
Abstract
Introduction
DNA methylation studies have associated methylation levels at different CpG sites or genomic regions with lung function. Moreover, genetic ancestry has been associated with lung function in Latinos. However, no epigenome-wide association study (EWAS) of lung function has been performed in this population. Here, we aimed to identify DNA methylation patterns associated with lung function in pediatric asthma among Latinos.
Results
We conducted an EWAS in whole blood from 250 Puerto Rican and 148 Mexican American children and young adults with asthma. A total of five CpGs exceeded the genome-wide significance threshold of p = 1.17 × 10−7 in the combined analyses from Puerto Ricans and Mexican Americans: cg06035600 (MAP3K6, p = 6.13 × 10−8) showed significant association with pre-bronchodilator Tiffeneau–Pinelli index, the probes cg00914963 (TBC1D16, p = 1.04 × 10−7), cg16405908 (MRGPRE, p = 2.05 × 10−8), and cg07428101 (MUC2, p = 5.02 × 10−9) were associated with post-bronchodilator forced vital capacity (FVC), and cg20515679 (KCNJ6) with post-bronchodilator Tiffeneau–Pinelli index (p = 1.13 × 10−8). However, these markers did not show significant associations in publicly available data from Europeans (p > 0.05). A methylation quantitative trait loci analysis revealed that methylation levels at these CpG sites were regulated by genetic variation in Latinos and the Biobank-based Integrative Omics Studies (BIOS) consortium. Additionally, two differentially methylated regions in REXOC and AURKC were associated with pre-bronchodilator Tiffeneau–Pinelli index (adjusted p < 0.05) in Puerto Ricans and Mexican Americans. Moreover, we replicated some of the previous differentially methylated signals associated with lung function in non-Latino populations.
Conclusions
We replicated previous associations of epigenetic markers with lung function in whole blood and identified novel population-specific associations shared among Latino subgroups.
Similar content being viewed by others

Introduction
Asthma is the most prevalent chronic inflammatory disorder among children and young adults worldwide [1]. It is characterized by reversible airflow obstruction and impaired lung function, with a larger decline in lung function among children and adults with asthma than in individuals without asthma [2]. Although many genotypic predictors of respiratory diseases have been identified, genetic effects do not fully explain the high prevalence of asthma worldwide [3]. Decreased lung function in childhood and early adulthood is an important predictor of wheezing [4], asthma severity [5], future reduced lung function [6], the development of chronic obstructive pulmonary disease (COPD), and even death [7]. Furthermore, lung function is also influenced by several factors, including smoking exposure, air pollution, socioeconomic factors, and prenatal exposures [8, 9]. These environmental factors can observably impact the epigenome.
The most studied epigenetic marker type is DNA methylation (DNAm), which consists of the addition of a methyl group to cytosine residues within 5′-cytosine-phosphate-guanine-3′ dinucleotide sequences (known as ‘CpG’ sites). Changes in DNAm can regulate gene function by modulating gene expression in response to a wide range of environmental factors or genetic determinants [10]. The association between different methylation patterns and lung function can be analyzed with epigenome-wide association studies (EWAS). However, all EWAS of lung function have been conducted in European-descent populations, except for a study in Korean adults with COPD [11].
The Hispanic/Latino populations are the largest minority group in the United States (US) [12]. Hispanics/Latinos are genetically diverse, with varying proportions of European, African, and Native American ancestries depending on each subgroup-specific historical event of genetic admixture [13]. Native American ancestry is associated with higher lung function in Mexican American children with asthma [14, 15], whereas African ancestry is associated with lower lung function among Mexican American and Puerto Rican children with asthma [15, 16].
Since methylation is also associated with genetic ancestry [17], we hypothesize that differential DNAm patterns in whole blood may contribute to the differences in lung function among Puerto Ricans and Mexicans. Therefore, we aimed to identify CpG sites and differentially methylated regions (DMRs) in which DNAm levels in blood associated with lung function in Latino children and young adults with asthma. For that purpose, we performed several EWAS of pulmonary function test (PFT) measurements including forced expiratory volume in one second (FEV1), forced vital capacity (FVC), and their ratio (FEV1/FVC) pre- and post-administration of albuterol in Puerto Rican and Mexican American children and youth with asthma separately. Then, we assessed the ethnic-specific results for replication across the other ethnic subgroup. Moreover, we attempted to replicate in Latinos single epigenetic markers and DMRs associated with lung function in non-Latino populations.
Results
Characteristics of study populations
Characteristics of the 250 Puerto Ricans and 148 Mexican Americans from the Genes-Environment and Admixture in Latino Americans (GALA II) study included in the analysis are shown in Table 1. Overall, Puerto Ricans had lower pre- and post-FEV1% predicted and FVC % predicted than Mexican Americans (p < 0.05), although similar values were observed for the FEV1/FVC ratios. Among Puerto Ricans, the percentage of overweight participants was slightly lower than in Mexicans. Approximately, half of Puerto Ricans had normal weight and 27% were obese, while less than 40% of Mexican Americans had normal weight and approximately 40% were obese. In both subethnic groups, underweight individuals represented less than 10% of the populations. Exposure to second-hand smoking (SHS) and in utero maternal smoking was higher among individuals profiled with the Illumina Infinium HumanMethylation450 BeadChip array (450K) [17], compared with those where methylation was measured with the Illumina Infinium MethylationEPIC (EPIC).
Identification of CpGs associated with PFT measurements
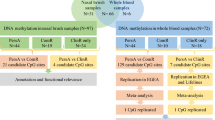
We performed an EWAS of PFT in Mexican Americans and Puerto Ricans separately by DNA methylation array, meta-analyzed individuals within subethnic group, and performed replication between populations, as detailed in Fig. 1. A total of 18 CpGs showed suggestive association with PFTs at a false discovery rate (FDR)-adjusted p ≤ 0.1 in Puerto Ricans (Additional file 5: Table S1; Additional file 1: Fig. S1). In Mexican Americans, we found 132 CpGs that were suggestively associated with PFT measurements at FDR-adjusted p ≤ 0.1 (Additional file 5: Table S1; Additional file 2: Fig. S2). The quantile–quantile plots indicated no major signs of inflation (Additional file 3: Fig. S3).

Study design of the EWAS of lung function in Mexican Americans and Puerto Ricans and post-EWAS analyses
From the CpGs identified in Puerto Ricans, the probes cg16405908 (MRGPRE, p = 1.64 × 10−3), cg07428101 (MUC2, p = 5.70 × 10−3) and cg00129273 (PRDM14, p = 1.17 × 10−2) had the same significant direction of the effect for post-FVC in Mexican Americans. From the CpGs associated with PFT in Mexican Americans, nine showed a significant (p < 0.05) and consistent direction of the effect (Table 2). The probes cg18573338 (TBC1D17) and cg18635207 (TMEM90A) were significantly associated with pre-FEV1 in Puerto Ricans (p = 4.64 × 10−2 and p = 4.37 × 10−2, respectively). One probe (cg10523319, DHRS3) showed association with pre-FVC in Puerto Ricans (p = 3.01 × 10−2). The probes cg00914963 (TBC1D16) and cg22467052 (CFTR) were associated with post-FVC in Puerto Ricans (p = 1.77 × 10−2 and p = 1.10 × 10−2, respectively). Likewise, the association of cg01408486 (CXXC5) and cg06035600 (MAP3K6) with pre-FEV1/FVC was replicated in Puerto Ricans (p = 2.45 × 10−2 and p = 3.69 × 10−3, respectively). The probes cg20515679 (KCNJ6) and cg25637972 (RBM24/STMND1) also exhibited a consistent significant association for post-FEV1/FVC in Puerto Ricans (p = 9.90 × 10−4 and p = 7.42 × 10−3, respectively).
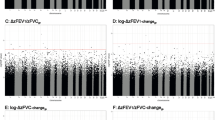
From the probes that were significant in the discovery stage and replicated across ethnic sub-groups, five exceeded the genome-wide significance threshold for significance based on the number of probes analyzed (p = 0.05/427,079 = 1.17 × 10−7) in the meta-analysis of Mexican Americans and Puerto Ricans. These genome-wide significant hits included cg06035600 (MAP3K6, p = 6.13 × 10−8) for pre-FEV1/FVC, cg20515679 (KCNJ6, p = 1.13 × 10−7) for post-FEV1/FVC, and the probes cg00914963 (TBC1D16, p = 1.04 × 10−7), cg16405908 (MRGPRE, p = 5.02 × 10−9), and cg07428101 (MUC2, p = 2.05 × 10−8) for post-FVC (Fig. 2). None of these five genome-wide significant probes was flagged as cross-reactive, multi-modal, or single nucleotide polymorphism (SNP)-containing probes (not flagged in Additional file 5: Table S1).

Correlation between lung function measurements and DNA methylation levels at the five genome-wide significant CpG sites in the meta-analysis of Puerto Ricans and Mexican Americans. The DNA methylation levels are shown as beta-values in the x-axis along the residuals of the regression of the lung function measurement adjusted by the covariates age, sex, height, in utero smoking exposure and genetic ancestry (represented in the y-axis)
Sensitivity analyses accounting for potential confounders
We next performed sensitivity analyses including body mass index (BMI) category, SHS exposure, use of asthma controller medication in the two weeks preceding the spirometry, insurance status, and maternal education level. However, minimal differences in the effect sizes for the five genome-wide significant CpG sites were detected (Table 3), which suggests no major effect of these factors. We also explored whether the effects were consistent across the two arrays used for methylation profiling and similar effects were found (Additional file 5: Table S2).
We next tested the association of the genome-wide significant probes with the exposure to air pollution during the past year and lifetime, including daily average of 1-h ozone (O3), sulfur dioxide (SO2), nitrogen dioxide (NO2), particulate matter ≤ 10 µm in diameter (PM10) or 2.5 µm (PM2.5) (Additional file 5: Table S3). The probe cg07428101 exhibited association at p < 0.05 with lifetime daily average of 1-h O3 (p = 1.77 × 10−2). Moreover, the probe cg00914963 showed significant association with PM2.5 and O3 exposure in the past year at p < 0.05 (p = 6.94 × 10−3 and p = 2.18 × 10−2, respectively), and with the participant’s lifetime exposure (p = 1.16 × 10−2 and p = 2.32 × 10−2, respectively). However, the association with PFT measurements was not confounded by air pollution exposure (Additional file 5: Table S4).
Assessment of genome-wide significant association hits for replication in Europeans
The replication in non-Latino populations of the 5 CpG sites that showed genome-wide significant association in the meta-analysis of Latinos was assessed in 2,043 European adults, including all individuals and a subset of never smokers. However, none of these were associated with PFT in publicly available data from European adults [18] (Additional file 5: Table S5).
Methylation quantitative trait loci analysis
A cis-methylation quantitative trait loci (meQTL) analysis was performed to test whether genetic variation was associated with DNAm levels at the associated CpGs. The five tested CpG sites were genetically regulated. From the 13,668 SNPs evaluated, 785 meQTLs exhibited association at Storey q-value < 0.05 (Additional file 5: Table S6). A total of 78 quasi-independent SNPs identified by linkage disequilibrium clumping of SNPs with pairwise r2 < 0.25 within 250 kilobases. These were distributed per CpG site as follows: cg00914963 (14 SNPs), cg06035600 (10 SNPs), cg07428101 (11 SNPs), cg16405908 (40 SNPs), and cg20515679 (3 SNPs). From these, 16 were replicated in the Biobank-based Integrative Omics Studies (BIOS) consortium [19] (FDR < 0.05) (Additional file 5: Table S7). From the 78 meQTLs, the SNPs rs61870478 (p = 3.01 × 10−3), rs74382103 (p = 2.69 × 10−2), rs2362396 (p = 3.51 × 10−2), and rs234850 (p = 4.60 × 10−2) showed association with post-FVC (%) predicted at p < 0.05 among Latinos in an analysis adjusted by age, sex, and the first three genotype principal components (Additional file 5: Table S8). As post-FVC is not available in the Pan-UK Biobank [20], we tested the association with pre-FVC-associated traits, but no meQTL was replicated (p > 0.05) (Additional file 5: Table S9).
Enrichment analyses in previous EWAS signals
Among the 100 most significant PFT associated probes for each subethnic group, there was a significant enrichment in previous EWAS signals for known factors involved in lung function, including respiratory diseases (asthma and COPD), allergic phenotypes (respiratory allergies and allergic sensitization), BMI, pollutants exposure, smoking, socioeconomic status, alcohol consumption, ancestry, or circadian rhythm (Additional file 4: Fig. S4). Additionally, enrichment in associations for traits related to autoimmune diseases, mortality, and preterm birth were also shared among several PFTs.
Identification of differentially methylated regions associated with PFT measurements
We next assessed lung function-related DMRs (Additional file 5: Table S10) for a total of 405,366 non-multi-modal, non-cross-reactive and non-SNP-containing CpG sites, as detailed in Additional file 6. Multiple DMRs in genes involved in airway remodeling or inflammation overlapped across phenotypes, such as pre- and post-FEV1 as well as pre- and post-FVC in Mexican Americans (e.g., TNFRSF14/HES5, MAN2B1/ZNF791, and MRPS23/VEZF1) and Puerto Ricans (e.g., CLMN/SYNE3, and GSDMD). Moreover, two regions that contain 3 CpGs (REXO1) and 8 CpGs (AURKC) were associated with pre-FEV1/FVC in both subethnic groups.
Replication of previous epigenetic loci for lung function
The analyses in Mexican Americans showed significant enrichment in previous PFT-associated epigenetic loci [11, 18, 21,22,23,24,25,26,27,28] for pre-FEV1 (p = 0.031), pre-FEV1/FVC (p = 0.009), and post-FEV1 (p = 0.006), whereas Puerto Ricans showed significant enrichment for pre-FEV1/FVC (p = 0.002) (Additional file 5: Table S11). Previous PFT-associated CpGs that were associated in either subethnic group at nominal level (p < 0.05) are shown in Additional file 5: Table S12. Among those probes that showed significant association in both subethnic groups separately, 19 probes also had p < 0.05 in the combined results of both subethnic groups, and four of these were associated with multiple PFT traits: cg16734845 (CTDSPL2), cg25634666 (FOLR3), cg07148038 (TNXB), and cg26206598 (PREX1). However, none exceeded the Bonferroni-corrected threshold, accounting for the number of probes assessed for replication (p = 0.05/554 tested probes = 9.02 × 10−5). Regarding the DMRs, a total of three out of the 54 DMRs previously associated with PFTs in Korean adults [11] showed Šidák-corrected p < 0.05 in Mexican Americans, but not in Puerto Ricans. Specifically, the DMR at the BHMT region associated with pre- and post-FEV1, the DMR at F2R was associated with pre- and post-FEV1/FVC, and the DMR at TACR3 was associated with post-FEV1/FVC. Additionally, a DMR at ZNF429 was associated with pre-FEV1, although the region limits differed from those reported previously for a DMR of FEV1/FVC [11]. However, one of the 37 DMRs from cord blood previously associated with childhood lung function in Europeans [18] was replicated in Puerto Ricans (Šidák-corrected p > 0.05).The DMR at this region was significantly associated with Pre- and Post-FVC and Post-FEV1.
Discussion
To our knowledge, this is the first EWAS of lung function in whole blood from Puerto Rican and Mexican American children and young adults with asthma. We identified five differentially methylated probes that showed genome-wide significant association with lung function, including one CpG site with evidence of being regulated by genetic variation. DNA methylation at these probes was genetically regulated according to the meQTL analysis. We also flagged two DMRs associated with lung function that were shared among both Puerto Ricans and Mexican Americans. Moreover, we validated five DMRs in Latinos (four were previously reported in Koreans and one in Europeans) and several CpG sites originally reported in European adults.
In the EWAS, we set a looser threshold for suggestive significance, with an FDR-adjusted p < 0.10, in order to identify potentially relevant sites for replication and established a stringent Bonferroni-corrected genome-wide significance threshold significance to control for false positives in the combined analysis of Mexican Americans and Puerto Ricans. This led to the identification of two CpG sites associated in Puerto Ricans that replicated in Mexican Americans and three CpGs associated in Mexican Americans that replicated in Puerto Ricans.
The two CpG sites discovered in Puerto Ricans were associated with post-FVC and annotated to genes that play a role in mucosal tissues. The probe cg16405908 was annotated to MRGPRE, which is expressed in whole-blood and lung [29]. Despite the fact that the role of MRGPRE is unknown, Mas-related G protein coupled receptors are involved in nociception homeostasis, bronchoconstriction, and airway hyperresponsiveness [30]. The other probe (cg07428101) was located in the intronic region of MUC2, a mucin expressed in the airway mucosa. In contrast to the genes encoding for other mucins (i.e., MUC5AC and MUC5B) located nearby MUC2, with an important role on mucus homeostasis and airway inflammation, the role of MUC2 is less known [31]. Still, it is plausible that DNA methylation levels at this CpG may exert regulatory effects over genes nearby. Another post-FVC-associated probe was cg00914963, annotated to TBC1D16, which encodes a RAB GTPase that promotes GTP hydrolysis by Rab4A, which in turn mediates VEGFR2 trafficking in endothelial cells and thereby regulates vascular permeability [32]. Interestingly, VEGFR2 is overexpressed in pulmonary tissue from patients with COPD and it is related to disease severity [29]. Indeed, among COPD patients, VEGFR2 expression in lung is negatively correlated with lung function [33].
The probe cg06035600 (MAP3K6) was associated with post-FEV1/FVC and has been previously related to aging and smoking [34]. However, the effect of the association was not modified by passive smoke exposure in our analyses. Reduced expression of MAP3K6 downregulates VEGF, which alters normal angiogenesis [35]. Moreover, MAP3K6 participates in cell signaling and subsequent regulation of gene expression [36].
The cg20515679 probe from KCNJ6, which was associated with post-FEV1/FVC, has also been associated with Crohn's disease and NO2 according to EWASatlas [34]. However, the association with NO2 described previously in an adult Dutch population-based cohort [37] was not replicated in our data. Moreover, KCNJ6 is also upregulated in mild and severe asthma in peripheral blood cells [38]. Although genetic variation in KCNJ6 has been associated with FVC [39], we did not find any significant SNP-CpG pairs in Latinos or Europeans.
The lack of replication of the genome-wide significant CpGs in European adults could be due to differences in age, population background, study design, and analytical methods. The association of reduced lung function and African ancestry among Puerto Ricans [14, 15] and the significant enrichment in ancestry-associated probes for several PFTs (Additional file 4: Fig. S4) suggests the existence of population-specific drivers among the Latino population. Nevertheless, several previously reported probes were associated at p < 0.05 in both subethnic groups.
The two DMRs consistently associated in Puerto Ricans and Mexican Americans were annotated to genes that were not known to be implicated in PFTs, but that are likely implicated in inflammatory or regulatory processes. AURKC encodes a serine/threonine kinase involved in histone phosphorylation and cell division, whose expression is induced by tumor necrosis factor-alpha in response to the inflammation-related transcription factor CEBPD [40]. Little is known about the protein encoded by the REXO1 gene, other than its participation in RNA polymerase II transcription via Elongin A [41] and its role promoting cervical cancer cell proliferation and progression [42].
It is worth noting that the association signals identified here were enriched in biologically relevant diseases and traits, including chronic inflammatory diseases and preterm birth. Interestingly, prematurity is inversely associated with airflow limitation in children [43] and adults [44]. However, despite DNAm may have a role in the interplay of gestational age and lung function in pediatric asthma, preterm birth did not explain the association of the identified CpG sites with PFTs.
Some limitations of this study must be acknowledged. First, the sample size of our study is modest in comparison to previous EWAS in individuals of European descent, especially to detect subethnic-specific in Mexican Americans and Puerto Ricans separately. Second, long-term longitudinal methylation changes could not be evaluated due to the cross-sectional design of GALA II. Third, since measured cell counts were not available, we used a reference-free method to adjust for effects of cell-type heterogeneity on DNAm patterns. Fourth, the fact that we analyzed the markers that were shared between two different arrays, one of them with a reduced number of markers, limited our genomic coverage. Despite these limitations, our study was strengthened by the fact that first, we focused on methylation profiling of whole blood from minority populations understudied in previous EWAS of PFTs. Second, we conducted both single-marker and region-based analyses and we evaluated whether genome-wide significant epigenetic loci were genetically regulated in Hispanics/Latinos with asthma. Third, we assessed several potential confounders on the association with epigenetic loci, including prematurity, BMI, in utero and current SHS exposure, medication use, air pollution exposure, and socioeconomic factors (insurance status and maternal education level).
Conclusions
In summary, we identified consistent DNA methylation patterns in whole blood associated with lung function in pediatric asthma among Mexican Americans and Puerto Ricans that may be population-specific for Latinos/Hispanics. Moreover, we replicated previous findings originally described in non-Latino/Hispanic populations. These results provide insights into the mechanisms involved in lung function.
Methods
Study participants
GALA II is a case–control study of pediatric asthma in Hispanics/Latinos that were recruited between 2006 and 2014 in five areas from the US (Chicago, Bronx, Houston, San Francisco Bay Area) and Puerto Rico (San Juan) [45]. Briefly, individuals were included if they were aged between 8 and 21 years old, self-identified as Hispanic or Latino, and had four Latino grandparents. Asthma was defined by physician diagnosis, use of controller or rescue medication, and report of two or more symptoms of coughing, wheezing, or shortness of breath. Exclusion criteria for the study included the following: (1) any smoking within one year of the recruitment date; (2) 10 or more pack-years of smoking; (3) pregnancy in the third trimester; (4) history of lung diseases other than chronic illness.
Pulmonary function tests
Pre- and post-bronchodilation spirometric data for FEV1, FVC, and FEV1/FVC were recorded with a KoKo® PFT Spirometer (nSpire Health Inc., Louisville, CO) according to the American Thoracic Society recommendations [46]. Post-bronchodilator PFT measurements were assessed 15 min after providing the participants a dose of albuterol, consisting of 4 puffs of albuterol (360 μg). Raw values were normalized as predicted percentages based on predicted values from the Global Lung Initiative 2012 (GLI-12) reference equation [47].
Methylation profiling and quality control
DNAm measurements were obtained from whole blood using the Infinium EPIC BeadChip or the Infinium HumanMethylation450 BeadChip array (Illumina, San Diego, CA, USA). Methylation profiling and quality control (QC) are detailed in Additional file 6. QC was performed with the ENmix (1.22.0) R package [48] (Additional file 5: Table S13). Low quality probes (beads < 3 or a detection p value > 1 × 10−6 for ≥ 5% of the samples) and samples with low quality data points for ≥ 5% of the CpG sites were removed along with samples with a total bisulfite intensity less than 3 standard deviations of the sample bisulfite control, outliers of the total bisulfite intensity or beta value distribution. We then performed background correction, dye-bias correction, inter-array normalization, and probe-type bias adjustment. Missing values were imputed after removal of samples with more than 10% of missing probes as well as probes with missing values in more than 5% of the samples. Samples with mismatched sex or mixed genotype distributions on the control SNP probes and probes on sex chromosomes were excluded. After QC, 427,079 probes that overlapped in both arrays were available for subsequent analysis.
Whole-genome sequencing
WGS was performed at the New York Genome Center and Northwest Genomics Center on an Illumina HiSeq X system. DNA processing, quality control, library construction, WGS, read processing, and sequence data quality control are described elsewhere [49]. Genotypes used in this study were based on TOPMed freeze 8 data with a minimal depth of 10 (DP10).
Statistical analysis
Genetic ancestry assessment was performed by a principal component (PC) analysis of genotype data, as detailed in Additional file 6. Cell-type heterogeneity was captured using ReFACTor [50] within the GLINT [51] v1.09 framework. The association of DNAm beta-values and raw PFT values (in liters) was tested by robust linear regressions with correction for age, sex, height, the first three genotype principal components (PCs), in utero maternal smoking exposure, the first six ReFACTor components, and batch, when appropriate, via limma R package [52]. Meta-analysis of fixed- or random-effects models, based on Cochran's Q p value, was conducted with METASOFT [53]. In silico replication of the significant probes in Europeans was performed using publicly available data [18].
Prior to downstream analysis, we removed previously reported cross-reactive probes, probes with a SNP at a single base extension (SBE) or at the CpG site with minor allele frequency (MAF) > 0.01, and multimodal probes.
Beta values, ranging from 0 to 1, were transformed to M-values as log2(β/(1 − β)) for downstream analysis. To assess the effects of genetic variation on DNA methylation, we conducted meQTL analysis using fastQTL [54]. Genetic variants located ± 500 kilobases of the probe site and with MAF ≥ 0.01 in at least 10 samples were considered. For each subethnic group and array, linear regression models were corrected for age, sex, genotype PCs, in utero maternal smoking exposure, ReFACTor components, and batch, when appropriate. Moreover blood meQTLs were assessed in the Biobank-based Integrative Omics Studies (BIOS) consortium data [19].
Additionally, we tested for enrichment in previous EWAS signals for the top 100 probes using EWAS toolkit [34] and enrichment in previous PFT signals in the full EWAS using Fisher’s exact test. DMRs were assessed using the uncorrected p values with comb-p [55]. Moreover, we assessed for replication CpG sites and DMRs previously associated with lung function. Further details are described in Additional file 6.
Availability of data and materials
Whole-genome sequencing is available at the database of Genotypes and Phenotypes (dbGaP) (Study Accession: phs000920.v1.p1, NHLBI TOPMed: Genes-environments and Admixture in Latino Americans Study). The summary statistics with the whole EWAS results are available at the Zenodo repository: 10.5281/zenodo.5236667. The scripts used for the analyses are available upon request to the corresponding author.
Abbreviations
- BMI:
-
Body Mass Index
- COPD:
-
Chronic obstructive pulmonary disease
- DMR:
-
Differentially methylated region
- DNAm:
-
DNA methylation
- EWAS:
-
Epigenome-wide association study
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- GALA II:
-
Study of Genes-Environment and Admixture in Latino Americans
- GLI- 12:
-
Global Lung Initiative 2012
- MAF:
-
Minor allele frequency
- meQTL:
-
Methylation quantitative trait loci
- NO2 :
-
Nitrogen dioxide
- O3 :
-
Ozone
- PCs:
-
Principal components
- PFT:
-
Pulmonary function test
- PM10 :
-
Particulate matter ≤ 10 µm in diameter
- PM2.5 :
-
Particulate matter ≤ 2.5 µm in diameter
- QC:
-
Quality control
- RNA:
-
Ribonucleic acid
- SHS:
-
Second-hand smoking
- SNP:
-
Single nucleotide polymorphism
- SO2 :
-
Sulfur dioxide
- US:
-
United States
- WGS:
-
Whole-genome sequencing
References
Global Initiative for Asthma. Global strategy for asthma management and prevention. 2021. https://ginasthma.org/. Accessed 21 September 2020.
Lange P, Parner J, Vestbo J, Schnohr P, Gensen G. A 15-year follow-up study of ventilatory function in adults with asthma. Pneumologie. 1999;53(1):24–5.
DeVries A, Vercelli D. Epigenetic mechanisms in asthma. In: Annals of the American Thoracic Society. American Thoracic Society; 2016. p. S48–50.
Sears MR, Greene JM, Willan AR, Wiecek EM, Taylor DR, Flannery EM, et al. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N Engl J Med. 2003;349(15):1414–22.
Moeller A, Carlsen KH, Sly PD, Baraldi E, Piacentini G, Pavord I, et al. Monitoring asthma in childhood: Lung function, bronchial responsiveness and inflammation. Eur Respir Rev. 2015;24(136):204–15.
Brand PLP, Mäkelä MJ, Szefler SJ, Frischer T, Price D, Baraldi E, et al. Monitoring asthma in childhood: symptoms, exacerbations and quality of life. Eur Respir Rev. 2015;24(136):187–93.
Agustí A, Noell G, Brugada J, Faner R. Lung function in early adulthood and health in later life: a transgenerational cohort analysis. Lancet Respir Med. 2017;5(12):935–45.
Hegewald MJ, Crapo RO. Socioeconomic status and lung function. Chest. 2007;132(5):1608–14.
Kurt OK, Zhang J, Pinkerton KE. Pulmonary health effects of air pollution. Curr Opin Pulm Med. 2016;22(2):138–43.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38.
Lee MK, Hong Y, Kim SY, Kim WJ, London SJ. Epigenome-wide association study of chronic obstructive pulmonary disease and lung function in Koreans. Epigenomics. 2017;9(7):971–84.
Bureau USC. Hispanics or Latino origin by race. 2019.
Bryc K, Durand EY, Macpherson JM, Reich D, Mountain JL. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am J Hum Genet. 2015;96(1):37–53.
Salari K, Choudhry S, Tang H, Naqvi M, Lind D, Avila PC, et al. Genetic admixture and asthma-related phenotypes in Mexican American and Puerto Rican asthmatics. Genet Epidemiol. 2005;29(1):76–86.
Pino-Yanes M, Thakur N, Gignoux CR, Galanter JM, Roth LA, Eng C, et al. Genetic ancestry influences asthma susceptibility and lung function among Latinos. J Allergy Clin Immunol. 2015;135(1):228–35.
Brehm JM, Acosta-Pérez E, Klei L, Roeder K, Barmada MM, Boutaoui N, et al. African ancestry and lung function in Puerto Rican children. J Allergy Clin Immunol. 2012;129(6):1484-1490.e6.
Rahmani E, Shenhav L, Schweiger R, Yousefi P, Huen K, Eskenazi B, et al. Genome-wide methylation data mirror ancestry information. Epigenetics Chromatin. 2017;10(1):1.
Imboden M, Wielscher M, Rezwan FI, Amaral AFS, Schaffner E, Jeong A, et al. Epigenome-wide association study of lung function level and its change. Eur Respir J. 2019;54(1):1900457.
Bonder MJ, Luijk R, Zhernakova DV, Moed M, Deelen P, Vermaat M, et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat Genet. 2017;49(1):131–8.
Pan-UKB team. Pan-UKB GWAS summary statistics. 2020. https://pan.ukbb.broadinstitute.org. Accessed 21 June 2021.
Qiu W, Baccarelli A, Carey VJ, Boutaoui N, Bacherman H, Klanderman B, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med. 2012;185(4):373–81.
Sunny SK, Zhang H, Rezwan FI, Relton CL, Henderson AJ, Merid SK, et al. Changes of DNA methylation are associated with changes in lung function during adolescence. Respir Res. 2020;21(1):80.
Wielscher M, Vierlinger K, Kegler U, Ziesche R, Gsur A, Weinhäusel A. Diagnostic performance of plasma DNA methylation profiles in lung cancer, pulmonary fibrosis and COPD. EBioMedicine. 2015;2(8):929–36.
de Vries M, Nedeljkovic I, van der Plaat DA, Zhernakova A, Lahousse L, Brusselle GG, et al. DNA methylation is associated with lung function in never smokers. Respir Res. 2019;20(1):268.
Nedeljkovic I, Lahousse L, Carnero-Montoro E, Faiz A, Vonk JM, de Jong K, et al. COPD GWAS variant at 19q13.2 in relation with DNA methylation and gene expression. Hum Mol Genet. 2018;27(2):396–405.
Carmona JJ, Barfield RT, Panni T, Nwanaji-Enwerem JC, Just AC, Hutchinson JN, et al. Metastable DNA methylation sites associated with longitudinal lung function decline and aging in humans: an epigenome-wide study in the NAS and KORA cohorts. Epigenetics. 2018;13(10–11):1039–55.
Bermingham ML, Walker RM, Marioni RE, Morris SW, Rawlik K, Zeng Y, et al. Identification of novel differentially methylated sites with potential as clinical predictors of impaired respiratory function and COPD. EBioMedicine. 2019;43:576–86.
de Vries M, van der Plaat DA, Nedeljkovic I, Verkaik-Schakel RN, Kooistra W, Amin N, et al. From blood to lung tissue: effect of cigarette smoke on DNA methylation and lung function. Respir Res. 2018;19(1):212.
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–30.
Han L, Limjunyawong N, Ru F, Li Z, Hall OJ, Steele H, et al. Mrgprs on vagal sensory neurons contribute to bronchoconstriction and airway hyper-responsiveness. Nat Neurosci. 2018;21(3):324–8.
Frey A, Lunding LP, Ehlers JC, Weckmann M, Zissler UM, Wegmann M. More than just a barrier: the immune functions of the airway epithelium in asthma pathogenesis. Front Immunol. 2020;11:761.
Kofler N, Corti F, Rivera-Molina F, Deng Y, Toomre D, Simons M. The Rab-effector protein RABEP2 regulates endosomal trafficking to mediate vascular endothelial growth factor receptor-2 (VEGFR2)-dependent signaling. J Biol Chem. 2018;293(13):4805–17.
Fu X, Zhang F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp Ther Med. 2018;16(6):4553–61.
Li M, Zou D, Li Z, Gao R, Sang J, Zhang Y, et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019;47(D1):D983–8.
Eto N, Miyagishi M, Inagi R, Fujita T, Nangaku M. Mitogen-activated protein 3 kinase 6 mediates angiogenic and tumorigenic effects via vascular endothelial growth factor expression. Am J Pathol. 2009;174(4):1553–63.
Ilinca A, Englund E, Samuelsson S, Truvé K, Kafantari E, Martinez-Majander N, et al. MAP3K6 mutations in a neurovascular disease causing stroke, cognitive impairment, and tremor. Neurol Genet. 2021;7(1):e548.
de F C Lichtenfels AJ, van der Plaat DA, de Jong K, van Diemen CC, Postma DS, Nedeljkovic I, et al. Long-term air pollution exposure, genome-wide DNA methylation and lung function in the lifelines cohort study. Environ Health Perspect. 2018;126(2):027004.
Alrashoudi RH, Crane IJ, Wilson HM, Al-Alwan M, Alajez NM. Gene expression data analysis identifies multiple deregulated pathways in patients with asthma. Biosci Rep. 2018;38(6):BSR20180548.
Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. 2019;104(1):65–75.
Wu S-R, Li C-F, Hung L-Y, Huang A-M, Tseng JT, Tsou J-H, et al. CCAAT/enhancer-binding protein delta mediates tumor necrosis factor alpha-induced Aurora kinase C transcription and promotes genomic instability. J Biol Chem. 2011;286(33):28662–70.
Ardehali MB, Damle M, Perea-Resa C, Blower MD, Kingston RE. Elongin A associates with actively transcribed genes and modulates enhancer RNA levels with limited impact on transcription elongation rate in vivo. J Biol Chem. 2021;296:100202.
Zhang Y, Li X, Zhang J, Mao L. Circ-CCDC66 upregulates REXO1 expression to aggravate cervical cancer progression via restraining miR-452-5p. Cancer Cell Int. 2021;21(1):20.
Den Dekker HT, Sonnenschein-Van Der Voort AMM, De Jongste JC, Anessi-Maesano I, Arshad SH, Barros H, et al. Early growth characteristics and the risk of reduced lung function and asthma: a meta-analysis of 25,000 children. J Allergy Clin Immunol. 2016;137(4):1026–35.
Näsänen-Gilmore P, Sipola-Leppänen M, Tikanmäki M, Matinolli H-M, Eriksson JG, Järvelin M-R, et al. Lung function in adults born preterm. PLOS ONE. 2018;13(10):e0205979.
Nishimura KK, Galanter JM, Roth LA, Oh SS, Thakur N, Nguyen EA, et al. Early-life air pollution and asthma risk in minority children. The GALA II and SAGE II studies. Am J Respir Crit Care Med. 2013;188(3):309–18.
Standardization of Spirometry, 1994 Update. American Thoracic Society. Am J Respir Crit Care Med. 1995;152(3):1107–36.
Lytras T. rspiro: Implementation of spirometry equations. R package version 2.0; 2020.
Xu Z, Niu L, Li L, Taylor JA. ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016;44(3):e20.
Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature. 2021;590(7845):290–9.
Rahmani E, Zaitlen N, Baran Y, Eng C, Hu D, Galanter J, et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13(5):443–5.
Rahmani E, Yedidim R, Shenhav L, Schweiger R, Weissbrod O, Zaitlen N, et al. GLINT: A user-friendly toolset for the analysis of high-throughput DNA-methylation array data. Bioinformatics. 2017;33(12):1870–2.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011;88(5):586–98.
Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics. 2016;32(10):1479–85.
Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28(22):2986–8.
Acknowledgements
We acknowledge the patients, families, recruiters, health care providers and community clinics for their participation. We thank Sandra Salazar for her support as GALA II study coordinator. The authors thank Soren Germer, Michael C. Zody, Lara Winterkorn and Catherine Reeves from NYGC and Deborah A. Nickerson from NWGC for overseeing the production of whole genome sequencing data for GALA II. We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. We also gratefully acknowledge the contributions of the investigators of the NHLBI TOPMed Consortium (https://www.nhlbiwgs.org/topmed--banner--authorship). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
Whole-genome sequencing data for the TOPMed (Trans-Omics in Precision Medicine) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Whole-genome sequencing for "NHLBI TOPMed: Gene-Environment, Admixture and Latino Asthmatics Study" (phs000920, GALA II) was performed were performed at the New York Genome Center (NYGC, 3R01HL117004-01S3) and Northwest Genomics Center (NWGC, HHSN268201600032I). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). WGS of part of GALA II was performed by New York Genome Center under The Centers for Common Disease Genomics of the Genome Sequencing Program (GSP) Grant (UM1 HG008901). The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. GSP is funded by the National Human Genome Research Institute, the National Heart, Lung, and Blood Institute, and the National Eye Institute. This work was supported in part by the Sandler Family Foundation, the American Asthma Foundation, the RWJF Amos Medical Faculty Development Program, Harry Wm. and Diana V. Hind Distinguished Professor in Pharmaceutical Sciences II, the National Heart, Lung, and Blood Institute (R01HL117004, R01HL128439, R01HL135156, X01HL134589), the National Institute of Health and Environmental Health Sciences (R01ES015794, R21ES24844), the National Institute on Minority Health and Health Disparities (R01MD010443, R56MD013312), and the Tobacco-Related Disease Research Program under Award Number (24RT-0025 and 27IR-0030). This was also funded by the Spanish Ministry of Science and Innovation MCIN/AEI/10.13039/50110001103 (grant PID2020-116274RB-I00) and by Instituto de Salud Carlos III, ISCIII (grant CB06/06/1088). MP-Y was funded by the Ramón y Cajal Program (RYC-2015–17205) by MCIN/AEI/10.13039/501100011033 and by the European Social Fund “ESF Investing in your future.” EH-L was supported by a fellowship awarded by MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future” (PRE2018-083837). JP-G was supported by a fellowship awarded by Spanish Ministry of Universities (FPU19/02175). KLK was additionally supported by a diversity supplement of NHLBI R01HL135156, the UCSF Bakar Computational Health Sciences Institute, the Gordon and Betty Moore Foundation grant GBMF3834, and the Alfred P. Sloan Foundation grant 2013–10-27 to UC Berkeley through the Moore-Sloan Data Sciences Environment initiative at the Berkeley Institute for Data Science (BIDS).
Author information
Authors and Affiliations
Contributions
EHL: Conceptualization (supporting), Data curation (equal), Formal analysis (lead), Methodology (equal), Writing original draft (lead), Writing review and editing (equal); AL: Data curation (equal), Methodology (supporting), Writing review and editing (equal); ACYM: Data curation (equal), Methodology (equal), Writing review and editing (equal); JPG: Data curation (equal), Methodology (equal), Writing original draft (supporting), Writing review and editing (equal); JRE: Data curation (supporting), Writing review and editing (equal); SSO: Investigation (supporting); LNB: Formal analysis (supporting), Methodology (supporting); DH: Data curation (equal); CE: Investigation (supporting); KBB: Investigation (supporting): KLK: Conceptualization (supporting), Methodology (supporting), Writing review and editing (supporting); SH: Data curation (equal), Methodology (supporting),JRS: Investigation (supporting); EGB: Conceptualization (supporting); Funding acquisition (lead); Supervision (supporting); MPY: Conceptualization (lead); Supervision (lead); Methodology (equal), Writing original draft (supporting), Writing review and editing (lead). All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
GALA II was approved by the Human Research Protection Program Institutional Review Board of the University of California, San Francisco (ethics approval numbers: 217802). Participants/parents provided written assent/consent, respectively.
Consent for publication
Not applicable.
Competing interests
MP-Y report grants from MCIN/AEI/10.13039/50110001103. MP-Y also reports grant support from GlaxoSmithKline, Spain through Fundación Canaria Instituto de Investigación Sanitaria de Canarias (FIISC) for a project outside the submitted work. MP-Y, EH-L and JP-G report funding from MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future.” EGB reports grants from the National Institutes of Health, the Tobacco-Related Disease Research Program, the Sandler Family Foundation, the American Asthma Foundation, the Amos Medical Faculty Development Program from the Robert Wood Johnson Foundation, and from the Harry Wm. and Diana V. Hind Distinguished Professorship in Pharmaceutical Sciences II. KLK reports grants from the National Institutes of Health, the Gordon and Betty Moore Foundation, and the Alfred P. Sloan Foundation. Additionally, KLK owns stock in Ambys Medicines.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1
. Manhattan-plot for the EWAS of lung function in Puerto Ricans. (A) pre-forced expiratory volume in 1 s (FEV1), (B) pre-forced vital capacity (FVC), (C) pre-FEV1/FVC ratio, (D) post-FEV1, (E) post-FVC, and (F) post-FEV1/FVC ratio. The statistical significance of association results (− log10 p value) is represented for each CpG site as a dot (y-axis) along the autosomal chromosomes (x-axis) from chromosome 1 (left) to chromosome 22 (right). The threshold for a false discovery rate less than 1% for each trait of lung function is indicated by the dashed gray line and the genome-wide threshold for significance is represented by the continuous gray line.
Additional file 2: Fig. S2
. Manhattan-plot for the EWAS of lung function in Mexican Americans. (A) pre-forced expiratory volume in 1 s (FEV1), (B) pre-forced vital capacity (FVC), (C) pre-FEV1/FVC ratio, (D) post-FEV1, (E) post-FVC, and (F) post-FEV1/FVC ratio. The statistical significance of association results (− log10 p value) is represented for each CpG site as a dot (y-axis) along the autosomal chromosomes (x-axis) from chromosome 1 (left) to chromosome 22 (right). The threshold for a false discovery rate less than 1% for each trait of lung function is indicated by the dashed gray line and the genome-wide threshold for significance is represented by the continuous gray line.
Additional file 3: Fig. S3
. Quantile–quantile plot for the EWAS of the association between each specific lung function measurement and DNA methylation in Mexican Americans (MEX) and Puerto Ricans (PR). For the EWAS in Mexican Americans, inflation factors were 1.02, 1.13, 1.24, 1.03, 1.12 and 1.24 for pre-forced expiratory volume in 1 s (FEV1), pre-forced vital capacity (FVC) and their pre-ratio (FEV1/FVC) and post-FEV1, post-FVC and post-FEV1/FVC, respectively. For the EWAS in Puerto Ricans, inflation factors were 1.19, 1.17, 1.09, 1.22, 1.21, 1.20 for pre-FEV1, pre-FVC, pre-FEV1/FVC, post-FEV1, post-FVC and post-FEV1/FVC, respectively. The observed p value (− log10 p value) is shown in the y-axis along the expected p value (− log10 p value) represented in the x-axis.
Additional file 4: Fig. S4
. Heatmap of the trait enrichment for the top 100 CpGs for each pulmonary function test and ethnicity. Significant results at an FDR < 0.05 for forced expiratory volume in 1 s (Pre-FEV1), forced vital capacity (FVC) and their ratio (FF) pre- and post-administration of albuterol are shown for Puerto Ricans (PR) and Mexican Americans (MX). The enrichment raw p value is colored on a scale from blue (less significant association) to red (more significant association). Non-significant p values are represented in gray.
Additional file 5.
Supplementary Tables S1-S13.
Additional file 6.
Supplementary Material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Herrera-Luis, E., Li, A., Mak, A.C.Y. et al. Epigenome-wide association study of lung function in Latino children and youth with asthma. Clin Epigenet 14, 9 (2022). https://doi.org/10.1186/s13148-022-01227-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-022-01227-5