMT3A expression was associated with decreased HIF2A promoter methylation and increased HIF2A expression [200]. The ectopic expression of DNMT3A in hypoxia (1% O2) was also shown to impair cell proliferation and viability by reducing HIF2A mRNA expression and protein activity.
DNA methylation may also impact hypoxia responses by modulating HIF binding across the genome (Fig. 2d). In one study, DNMT-triple knockout MCF7 breast cancer cells demonstrated preferential binding of HIF1β to promoter and enhancer regions containing unmethylated HREs [201]. HIF1β ChIP-seq also demonstrated that methylation of these HRE sites reduced HIF1β binding 12.4-fold.
Overall, evidence suggests that there are complex relationships between DNMTs, DNA methylation, and hypoxia in cancer. HIF heterodimers have a higher affinity for unmethylated HRE motifs, implying that DNA methylation can limit hypoxia responses. Further, transcriptional changes induced by DNMT and HIF enzymes can be modulated through negative feedback. Therefore, a greater understanding of these interactions will be critical for optimal use of epigenetic therapies in AML.
Effect of hypoxia on DNA methylation therapies in AML
Since hypoxia and DNA methylation both play important roles in AML, the bidirectional crosstalk described above is likely to have important clinical implications. For example, the hypoxic BMME could influence the efficacy of AML therapies targeting DNA methylation.
Given that HMAs are incorporated into DNA during replication, quiescent or low-cycling LSCs in the hypoxic BM niche may not respond to treatment. Consistent with this idea, MDS and AML patients who did not respond to AZA had a higher proportion of quiescent progenitor cells in the BM [87,88,89,90,91,92, 202, 203]. These LSCs could later resume cycling and promote relapse, suggesting that the long-term efficacy of HMAs depends upon uptake in cycling AML cells [127, 204,205,206]. One interesting pre-clinical study altered administration schedules to increase the proportion of cells exposed to HMA treatment during S-phase [206]. Continuous, twice-weekly administration of DAC was found to be more effective than 5 consecutive days of treatment followed by 3 weeks off therapy, suggesting that the treatment schedule used in current clinical practice may be sub-optimal.
HMA co-treatment schedules that increase AML cell cycling are also being considered. For example, E-selectin inhibitors may enhance HMA efficacy by forcing AML cells to move out of the hypoxic BM and resume cycling [207], encouraging the uptake of HMAs and a better treatment response. IDH1 inhibitors combined with AZA have also shown promise in treating AML by increasing the cycling of LSCs, allowing AZA to more effectively target these cells [208].
The key to improving treatment efficacy and long-term survival for AML patients may lie in combination therapies that target not only cancer cell proliferation, survival, or DNA methylation changes, but also their interactions with the hypoxic BM microenvironment. Such a treatment regime may aid in preventing or at least delaying relapse in AML.
Conclusion
Hypoxia signalling within the BMME, and dysregulation of DNA methylation, both contribute to the development of AML. As outlined above, complex bidirectional interactions between hypoxia and DNA methylation are likely to influence AML cell proliferation, with important clinical implications. Specifically, epigenetic therapies such as HMAs may have limited efficacy in the hypoxic BM due to reduced cell cycling in this microenvironment. Consideration of the interactions between the epigenome and the microenvironment in AML will lead to improved outcomes for patients.
Availability of data and materials
Not applicable.
Abbreviations
- BM:
-
Bone Marrow
- AML:
-
Acute Myeloid Leukaemia
- BMME:
-
Bone Marrow Microenvironment
- 5mC:
-
5’-Methylcytosine
- CGIs:
-
CpG Islands
- DNMT:
-
DNA Methyltransferases
- TET:
-
Ten–Eleven Translocases
- 5hmC:
-
5’-Hydroxymethylcytosine
- α-KG:
-
α-Ketoglutarate
- IDH:
-
Isocitrate Dehydrogenases
- 2-HG:
-
2-Hydroxyglutarate
- PHD:
-
Prolyl Hydroxylase Domains
- HIF:
-
Hypoxia-Inducible Factors
- ROS:
-
Reactive Oxygen Species
- HMAs:
-
Hypomethylating Agents
- DAC:
-
Decitabine; 2’-deoxy-5-azacytidine
- AZA:
-
Azacytidine; 5-azacytidine
- MDS:
-
Myelodysplastic Syndrome
- OS:
-
Overall Survival
- ERV:
-
Endogenous Retroviral
- ORR:
-
Overall Response Rates
- CR:
-
Complete Remission
- r/r:
-
Relapsed or Refractory
- HSCs:
-
Haematopoietic Stem Cells
- HRE:
-
Hypoxia-Responsive Element
- VHL:
-
Von Hippel–Lindau
- FIH-1:
-
Factor-Inhibiting HIF1
- Ara-C:
-
Cytarabine
- OXPHOS:
-
Oxidative Phosphorylation
- PDK:
-
Pyruvate Dehydrogenase Kinase
- PDH:
-
Pyruvate Dehydrogenase
- ATP:
-
Adenosine-5’-Triphosphate
- SOD:
-
Superoxide Dismutase
- HSPCs:
-
Haematopoietic Stem and Progenitor Cells
- LSC:
-
Leukaemic Stem Cell
- TCA:
-
Tricarboxylic Acid
- COX:
-
Cytochrome C Oxidase
- ETC:
-
Electron Transport Chain
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48.
Beckmann K, Kearney AMBJ, Yeung D, Hiwase D, Li M, Roder DM. Changes in five-year survival for people with acute leukaemia in South Australia, 1980–2016. Med J Aust. 2022;216(6):296–302.
Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev. 2019;36:70–87.
Murphy T, Yee KWL. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert Opin Pharmacother. 2017;18(16):1765–80.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–52.
Brandwein JM, Saini L, Geddes MN, Yusuf D, Liu F, Schwann K, Billawala A, Westcott C, Kurniawan JA, Cheung WY. Outcomes of patients with relapsed or refractory acute myeloid leukemia: a population-based real-world study. Am J Blood Res. 2020;10(4):124–33.
Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, Perry MD, Nahal-Bose HK, Ouellette BFF, Li CH, et al. Pan-cancer analysis of whole genomes. Nature. 2020;578(7793):82–93.
Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–33.
Yao Y, Li F, Huang J, Jin J, Wang H. Leukemia stem cell-bone marrow microenvironment interplay in acute myeloid leukemia development. Exp Hematol Oncol. 2021;10(1):39.
Pimenta DB, Varela VA, Datoguia TS, Caraciolo VB, Lopes GH, Pereira WO. The bone marrow microenvironment mechanisms in acute myeloid leukemia. Front Cell Dev Biol. 2021;9:764698.
Liu Z, Ren Y, Meng L, Li L, Beatson R, Deng J, Zhang T, Liu J, Han X. Epigenetic signaling of cancer stem cells during inflammation. Front Cell Dev Biol. 2021;9:772211.
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–20.
Schubeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321–6.
Yagi M, Kabata M, Tanaka A, Ukai T, Ohta S, Nakabayashi K, Shimizu M, Hata K, Meissner A, Yamamoto T, et al. Identification of distinct loci for de novo DNA methylation by DNMT3A and DNMT3B during mammalian development. Nat Commun. 2020;11(1):3199.
Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19(2):81–92.
Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156(1–2):45–68.
Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18(9):517–34.
Guo J, Zhang R, Yang Z, Duan Z, Yin D, Zhou Y. Biological roles and therapeutic applications of IDH2 mutations in human cancer. Front Oncol. 2021;11:644857.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67.
Koya J, Kataoka K, Sato T, Bando M, Kato Y, Tsuruta-Kishino T, Kobayashi H, Narukawa K, Miyoshi H, Shirahige K, et al. DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat Commun. 2016;7:10924.
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44(1):23–31.
Feng Y, Li X, Cassady K, Zou Z, Zhang X. TET2 function in hematopoietic malignancies, immune regulation, and DNA repair. Front Oncol. 2019;9:210.
Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, Adès L, Fenaux P, Platzbecker U, Gagey O, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186–98.
Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, Challen GA, Li W, Wheeler D, Rebel VI, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015;125(4):629–38.
Jeong M, Park HJ, Celik H, Ostrander EL, Reyes JM, Guzman A, Rodriguez B, Lei Y, Lee Y, Ding L, et al. Loss of Dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep. 2018;23(1):1–10.
Morinishi L, Kochanowski K, Levine RL, Wu LF, Altschuler SJ. Loss of TET2 affects proliferation and drug sensitivity through altered dynamics of cell-state transitions. Cell Syst. 2020;11(1):86-94.e85.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24.
Gu Y, Yang R, Yang Y, Zhao Y, Wakeham A, Li WY, Tseng A, Leca J, Berger T, Saunders M, et al. IDH1 mutation contributes to myeloid dysplasia in mice by disturbing heme biosynthesis and erythropoiesis. Blood. 2021;137(7):945–58.
Tovy A, Reyes JM, Gundry MC, Brunetti L, Lee-Six H, Petljak M, Park HJ, Guzman AG, Rosas C, Jeffries AR, et al. Tissue-biased expansion of DNMT3A-mutant clones in a mosaic individual is associated with conserved epigenetic erosion. Cell Stem Cell. 2020;27(2):326-335.e324.
Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, Samuel M, Ritchie EK, Guzman ML, Ballman KV, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018;24(7):1015–23.
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–87.
Lin M-E, Hou H-A, Tsai C-H, Wu S-J, Kuo Y-Y, Tseng M-H, Liu M-C, Liu C-W, Chou W-C, Chen C-Y, et al. Dynamics of DNMT3A mutation and prognostic relevance in patients with primary myelodysplastic syndrome. Clin Epigenet. 2018;10(1):42.
Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17(1):5–19.
Ogawa S. Genetics of MDS. Blood. 2019;133(10):1049–59.
Patnaik MM, Hanson CA, Hodnefield JM, Lasho TL, Finke CM, Knudson RA, Ketterling RP, Pardanani A, Tefferi A. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic Study of 277 patients. Leukemia. 2012;26(1):101–5.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–8.
Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, Dicker F, Fasan A, Haferlach C, Haferlach T, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26(5):934–42.
Thol F, Damm F, Lüdeking A, Winschel C, Wagner K, Morgan M, Yun H, Göhring G, Schlegelberger B, Hoelzer D, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol. 2011;29(21):2889–96.
Ribeiro AFT, Pratcorona M, Erpelinck-Verschueren C, Rockova V, Sanders M, Abbas S, Figueroa ME, Zeilemaker A, Melnick A, Löwenberg B, et al. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012;119(24):5824–31.
Shivarov V, Gueorguieva R, Stoimenov A, Tiu R. DNMT3A mutation is a poor prognosis biomarker in AML: results of a meta-analysis of 4500 AML patients. Leuk Res. 2013;37(11):1445–50.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33.
Jawad M, Afkhami M, Ding Y, Zhang X, Li P, Young K, Xu ML, Cui W, Zhao Y, Halene S, et al. DNMT3A R882 mutations confer unique clinicopathologic features in MDS including a high risk of AML transformation. Front Oncol. 2022;12:849376.
Singh RR, Bains A, Patel KP, Rahimi H, Barkoh BA, Paladugu A, Bisrat T, Ravandi-Kashani F, Cortes JE, Kantarjian HM, et al. Detection of high-frequency and novel DNMT3A mutations in acute myeloid leukemia by high-resolution melting curve analysis. J Mol Diagn. 2012;14(4):336–45.
Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, Meyer MR, Erdmann-Gilmore P, Townsend RR, Wilson RK, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014;25(4):442–54.
Yuan X-Q, Peng L, Zeng W-J, Jiang B-Y, Li G-C, Chen X-P. DNMT3A R882 mutations predict a poor prognosis in AML: a meta-analysis from 4474 patients. Medicine (Baltimore). 2016;95(18):e3519–e3519.
Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RS, Fronick CC, O’Laughlin M, Heath SE, Shinawi M, et al. CpG island hypermethylation mediated by DNMT3A Is a consequence of AML progression. Cell. 2017;168(5):801–16.
The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Nam AS, Dusaj N, Izzo F, Murali R, Myers RM, Mouhieddine T, Sotelo J, Benbarche S, Waarts M, Gaiti F, et al. Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation. Nat Genet. 2022;54:1514–26.
Chou W-C, Chou S-C, Liu C-Y, Chen C-Y, Hou H-A, Kuo Y-Y, Lee M-C, Ko B-S, Tang J-L, Yao M, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118(14):3803–10.
Shaikh ARK, Ujjan I, Irfan M, Naz A, Shamsi T, Khan MTM, Shakeel M. TET2 mutations in acute myeloid leukemia: a comprehensive study in patients of Sindh, Pakistan. PeerJ. 2021;9:e10678.
Wang R, Gao X, Yu L. The prognostic impact of tet oncogene family member 2 mutations in patients with acute myeloid leukemia: a systematic-review and meta-analysis. BMC Cancer. 2019;19(1):389.
Kroeze LI, Aslanyan MG, van Rooij A, Koorenhof-Scheele TN, Massop M, Carell T, Boezeman JB, Marie JP, Halkes CJ, de Witte T, et al. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood. 2014;124(7):1110–8.
Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, Ghesquière B, Van Dyck L, Boeckx B, Schoonjans L, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016;537(7618):63–8.
Wang J, He N, Wang R, Tian T, Han F, Zhong C, Zhang C, Hua M, Ji C, Ma D. Analysis of TET2 and EZH2 gene functions in chromosome instability in acute myeloid leukemia. Sci Rep. 2020;10(1):2706.
Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 2017;170(6):1079-1095.e1020.
Tulstrup M, Soerensen M, Hansen JW, Gillberg L, Needhamsen M, Kaastrup K, Helin K, Christensen K, Weischenfeldt J, Grønbæk K. TET2 mutations are associated with hypermethylation at key regulatory enhancers in normal and malignant hematopoiesis. Nat Commun. 2021;12(1):6061.
Liu X, Gong Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark Res. 2019;7:22–22.
Chotirat S, Thongnoppakhun W, Promsuwicha O, Boonthimat C, Auewarakul CU. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J Hematol Oncol. 2012;5(1):5.
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30.
Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. 2014;28(3):485–96.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-Hydroxyglutarate. Cancer Cell. 2010;17(3):225–34.
Wilson ER, Helton NM, Heath SE, Fulton RS, Payton JE, Welch JS, Walter MJ, Westervelt P, DiPersio JF, Link DC, et al. Focal disruption of DNA methylation dynamics at enhancers in IDH-mutant AML cells. Leukemia. 2022;36(4):935–45.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell. 2017;31(1):127–41.
Durán RV, MacKenzie ED, Boulahbel H, Frezza C, Heiserich L, Tardito S, Bussolati O, Rocha S, Hall MN, Gottlieb E. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene. 2013;32(38):4549–56.
Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, Small D, Rassool F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111(6):3173–82.
Sillar JR, Germon ZP, DeIuliis GN, Dun MD. The role of reactive oxygen species in acute myeloid leukaemia. Int J Mol Sci. 2019;20(23):6003.
Germon ZP, Sillar JR, Mannan A, Duchatel RJ, Staudt D, Murray HC, Findlay IJ, Jackson ER, McEwen HP, Douglas AM, et al. Blockade of ROS production inhibits oncogenic signaling in acute myeloid leukemia and amplifies response to recision therapies. Sci Signaling. 2023;16(778):eabp9586.
Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. 2010;15(2):551–89.
Kietzmann T, Petry A, Shvetsova A, Gerhold JM, Görlach A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br J Pharmacol. 2017;174(12):1533–54.
Rang FJ, Boonstra J. Causes and consequences of age-related changes in DNA methylation: a role for ROS? Biology (Basel). 2014;3(2):403–25.
Turk PW, Laayoun A, Smith SS, Weitzman SA. DNA adduct 8-hydroxyl-2’-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis. 1995;16(5):1253–5.
O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20(5):606–19.
Bullinger L, Ehrich M, Döhner K, Schlenk RF, Döhner H, Nelson MR, van den Boom D. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood. 2010;115(3):636–42.
Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13–27.
Li S, Chen X, Wang J, Meydan C, Glass JL, Shih AH, Delwel R, Levine RL, Mason CE, Melnick AM. Somatic mutations drive specific, but reversible, epigenetic heterogeneity states in AML. Cancer Discov. 2020;10(12):1934–49.
Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, Patel J, Dillon R, Vijay P, Brown AL, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. 2016;22(7):792–9.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–32.
Jabbour E, Short NJ, Montalban-Bravo G, Huang X, Bueso-Ramos C, Qiao W, Yang H, Zhao C, Kadia T, Borthakur G, et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood. 2017;130(13):1514–22.
Fenaux P, Mufti GJ, Hellström-Lindberg E, Santini V, Gattermann N, Germing U, Sanz G, List AF, Gore S, Seymour JF, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28(4):562–9.
Kantarjian HM, O’Brien S, Huang X, Garcia-Manero G, Ravandi F, Cortes J, Shan J, Davisson J, Bueso-Ramos CE, Issa JP. Survival advantage with decitabine versus intensive chemotherapy in patients with higher risk myelodysplastic syndrome: comparison with historical experience. Cancer. 2007;109(6):1133–7.
Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, Gau JP, Chou WC, Buckstein R, Cermak J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30(21):2670–7.
Lee JH, Choi Y, Kim SD, Kim DY, Lee JH, Lee KH, Lee SM, Cho SH, Lee WS, Joo YD. Comparison of 7-day azacitidine and 5-day decitabine for treating myelodysplastic syndrome. Ann Hematol. 2013;92(7):889–97.
Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, Kumar R, Cavenagh J, Schuh AC, Candoni A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126(3):291–9.
Helbig G, Chromik K, Woźniczka K, Kopińska AJ, Boral K, Dworaczek M, Koclęga A, Armatys A, Panz-Klapuch M, Markiewicz M. Real life data on efficacy and safety of azacitidine therapy for myelodysplastic syndrome, chronic myelomonocytic leukemia and acute myeloid leukemia. Pathol Oncol Res. 2019;25(3):1175–80.
Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, Blum W, Marcucci G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26(5):1106–7.
Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, Quesnel B, Vey N, Gelsi-Boyer V, Raynaud S, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25(7):1147–52.
Emadi A, Faramand R, Carter-Cooper B, Tolu S, Ford LA, Lapidus RG, Wetzler M, Wang ES, Etemadi A, Griffiths EA. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol. 2015;90(5):E77–9.
Stomper J, Lubbert M. Can we predict responsiveness to hypomethylating agents in AML? Semin Hematol. 2019;56(2):118–24.
Cabezón M, Malinverni R, Bargay J, Xicoy B, Marcé S, Garrido A, Tormo M, Arenillas L, Coll R, Borras J, et al. Different methylation signatures at diagnosis in patients with high-risk myelodysplastic syndromes and secondary acute myeloid leukemia predict azacitidine response and longer survival. Clin Epigenet. 2021;13(1):9.
Unnikrishnan A, Papaemmanuil E, Beck D, Deshpande NP, Verma A, Kumari A, Woll PS, Richards LA, Knezevic K, Chandrakanthan V, et al. Integrative genomics identifies the molecular basis of resistance to azacitidine therapy in myelodysplastic syndromes. Cell Rep. 2017;20(3):572–85.
Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123(1):8–13.
Schermelleh L, Spada F, Easwaran HP, Zolghadr K, Margot JB, Cardoso MC, Leonhardt H. Trapped in action: direct visualization of DNA methyltransferase activity in living cells. Nat Methods. 2005;2(10):751–6.
Patel K, Dickson J, Din S, Macleod K, Jodrell D, Ramsahoye B. Targeting of 5-aza-2’-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010;38(13):4313–24.
Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, Krushel L, Aukerman SL, Heise C, MacBeth KJ. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE. 2010;5(2):e9001–e9001.
Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, Vyas P, Cavenagh J, Stankovic T, Moss P, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116(11):1908–18.
Sutherland MK, Yu C, Anderson M, Zeng W, van Rooijen N, Sievers EL, Grewal IS, Law CL. 5-azacytidine enhances the anti-leukemic activity of lintuzumab (SGN-33) in preclinical models of acute myeloid leukemia. MAbs. 2010;2(4):440–8.
Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21(3):430–46.
Du F, Jin T, Wang L. Mechanism of action of decitabine in the treatment of acute myeloid leukemia by regulating LINC00599. Anal Cell Pathol (Amst). 2023;2023:2951519.
Leung KK, Nguyen A, Shi T, Tang L, Ni X, Escoubet L, MacBeth KJ, DiMartino J, Wells JA. Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proc Natl Acad Sci. 2019;116(2):695–700.
Klco JM, Spencer DH, Lamprecht TL, Sarkaria SM, Wylie T, Magrini V, Hundal J, Walker J, Varghese N, Erdmann-Gilmore P, et al. Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood. 2013;121(9):1633–43.
Flotho C, Claus R, Batz C, Schneider M, Sandrock I, Ihde S, Plass C, Niemeyer CM, Lübbert M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23(6):1019–28.
Chiappinelli Katherine B, Strissel Pamela L, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote Neal S, Cope Leslie M, Snyder A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–86.
Liu M, Ohtani H, Zhou W, Orskov AD, Charlet J, Zhang YW, Shen H, Baylin SB, Liang G, Gronbaek K, et al. Vitamin C increases viral mimicry induced by 5-aza-2’-deoxycytidine. Proc Natl Acad Sci U S A. 2016;113(37):10238–44.
Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–73.
Ohtani H, Ørskov AD, Helbo AS, Gillberg L, Liu M, Zhou W, Ungerstedt J, Hellström-Lindberg E, Sun W, Liang G, et al. Activation of a subset of evolutionarily young transposable elements and innate immunity are linked to clinical responses to 5-azacytidine. Can Res. 2020;80(12):2441–50.
Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, Cianchetta G, Cai Z, Zhou D, Cui D, et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett. 2018;9(4):300–5.
Yen K, Travins J, Wang F, David MD, Artin E, Straley K, Padyana A, Gross S, DeLaBarre B, Tobin E, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7(5):478–93.
DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, Swords R, Collins RH, Mannis GN, Pollyea DA, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–98.
Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, Altman JK, Arellano ML, Donnellan W, Erba HP, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2020;135(7):463–71.
Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–31.
de Botton S, Montesinos P, Schuh AC, Papayannidis C, Vyas P, Wei AH, Ommen H, Semochkin S, Kim H-J, Larson RA, et al. Enasidenib vs conventional care in older patients with late-stage mutant-IDH2 relapsed/refractory AML: a randomized phase 3 trial. Blood. 2023;141(2):156–67.
DiNardo CD, Stein AS, Stein EM, Fathi AT, Frankfurt O, Schuh AC, Döhner H, Martinelli G, Patel PA, Raffoux E, et al. Mutant isocitrate dehydrogenase 1 inhibitor ivosidenib in combination with azacitidine for newly diagnosed acute myeloid leukemia. J Clin Oncol. 2021;39(1):57–65.
Montesinos P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, Heuser M, Calado RT, Schuh AC, Yeh S-P, et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med. 2022;386(16):1519–31.
DiNardo CD, Schuh AC, Stein EM, Montesinos P, Wei AH, de Botton S, Zeidan AM, Fathi AT, Kantarjian HM, Bennett JM, et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukaemia (AG221-AML-005): a single-arm, phase 1b and randomised, phase 2 trial. Lancet Oncol. 2021;22(11):1597–608.
Venugopal S, Takahashi K, Daver N, Maiti A, Borthakur G, Loghavi S, Short NJ, Ohanian M, Masarova L, Issa G, et al. Efficacy and safety of enasidenib and azacitidine combination in patients with IDH2 mutated acute myeloid leukemia and not eligible for intensive chemotherapy. Blood Cancer J. 2022;12(1):10.
Chaturvedi A, Gupta C, Gabdoulline R, Borchert NM, Goparaju R, Kaulfuss S, Görlich K, Schottmann R, Othman B, Welzenbach J, et al. Synergistic activity of IDH1 inhibitor BAY1436032 with azacitidine in IDH1 mutant acute myeloid leukemia. Haematologica. 2021;106(2):565–73.
Griffioen MS, de Leeuw DC, Janssen JJWM, Smit L. Targeting acute myeloid leukemia with venetoclax; biomarkers for sensitivity and rationale for venetoclax-based combination therapies. Cancers. 2022;14(14):3456.
Aldoss I, Yang D, Aribi A, Ali H, Sandhu K, Al Malki MM, Mei M, Salhotra A, Khaled S, Nakamura R, et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica. 2018;103(9):e404–7.
Morsia E, McCullough K, Joshi M, Cook J, Alkhateeb HB, Al-Kali A, Begna K, Elliott M, Hogan W, Litzow M, et al. Venetoclax and hypomethylating agents in acute myeloid leukemia: Mayo Clinic series on 86 patients. Am J Hematol. 2020;95(12):1511–21.
Schmalbrock LK, Braitsch K, Jung P, Bumeder I, Kiewe P, Westermann J, Bullinger L, Keller U, Bassermann F, Krönke J, et al. Combination treatment of venetoclax and hypomethylating agents (HMA) or low-dose cytarabine (LDAC) for patients with acute myeloid leukemia (AML)—real-world data from two German academic centers. Blood. 2021;138(Supplement 1):1257–1257.
DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH, Kantarjian HM, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17.
DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, Konopleva M, Döhner H, Letai A, Fenaux P, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–29.
Ball BJ, Famulare CA, Stein EM, Tallman MS, Derkach A, Roshal M, Gill SI, Manning BM, Koprivnikar J, McCloskey J, et al. Venetoclax and hypomethylating agents (HMAs) induce high response rates in MDS, including patients after HMA therapy failure. Blood Adv. 2020;4(13):2866–70.
Zeidan AM, Borate U, Pollyea DA, Brunner AM, Roncolato F, Garcia JS, Filshie R, Odenike O, Watson AM, Krishnadasan R, et al. A phase 1b study of venetoclax and azacitidine combination in patients with relapsed or refractory myelodysplastic syndromes. Am J Hematol. 2023;98(2):272–81.
Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, D’Alessandro A, Culp-Hill R, Riemondy KA, Gillen AE, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859–66.
Tenold ME, Moskoff BN, Benjamin DJ, Hoeg RT, Rosenberg AS, Abedi M, Tuscano JM, Jonas BA. Outcomes of adults with relapsed/refractory acute myeloid leukemia treated with venetoclax plus hypomethylating agents at a comprehensive cancer center. Front Oncol. 2021;11:649209.
Barbier V, Erbani J, Fiveash C, Davies JM, Tay J, Tallack MR, Lowe J, Magnani JL, Pattabiraman DR, Perkins AC, et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. 2020;11(1):2042.
DeAngelo DJ, Jonas BA, Liesveld JL, Bixby DL, Advani AS, Marlton P, Magnani JL, Thackray HM, Feldman EJ, O’Dwyer ME, et al. Phase 1/2 study of uproleselan added to chemotherapy in patients with relapsed or refractory acute myeloid leukemia. Blood. 2022;139(8):1135–46.
Chang KH, Zhang W, Basyal M, Ostermann L, Fogler W, Magnani J, Andreeff M. AML-337: targeting E-selectin with GMI-1271 overcomes microenvironment-mediated resistance to venetoclax/HMA therapy. Clin Lymphoma Myeloma Leuk. 2020;20:S205.
Jonas BA, Welborn JL, Esteghamat NS, Hoeg RT, Rosenberg AS, Molnar L, Dang-Chu AL, Stewart SL, Tuscano JM. A phase I study of uproleselan combined with azacitidine and venetoclax for the treatment of older or unfit patients with treatment naïve acute myeloid leukemia. Blood. 2022;140(Supplement 1):6213–4.
Ramasamy SK. Structure and functions of blood vessels and vascular niches in bone. Stem Cells Int. 2017;2017:5046953–5046953.
Lebedev AY, Troxler T, Vinogradov SA. Design of metalloporphyrin-based dendritic nanoprobes for two-photon microscopy of oxygen. J Porphyr Phthalocyanines. 2008;12(12):1261–9.
Gao S, Zhou J, Zhao Y, Toselli P, Li W. Hypoxia-response element (HRE)-directed transcriptional regulation of the rat lysyl oxidase gene in response to cobalt and cadmium. Toxicol Sci. 2013;132(2):379–89.
Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005(306):re12.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–97.
Suzuki N, Yamamoto M. Roles of renal erythropoietin-producing (REP) cells in the maintenance of systemic oxygen homeostasis. Pflügers Arch Eur J Physiol. 2016;468(1):3–12.
Dai Y, Xu M, Wang Y, Pasha Z, Li T, Ashraf M. HIF-1alpha induced-VEGF overexpression in bone marrow stem cells protects cardiomyocytes against ischemia. J Mol Cell Cardiol. 2007;42(6):1036–44.
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481(7381):380–4.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–22.
Min IM, Pietramaggiori G, Kim FS, Passegué E, Stevenson KE, Wagers AJ. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell. 2008;2(4):380–91.
Wang Z, Li G, Tse W, Bunting KD. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood. 2009;113(20):4856–65.
Rani S, Roy S, Singh M, Kaithwas G. Regulation of transactivation at C-TAD domain of HIF-1α by factor-inhibiting HIF-1α (FIH-1): a potential target for therapeutic intervention in cancer. Oxid Med Cell Longev. 2022;2022:2407223.
Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. Embo J. 2005;24(22):3846–58.
Strowitzki MJ, Cummins EP, Taylor CT. Protein hydroxylation by hypoxia-inducible factor (HIF) hydroxylases: unique or ubiquitous? Cells. 2019;8(5):384.
Wierenga ATJ, Cunningham A, Erdem A, Lopera NV, Brouwers-Vos AZ, Pruis M, Mulder AB, Günther UL, Martens JHA, Vellenga E, et al. HIF1/2-exerted control over glycolytic gene expression is not functionally relevant for glycolysis in human leukemic stem/progenitor cells. Cancer Metab. 2019;7(1):11.
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271(51):32529–37.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–85.
van Noorden CJF, Breznik B, Novak M, van Dijck AJ, Tanan S, Vittori M, Bogataj U, Bakker N, Khoury JD, Molenaar RJ, et al. Cell biology meets cell metabolism: energy production is similar in stem cells and in cancer stem cells in brain and bone marrow. J Histochem Cytochem. 2022;70(1):29–51.
Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15(6):1583–606.
Tirichen H, Yaigoub H, Xu W, Wu C, Li R, Li Y. Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front Physiol. 2021;12:398.
Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–63.
Xie Y, Yin T, Wiegraebe W, He XC, Miller D, Stark D, Perko K, Alexander R, Schwartz J, Grindley JC, et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature. 2009;457(7225):97–101.
Zhang YL, Xu L, Qiu J, Li ZL, Wang JQ, Li R, Liu H, Zhu HM. Effect of hypoxia on the proliferation and hypoxia inducible factor-1α expression in human leukemia HL-60 cells. Nan Fang Yi Ke Da Xue Xue Bao. 2011;31(11):1890–4.
Tripathi VK, Subramaniyan SA, Hwang I. Molecular and cellular response of co-cultured cells toward cobalt chloride (CoCl(2))-induced hypoxia. ACS Omega. 2019;4(25):20882–93.
Drolle H, Wagner M, Vasold J, Kütt A, Deniffel C, Sotlar K, Sironi S, Herold T, Rieger C, Fiegl M. Hypoxia regulates proliferation of acute myeloid leukemia and sensitivity against chemotherapy. Leuk Res. 2015;39(7):779–85.
Akinduro O, Weber TS, Ang H, Haltalli MLR, Ruivo N, Duarte D, Rashidi NM, Hawkins ED, Duffy KR, Lo Celso C. Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat Commun. 2018;9(1):519–519.
Jensen PO, Mortensen BT, Hodgkiss RJ, Iversen PO, Christensen IJ, Helledie N, Larsen JK. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif. 2000;33(6):381–95.
Benito J, Ramirez MS, Millward NZ, Velez J, Harutyunyan KG, Lu H, Shi Y-X, Matre P, Jacamo R, Ma H, et al. Hypoxia-activated prodrug TH-302 targets hypoxic bone marrow niches in preclinical leukemia models. Clin Cancer Res. 2016;22(7):1687–98.
Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861–5.
Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, Levis M, Rubin JB, Negrin RR, Estey EH, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113(24):6215–24.
Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, Prior JL, Piwnica-Worms D, Bridger G, Ley TJ, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206–14.
Jabari M, Allahbakhshian Farsani M, Salari S, Hamidpour M, Amiri V, Mohammadi MH. Hypoxia-inducible factor1-Α (HIF1α) and vascular endothelial growth factor-A (VEGF-A) expression in de novo AML patients. Asian Pac J Cancer Prev. 2019;20(3):705–10.
Song K, Li M, Xu XJ, Xuan L, Huang GN, Song XL, Liu QF. HIF-1α and GLUT1 gene expression is associated with chemoresistance of acute myeloid leukemia. Asian Pac J Cancer Prev. 2014;15(4):1823–9.
Zhe N, Chen S, Zhou Z, Liu P, Lin X, Yu M, Cheng B, Zhang Y, Wang J. HIF-1α inhibition by 2-methoxyestradiol induces cell death via activation of the mitochondrial apoptotic pathway in acute myeloid leukemia. Cancer Biol Ther. 2016;17(6):625–34.
Gao XN, Yan F, Lin J, Gao L, Lu XL, Wei SC, Shen N, Pang JX, Ning QY, Komeno Y, et al. AML1/ETO cooperates with HIF1α to promote leukemogenesis through DNMT3a transactivation. Leukemia. 2015;29(8):1730–40.
Mesbahi Y, Trahair TN, Lock RB, Connerty P. Exploring the metabolic landscape of AML: from haematopoietic stem cells to myeloblasts and leukaemic stem cells. Front Oncol. 2022;12:807266.
Lodi A, Tiziani S, Khanim FL, Drayson MT, Günther UL, Bunce CM, Viant MR. Hypoxia triggers major metabolic changes in AML cells without altering indomethacin-induced TCA cycle deregulation. ACS Chem Biol. 2011;6(2):169–75.
He P, Lei J, Zou L-X, Zhou G-Z, Peng L, Deng Q, Liu X-L. Effects of hypoxia on DNA hydroxymethylase Tet methylcytosine dioxygenase 2 in a KG-1 human acute myeloid leukemia cell line and its mechanism. Oncol Lett. 2021;22(4):692–692.
Goto M, Miwa H, Suganuma K, Tsunekawa-Imai N, Shikami M, Mizutani M, Mizuno S, Hanamura I, Nitta M. Adaptation of leukemia cells to hypoxic condition through switching the energy metabolism or avoiding the oxidative stress. BMC Cancer. 2014;14(1):76.
Erdem A, Marin S, Pereira-Martins DA, Cortés R, Cunningham A, Pruis MG, de Boer B, van den Heuvel FAJ, Geugien M, Wierenga ATJ, et al. The Glycolytic Gatekeeper PDK1 defines different metabolic states between genetically distinct subtypes of human acute myeloid leukemia. Nat Commun. 2022;13(1):1105.
Chen WL, Wang JH, Zhao AH, Xu X, Wang YH, Chen TL, Li JM, Mi JQ, Zhu YM, Liu YF, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. 2014;124(10):1645–54.
Griessinger E, Anjos-Afonso F, Pizzitola I, Rouault-Pierre K, Vargaftig J, Taussig D, Gribben J, Lassailly F, Bonnet D. A niche-like culture system allowing the maintenance of primary human acute myeloid leukemia-initiating cells: a new tool to decipher their chemoresistance and self-renewal mechanisms. Stem Cells Transl Med. 2014;3(4):520–9.
Yook Y-J, Seo Y-J, Kang HJ, Ko S-H, Shin HY, Lee JJ, Jeong G, Ahn HS. Induction of hypoxia-inducible factor-1α inhibits drug-induced apoptosis in the human leukemic cell line HL-60. KJH. 2010;45(3):158–63.
Hannah Å, Jesper SH, Peng H, Raminta V, Axel H-W, Giulio P, Tiziano T, Carlotta G, Filippo M, Anna KH-A, et al. Targeting GLUT1 in acute myeloid leukemia to overcome cytarabine resistance. Haematologica. 2020;106(4):1163–6.
Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25(11):1315–21.
Qiu S, Sheth VS, Yan C, Liu J, Chacko BK, Li H, Crossman DK, Fortmann SD, Aryal S, Rennhack A, et al. Metabolic adaptation to tyrosine kinase inhibition in leukemia stem cells. Blood. 2023. https://doi.org/10.1182/blood.2022018196/495850.
Mannan A, Germon ZP, Chamberlain J, Sillar JR, Nixon B, Dun MD. Reactive oxygen species in acute lymphoblastic leukaemia: reducing radicals to refine responses. Antioxidants (Basel). 2021;10(10):1616.
Forristal CE, Brown AL, Helwani FM, Winkler IG, Nowlan B, Barbier V, Powell RJ, Engler GA, Diakiw SM, Zannettino ACW, et al. Hypoxia inducible factor (HIF)-2α accelerates disease progression in mouse models of leukemia and lymphoma but is not a poor prognosis factor in human AML. Leukemia. 2015;29(10):2075–85.
Rouault-Pierre K, Lopez-Onieva L, Foster K, Anjos-Afonso F, Lamrissi-Garcia I, Serrano-Sanchez M, Mitter R, Ivanovic Z, de Verneuil H, Gribben J, et al. HIF-2α protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell. 2013;13(5):549–63.
Harris B, Saleem S, Cook N, Searle E. Targeting hypoxia in solid and haematological malignancies. J Exp Clin Cancer Res. 2022;41(1):318.
Fischer AP, Miles SL. Silencing HIF-1α induces TET2 expression and augments ascorbic acid induced 5-hydroxymethylation of DNA in human metastatic melanoma cells. Biochem Biophys Res Commun. 2017;490(2):176–81.
Mariani CJ, Vasanthakumar A, Madzo J, Yesilkanal A, Bhagat T, Yu Y, Bhattacharyya S, Wenger RH, Cohn SL, Nanduri J, et al. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 2014;7(5):1343–52.
Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG Jr, Godley LA, Koivunen P. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J Biol Chem. 2016;291(8):4256–65.
Lin G, Sun W, Yang Z, Guo J, Liu H, Liang J. Hypoxia induces the expression of TET enzymes in HepG2 cells. Oncol Lett. 2017;14(6):6457–62.
Cao JZ, Liu H, Wickrema A, Godley LA. HIF-1 directly induces TET3 expression to enhance 5-hmC density and induce erythroid gene expression in hypoxia. Blood Adv. 2020;4(13):3053–62.
Ricoult SJ, Dibble CC, Asara JM, Manning BD. Sterol regulatory element binding protein regulates the expression and metabolic functions of wild-type and oncogenic IDH1. Mol Cell Biol. 2016;36(18):2384–95.
Reitman ZJ, Duncan CG, Poteet E, Winters A, Yan LJ, Gooden DM, Spasojevic I, Boros LG, Yang SH, Yan H. Cancer-associated isocitrate dehydrogenase 1 (IDH1) R132H mutation and d-2-hydroxyglutarate stimulate glutamine metabolism under hypoxia. J Biol Chem. 2014;289(34):23318–28.
Wise DR, Ward PS, Shay JES, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci. 2011;108(49):19611–6.
Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015;22(2):304–11.
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science. 2009;324(5924):261–5.
Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–8.
Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020;37:101674.
Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, Zhao F, Medeiros BC, Tyvoll DA, Majeti R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–84.
McClintock DS, Santore MT, Lee VY, Brunelle J, Budinger GR, Zong WX, Thompson CB, Hay N, Chandel NS. Bcl-2 family members and functional electron transport chain regulate oxygen deprivation-induced cell death. Mol Cell Biol. 2002;22(1):94–104.
Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C, Phelan D, Ledwidge MT, McDonald KM, McCann A, et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet. 2014;23(8):2176–88.
Gao X, Hicks KC, Neumann P, Patel TB. Hypoxia inducible factors regulate the transcription of the sprouty2 gene and expression of the sprouty2 protein. PLoS ONE. 2017;12(2):e0171616–e0171616.
Xu XH, Bao Y, Wang X, Yan F, Guo S, Ma Y, Xu D, Jin L, Xu J, Wang J. Hypoxic-stabilized EPAS1 proteins transactivate DNMT1 and cause promoter hypermethylation and transcription inhibition of EPAS1 in non-small cell lung cancer. FASEB J. 2018;32:6694–705.
Lachance G, Uniacke J, Audas TE, Holterman CE, Franovic A, Payette J, Lee S. DNMT3a epigenetic program regulates the HIF-2α oxygen-sensing pathway and the cellular response to hypoxia. Proc Natl Acad Sci. 2014;111(21):7783–8.
D’Anna F, Van Dyck L, Xiong J, Zhao H, Berrens RV, Qian J, Bieniasz-Krzywiec P, Chandra V, Schoonjans L, Matthews J, et al. DNA methylation repels binding of hypoxia-inducible transcription factors to maintain tumor immunotolerance. Genome Biol. 2020;21(1):182.
Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, et al. Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108(10):3271–9.
Craddock C, Quek L, Goardon N, Freeman S, Siddique S, Raghavan M, Aztberger A, Schuh A, Grimwade D, Ivey A, et al. Azacitidine fails to eradicate leukemic stem/progenitor cell populations in patients with acute myeloid leukemia and myelodysplasia. Leukemia. 2013;27(5):1028–36.
Riether C, Pabst T, Höpner S, Bacher U, Hinterbrandner M, Banz Y, Müller R, Manz MG, Gharib WH, Francisco D, et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med. 2020;26(9):1459–67.
Gruber E, Franich RL, Shortt J, Johnstone RW, Kats LM. Distinct and overlapping mechanisms of resistance to azacytidine and guadecitabine in acute myeloid leukemia. Leukemia. 2020;34(12):3388–92.
Gu X, Tohme R, Tomlinson B, Sakre N, Hasipek M, Durkin L, Schuerger C, Grabowski D, Zidan AM, Radivoyevitch T, et al. Decitabine- and 5-azacytidine resistance emerges from adaptive responses of the pyrimidine metabolism network. Leukemia. 2021;35(4):1023–36.
Erbani J, Tay J, Barbier V, Levesque JP, Winkler IG. Acute myeloid leukemia chemo-resistance is mediated by e-selectin receptor CD162 in bone marrow niches. Front Cell Dev Biol. 2020;8:668.
Gruber E, So J, Lewis AC, Franich R, Cole R, Martelotto LG, Rogers AJ, Vidacs E, Fraser P, Stanley K, et al. Inhibition of mutant IDH1 promotes cycling of acute myeloid leukemia stem cells. bioRxiv 2022:2022.2004.2006.487420.
Acknowledgements
None.
Funding
H.J.L. and M.D.D. are supported by the National Health and Medical Research Council of Australia (GNT2016283 and GNT1173892). The contents of the published material are solely the responsibility of the research institutions involved or individual authors and do not reflect the views of NHMRC.
Author information
Authors and Affiliations
Contributions
SH, DRB, ZPG, and HJL prepared the original draft. DRB and HJL designed and prepared figures. MDD and HJL supervised work and acquired funding. HJL conceived the scope of the review and oversaw the work. All authors reviewed and edited the manuscript and approved the final version. Correspondence should be addressed to HJL.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Humphries, S., Bond, D.R., Germon, Z.P. et al. Crosstalk between DNA methylation and hypoxia in acute myeloid leukaemia. Clin Epigenet 15, 150 (2023). https://doi.org/10.1186/s13148-023-01566-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-023-01566-x