
Abstract
Duchenne muscular dystrophy (DMD) is a multisystemic disorder that affects 1:5000 boys. The severity of the phenotype varies dependent on the mutation site in the DMD gene and the resultant dystrophin expression profile. In skeletal muscle, dystrophin loss is associated with the disintegration of myofibers and their ineffective regeneration due to defective expansion and differentiation of the muscle stem cell pool. Some of these phenotypic alterations stem from the dystrophin absence-mediated serine–threonine protein kinase 2 (MARK2) misplacement/downregulation in activated muscle stem (satellite) cells and neuronal nitric oxide synthase loss in cells committed to myogenesis. Here, we trace changes in DNA methylation, histone modifications, and expression of regulatory noncoding RNAs during muscle regeneration, from the stage of satellite cells to myofibers. Furthermore, we describe the abrogation of these epigenetic regulatory processes due to changes in signal transduction in DMD and point to therapeutic treatments increasing the regenerative potential of diseased muscles based on this acquired knowledge.

Similar content being viewed by others

Introduction
Duchenne muscular dystrophy (DMD, OMIM 310200), the most severe and the most common adult form of muscular dystrophy in humans, is caused by a lack of functional dystrophin due to mutations in the dystrophin gene (DMD) [1, 2]. This largest gene in the human genome (> 2.5 Mbp) gives rise to several transcripts that encode protein isoforms ranging in size from 40 to 427 kDa, which are variously distributed in many cell types [3,4,5,6,7,8,9,10,11]. Because DMD is located on the X chromosome (Xp21.2 region), the disease primarily affects boys with estimates of incidence ranging from one in every 3500 to more recent estimates of 1:5000 live births [12,13,14]. Interestingly, females also suffer from DMD in rare instances (1:50,000,000 live births) [15] and approximately 8% of female mutation carriers experience muscle weakness [16]. The genetic defects in the DMD gene include deletions (65%), duplications (5–10%), and point mutations (10–15%) [17,18,19].
The first sign of the disease is muscle weakness, which starts in boys at around the age of four and progresses quickly (Fig. 1a). It is usually accompanied by a loss of muscle contraction in the thigh and pelvis muscles followed by arm muscle weakness. At this point, the classic DMD symptoms begin to emerge, i.e., Gowers’ sign (patients use their hands and arms to ‘walk-up’ their body to stand from a sitting or squatting position), rocking gait, or walking on toes. In the second phase of the disease progression, movement causes increasing problems as patients experience difficulty walking, climbing stairs, or getting up from the floor. At this stage, cooperation with physiotherapists is needed, but also with a psychologist who will prepare the child for loss of mobility. As a result of the disease progression, the function of the lower extremities deteriorates and it becomes more difficult for the patient to maintain the correct body position. With time, the affected boys are forced to use a wheelchair. The consequence of the fragility and progressive loss of DMD myofibers is the accumulation of fibrotic and adipose tissue, thus contributing to skeletal muscle mass loss and function [20]. Ultimately, DMD leads to premature death in the twenties or thirties due to diaphragm dysfunction or cardiac failure [21, 22].

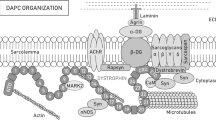
DMD—the disease of satellite cells and myofibers. a A timeline showing the progression of DMD symptoms. The affected boys develop motor skills until the age of 4–6, however, at a lower rate than their peers. Muscle weakness and Gowers’ sign are apparent from the age of 4. The condition of the muscles deteriorates quickly and the patients are forced to use a wheelchair in their teens. Typically, in the late teens, they need to start to use a temporary and then 24-h ventilation aid as a consequence of dysfunctional respiratory muscles. The boys die usually in their twenties/thirties, due to respiratory or cardiac failure. b In response to damage, QSCs (marked sky blue) that reside between the basal lamina and the plasmalemma, are activated and divide asymmetrically to generate SCs that return to the quiescent state (marked orange) and SCs undergoing differentiation into myoblasts that participate in muscle repair (marked pink). The asymmetric division is driven by dystrophin in combination with its binding partner, MARK2 (see in c). Lack of dystrophin leads to diminished levels of MARK2 and β-syntrophin in satellite cells (and α-syntrophin in skeletal muscle and NMJs), lower amounts of asymmetric divisions, and an increase in abnormal mitotic divisions. Also, note the elevated numbers of satellite cells in DMD muscles that are generated through symmetric divisions as well as increased fibrosis (marked red) and infiltration of immune cells (marked yellow). c DGC in the plasmalemma of satellite cells and myofibers performs structural and signal transduction functions, including those that pertain to NO production. In DMD muscles, the loss of dystrophin results in partial compensatory assembly of the utrophin-based complex (UGC) as well as other proteins and protein complexes (not shown). In neither case, the correct signal transduction functions are restored
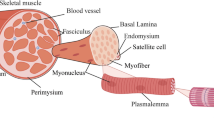
The skeletal muscle has the intrinsic ability to regenerate damaged myofibers after injury or as a consequence of a disease process. High regenerative capacity is directly linked to the presence of satellite cells (SCs) (Fig. 1b) [23], which are undifferentiated skeletal muscle precursor cells residing in a niche between the muscle fiber membrane (sarcolemma) and the basement membrane surrounding each muscle fiber [24,25,26,27]. In principle, these cells are required not only for myofiber regeneration but also for the growth and maintenance of skeletal muscle. SCs are normally mitotically quiescent (QSCs) but are poised to act and enter the cell cycle in response to stress stimuli such as injury [28]. Activated satellite cells (ASCs) undergo asymmetric division, myogenic differentiation, and self-renewal to restore the pool of QSCs (Fig. 1b, left panel). Defects in SCs have been shown to contribute to the etiology of some muscle diseases [29]. Specifically, DMD progression has been linked to a failure of SCs to divide asymmetrically and maintain the damage-repair cycle (Fig. 1b, right panel). This is due to the fact that in the dystrophin–glycoprotein complex (DGC), dystrophin is associated with the serine–threonine protein kinase 2 (MARK2), which plays a pivotal role in establishing cell polarity (Fig. 1c). Moreover, over time, the SC pool in diseased tissues undergoes exhaustion [30, 31] and cannot replenish damaged myofibers as underscored by the progressive loss of muscle mass in DMD patients. Inflammatory processes downstream of dystrophin deficiency, as well as metabolic abnormalities and defective autophagy, additionally contribute to muscle pathology in DMD. Particularly, chronic inflammation caused by muscle damage in DMD patients has an important impact on disease progression [32, 33].
The structural role of dystrophin is closely related to its participation in the DGC, which can be subdivided into three smaller subcomplexes: the dystroglycan complex (α- and β-dystroglycan, generated by proteolytic cleavage of a single precursor protein); the sarcoglycan complex (α-, β-, δ-, γ-sarcoglycan); and the complex located in the cytoplasm (Fig. 1c) [34]. The generally accepted role for the DGC is to stabilize the plasma membrane during muscle contraction, signified by the fact that disruption of the linkage between the cytoskeleton and the extracellular matrix (ECM) occurs in DMD [35,36,37]. Furthermore, the DGC also plays a pivotal role in the organization of neuromuscular junctions, where it stabilizes the postsynaptic machinery, including receptors for the neurotransmitter acetylcholine [38, 39]. However, the role of dystrophin is not limited to the structural function. Growing evidence suggests that dystrophin, through its multiple protein connections, plays a major role in gene expression via regulating signal transduction (Fig. 1c), including pathways that activate nitric oxide (NO) production, Ca2+ entry, and the production of reactive oxygen species (ROS) [40].
Despite the undoubted progress in the development of experimental therapeutic approaches, DMD is still incurable. Symptomatic treatment of the progressive loss of muscle tissue, caused by myofiber degeneration and their inefficient regeneration, is currently limited to corticosteroids that alleviate secondary inflammatory processes in DMD. In recent years, it has become more evident that epigenetic mechanisms such as DNA methylation or histone modification have a pivotal role in regulating muscle regeneration and regenerative medicine is providing novel therapeutic strategies by developing epigenetic drugs aimed to manipulate the chromatin targets of individual signaling pathways. In this review, we discuss the epigenetic regulation of various stages of skeletal muscle regeneration and the interplay of numerous factors that define the specific state of epigenetic homeostasis in health and DMD.
Epigenetics: the basics
The term epigenetics was first used by a British embryologist, Conrad H. Waddington, in 1942 [41]. At that time, epigenesis referred to the differentiation of cells from their initial totipotent state during embryonic development. Today, the term epigenetics indicates a field of science that studies the relationship between the genetic code and the living environment—mental and physical and, more specifically, that describes the mechanisms and effects of biochemical modification of genome expression without changing the DNA sequence [42]. Genome expression can be modified by environmental factors, lifestyle, upbringing, or emotions, and to some extent, these modifications can be inherited. At any stage of human life, from conception to death, intracellular stimuli and environmental factors such as nutrition, physical activity, environment pollution, stress, and bacterial infections also regulate the expression of our genes [43, 44].
In general, the major epigenetic signals include modifications related to (1) covalent posttranslational reversible modifications of histone proteins, such as methylation, acetylation, phosphorylation, ubiquitination, or incorporation of histone variants (Fig. 2a), (2) DNA methylation and demethylation (Fig. 2b), and (3) gene regulation by noncoding RNAs (ncRNAs), (Fig. 3) [45]. It is important to realize that epigenetic changes occur naturally in normal development and health as well as in aging and disease. Below we describe the selected epigenetic mechanisms.

Basic principles of epigenetic modifications. a Epigenetic mechanisms regulating gene expression via histone modifications. Chromatin transcription permissive histone modifications such as H3K9ac/H4K4ac (acetylation) and H3K4me3/H4K4me3 (methylation) are catalyzed by HATs and HMTs, while repressive, including H3K27me3, H3K9me2/3, H4K20me2/3 (methylation), and deacetylation, through the action of HMTs and HDACs. Histone modifications and enzymes catalyzing the corresponding reactions are marked with matching colors. b Epigenetic mechanisms regulating gene expression via DNA modifications. Methylation and demethylation of CpG islands in the promoter regions of genes cause gene silencing and transcriptional activation, respectively. DNMTs and the process of CpG demethylation are, respectively, color-matched to methylated and demethylated CpGs

Noncoding RNAs in chromatin modifications. a miRNAs induce degradation of target mRNAs containing matching sequences via the RNA–protein complex. b, c lncRNAs can recruit or inhibit various enzymes or protein complexes to induce transcription repressive and permissive DNA (b) or histone modifications (c)
Histone modifications
Chromatin activity is regulated by chromatin-modifying multiprotein complexes, whose catalytic subunits induce reversible posttranslational histone modifications such as acetylation, methylation, phosphorylation, or ubiquitination and are associated with permissive and repressive chromatin states [46,47,48,49,50,51]. Among them, histone acetylation and methylation are the most common mechanisms in myogenesis regulation, while phosphorylation and ubiquitination occur less frequently. In general, acetylation of lysine residues of histones H3 and H4 (H3K9ac, H4K4ac) and trimethylation of lysine 4 of histones H3 or H4 (H3K4me3, H4K4me3) are associated with activation of transcription (permissive chromatin), whereas trimethylation of lysine 27 of histone H3 (H3K27me3), di-/trimethylation of lysine 9 of histone H3 (H3K9me2/3), and di-/trimethylation of lysine 20 of histone H4 (H4K20me2/3) cause gene repression by chromatin condensation (Fig. 2a) [52].
Histone acetylation is dynamically regulated by the antagonistic action of histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Fig. 2a) that operate as catalytic centers of multiprotein assemblies. HATs are classified based on their cellular localization in the nucleus (type A) and the cytoplasm (type B). While not much is known about cytoplasmic HATs, several nuclear HATs have been identified and further divided into three major families that differ in their primary structure homology, represented by (1) p300/CBP-associated factor (PCAF), (2) the p300/CBP family, including p300 and cAMP response element-binding (CREB) protein (CBP), and (3) the MYST family [53, 54]. Generally, histone acetylation is achieved by the addition of an acetyl group (–CH3CO) transferred from acetylcoenzyme to one or more lysine residues in the ϵ-amino group of histones, resulting in the relaxation of chromosomal DNA [55]. Conversely, mammalian HDAC enzymes remove the acetyl group from histone proteins, causing chromosomal DNA to be less accessible to transcription factors [56, 57]. There are currently 18 known human HDACs grouped into four classes. Classes I, II, and IV are zinc-dependent HDACs, while class III (also called Sirtuins, SIRT) comprises HDACs that require NAD+ [58]. Interestingly, class I and IV HDACs are predominantly sublocalized in the nucleus, whereas class II HDACs shuttle between the nucleus and the cytoplasm [59]. Similar to class II, class III HDACs can occur in the nucleus and cytoplasm; however, enzymes from this class can also act in the mitochondria [60].
Histone methylation occurs on lysine and arginine residues and is generated by the activity of histone lysine methyltransferases (HKMTs), (Fig. 2a), and protein arginine methyltransferases (PRMTs). The first group includes, among others, the following families: suppressor of variegation 3–9 (SUV39), e.g., G9a methyltransferase that methylates lysine 9 of histone H3 (H3K9); enhancer of zeste homolog (EZH); SET1, which includes lysine methyltransferase 2A (MLL); SET2; SET7 and suppressor of variegation 4–20 (SUV4-20). The group of arginine methyltransferases comprises of ten mammalian PRMTs (PRMT1-10) that have been identified to date [61]. Here, PRMT4 or coactivator-associated arginine methyltransferase 1 (CARM1) was the first PRMT characterized as an activator of transcription by methylating histone H3 [62]. Conversely, the methyl groups from histones are removed by the action of two classes of histone demethylases (HDMs; not shown) [63], a lysine-specific histone demethylase 1 (LSD1), which has mono- and di-demethylating histone H3 activity (H3K4, H3K9) and the Jumonji C (JmjC) domain-containing family of HDMs, which unlike LSD1, is capable of removal of trimethylation [64].
Functional protein complexes are often required for HMTs to exert their catalytic activities, for example polycomb repressive complex 2 (PRC2) targets H3K27me3 addition to developmentally regulated genes. In humans, PRC2 consists of four core subunits required for its optimal functioning: EZH1 or EZH2, suppressor of zeste 12 (SUZ12), embryonic ectoderm development (EED), and retinoblastoma suppressor-associated proteins 46/48 (RbAp46/48) (Fig. 4). EZH1 and EZH2 are Su(var)3–9, enhancer of zeste, and trithorax catalytic (SET) domain-containing proteins harboring HKMT activity, while SUZ12 and EED are involved in the PRC2 stability and are required for the EZH1/2 catalytic activities [65, 66]. In turn, RbAp46/48 that constitute the fourth core subunit of PRC2 are histone chaperones that play a pivotal role in establishing and maintaining the chromatin structure and are not required for the enzymatic activity of EZH [67, 68]. Moreover, protein complexes that catalyze the repressive state of chromatin often cooperate. Boros et al., for example, proposed a model, in which H3K27me3-bound PRC2 stabilizes H3K9me3-anchored heterochromatin protein 1α (HP1α), a structural adapter necessary to form and maintain a condensed structure of heterochromatin in two ways: directly by interaction with SUZ12 or indirectly through an unknown factor [69], as was recently suggested by Canzio et al. [70]. In contrast, the main complex involved in permissive chromatin changes, the trithorax group protein (TrxG), is most commonly linked to gene activation by inducing H3K4me3 due to MLL1/2 HKMT activity (Fig. 4) [71].

Epigenetic regulation of the quiescent and proliferative state of SCs. a In QSC and during self-renewal, the PAX7 promoter is active, holding permissive chromatin marks through the TrxG activity, while MYF5/MYOD expression is repressed despite containing both permissive and repressive marks, including H3K4me3 (TrxG) and H3K27me3 (PRC2, YY1-EZH2 complex), respectively. Repression of MYF5 and MYOD is also induced by SUV39H1 (H3K9me2/3) and by removal of acetylation marks carried by HDACs. Furthermore, note that MYOD expression is repressed by ID, SIR2 as well as MEF2 and SUV4-20H1/H2 (H4K20me2/3). The MYOG gene expression is inactivated via the cooperative action of PRC2 and HDACs. b In proliferating myoblasts, CARM1 targets and methylates PAX7 protein, which facilitates recruitment of TrxG to the MYF5 promoter. In addition, PRC2 and HDACs are removed from the MYOD promoter, which enables binding of TrxG, PCAF-p300/CBP, SRF and phosphorylated MEF2 as well as the SWI/SNF complex that induces chromatin relaxation and MYOD transcription. Also note that at this stage of myogenesis, phosphorylated MYOD as well as SUV39H1 halt expression of MYOG in addition to PRC2 and HDACs
Changes in chromatin conformation require energy, which is obtained during the ATP hydrolysis reaction. In the remodeling process that activates transcription, the DNA-histone contact is loosened, allowing the nucleosomes to move along a specific DNA sequence [72, 73]. An important multisubunit enzymatic complex involved in this process is the SWItch/sucrose non-fermentable (SWI/SNF) (Figs. 4b and 5), which in humans consists of complexes that contain either Brahma (BRM) or Brahma-related gene 1 (BRG1) ATPases, associated with BRG1-associated factors (BAFs) [74]. The function of the SWI/SNF complex is to form the RNA polymerase II preinitiation complex that promotes transcriptional elongation. In detail, the ATPase subunits contain bromodomains that can recognize acetylated lysine on histone proteins and are responsible for nucleosome remodeling [75]. SWI/SNF enzymes can also physically interact with HATs and HDACs, showing the potential for coordination of chromatin remodeling activities [76].

Epigenetic regulation in differentiating myogenic cells. a In the initial stage of differentiation, PAX7 and MYF5 expression is inhibited via H3K27me3 catalyzed by PRC2 (YY1-EZH2 complex) and additionally, other unknown factors (inhibition of MYF5). Conversely, MYOD gains active transcription marks (H3K4me3, acetylation) through the combined action of TrxG, MEF2, PCAF-p300/CBP, SRF, as well as the SWI/SNF complex. The high level of MYOD contributes to an increase in the production of MYOG and via the action of KDM4A, TrxG, phosphorylated MEF2, UTX, SET7 and also by SWI/SNF complex. Additionally, EZH1 must be present on the MYOG promoter to enable its transcription. The combined action of MYOD and MYOG leads to the expression of genes characteristic for late differentiation, such as MRF4 (b), then levels of MYOD and MYOG decrease in response to the G9a HKMT and the PRC2-YY1 complex, respectively, and due to other unknown factors. c In the terminal differentiation, the level of MRF4 remains high and proteins characteristic for mature skeletal muscle such as MYHC, CKM, and ACNT1 are generated
DNA methylation
DNA methylation is a heritable yet reversible epigenetic modification and increasing evidence shows that methylated DNA is an important regulator in many biological processes, including X-chromosome inactivation, genomic imprinting, and gene expression [77,78,79,80,81,82], signified by the fact that aberrant DNA methylation patterns are often observed in many diseases [81]. In mammalians, the most often methylated nucleotide is cytosine (5-methylcytosine) in cytosine-guanine (CpG) adjacent sites (Fig. 2b) and also adenine at the 6-nitrogen position of the purine ring in the symmetric tetranucleotide motif 5′-G-A-T-C-3′ [82]. In adult mammals, palindromic CpGs of both DNA strands are methylated at the level of 3.5–4.5% in a cell- and tissue-dependent manner [83, 84]. Interestingly, about 70% of promoters within the human genome contain CpG sequences and their methylation is related to gene expression silencing (Fig. 2b). Additionally, asymmetric methylation at non-GpG sites is also observed [85,86,87].
The methyl mark is deposited by DNA methyltransferases (DNMTs), enzymes that transfer a methyl group from S-adenosyl-L-methionine (SAM) to the cytosine at the 5′ position (Fig. 2b) [88]. To date, three active methyltransferases have been identified in humans: DNMT1, DNMT3A, and DNMT3B. All of them require accessory proteins for their biological function, such as ubiquitin-like containing plant homeodomain (PHD) and ubiquitin-like with PHD and ring finger domain 1 (UHRF1) or DNMT3-like (DNMT3L) [89]. Interestingly, the previously considered methyltransferase, DNMT2, turned out not to methylate DNA, but instead position 38 in aspartic acid tRNA [90]. Thus, to better reflect its biological function, its name has been changed from DNMT2 to TRDMT1 (tRNA aspartic acid methyltransferase 1) [91]. DNMT3L was also previously considered as a DNA methylating enzyme, related to DNMT3A and DNMT3B in structure; however, unlike the other DNMTs, it does not possess any inherent enzymatic activity, despite its critical role in the DNA methylation process [92]. Human DNMT1 has a high preference for hemimethylated DNA and thus is called maintenance DNA methyltransferase, while DNMT3A and DNMT3B can place methylation marks on previously unmethylated CpGs and thus are mainly responsible for the de novo DNA methylation [93, 94]. Importantly, a relation between DNA methylation and the histone-modifying machinery was observed. Specifically, methyl-CpG-binding domain (MBD) proteins, which are necessary to recognize methylated CpGs, recruit HDACs and HMTs (e.g., PRC2) to methylated DNA regions [95, 96].
Gene regulation by noncoding RNA
The expression of genetic information may be affected by a specific group of regulatory molecules, so-called regulatory noncoding RNAs (rncRNAs) (Fig. 3) [97]. As the name indicates, rncRNAs do not encode proteins; however, often control gene transcription and can also directly influence the structural properties of chromatin via alterations in DNA methylation and histone modification [98]. Importantly, their functional roles are signified by the fact that their expression is strictly controlled and depends on the developmental stage as well as the differentiation level of the cell [99, 100]. The classification of rncRNA molecules is based on their size, and thus, a distinction can be made between small noncoding RNAs (sncRNAs; < 200 nt) and long noncoding RNAs (lncRNAs; > 200 nt). sncRNAs include microRNAs (miRNAs), piwiRNAs (piRNAs), or short interfering (siRNAs) [101, 102]. In particular, miRNAs (18–25 nt long) are highly conserved molecules across species that act as negative regulators of about 60% of mRNAs through their degradation (Fig. 3a) [103,104,105,106].
miRNAs can be viewed as a part of a larger genome expression feedback loop as they target the expression of key epigenetic enzymes such as DNMTs, HDACs, or EZHs [107, 108] and, on the contrary, the expression of miRNAs is regulated by the epigenetic machinery, such as DNA methylation, RNA, and histone modifications [109]. In contrast to miRNAs, piRNAs are involved in de novo DNA methylation [110], while siRNAs are necessary for the RNA-induced transcriptional silencing (RITS) ribonucleoprotein complex to be located in a specific region of chromatin, which leads to the formation of heterochromatin by cytosine and H3K9 methylation [111].
The most accepted categories of lncRNAs are sense and antisense, transcribed on the same or opposite strand of a gene; intronic; intragenic arising from an intron of a protein-coding gene or a region located between two protein-coding genes, and circular RNAs (circRNAs) [112]. Based on their location, lncRNAs can be also distinguished as nuclear or cytoplasmic [113]. In the nucleus, lncRNAs play a crucial role as modifiers of chromatin where they are involved in the spatial localization of DNA-associated proteins to genomic loci, positioning of nucleosomes, and formation of chromatin loops [114]. For instance, direct physical associations between lncRNAs and DNMTs have been attributed to gene expression inhibition [115, 116] or transcription enhancement (Fig. 3b) [117]. Furthermore, lncRNAs can also affect the chromatin structure through the interaction with chromatin-modifying complexes catalyzing repressive H3K27me3 or permissive H3K4me3 marks (Fig. 3c).
Myogenesis and epigenetic regulation of muscle gene expression
Myogenesis is the formation of muscle tissue, either during embryonic development or in response to myofiber damage that is observed in DMD patients (Fig. 1b) [118]. In the latter case, the myogenic process can be distinguished into three different stages: (1) an inflammatory process involving macrophages, (2) activation and division of SCs, and (3) formation and development of new muscle fibers [119]. In the first stage, leukocytes, neutrophils, and then macrophages, which play the most important role in the initial phase of regeneration, start to accumulate at the site of the damage. There are two subpopulations of macrophages, M1 (pro-inflammatory) and M2 (anti-inflammatory) [120]. In the M1 group, the cells show expression of neural cell adhesion molecule 1 (CD65) protein, secrete pro-inflammatory cytokines such as tumor necrosis factor alfa (TNF-α) and interleukin 1 beta (IL-1β), and are responsible for the removal of damaged fiber fragments during phagocytosis. Macrophages of the M2 group show expression of CD163 protein and secrete anti-inflammatory cytokines, interleukin 10 (IL-10), among others, which inhibits further development of the inflammatory process [121]. Additionally, macrophages from this group stimulate the activation, proliferation, and division of SCs, which initiates the next stage of muscle fiber regeneration [122]. Generally, on the second day after damage, QSCs defined by the expression of transcription factor paired box 7 (PAX7) become activated, start to multiply, and then divide and differentiate to create new skeletal muscle fibers. Interestingly, SCs proliferation and migration to the regeneration site have been observed along the entire fibers of injured muscle [123].
It is noteworthy that in the regenerating muscle, the largest pool of SCs consists of cells that express PAX7 and myogenic factor 5 (MYF5) (PAX7+/MYF5+). These cells divide, differentiate into myocytes, and either generate new fibers de novo or fuse and repair the damaged fiber (Fig. 1b). However, there is also a population of SCs that expresses PAX7 but is MYF5 negative (PAX7+/MYF5−). This type of cell undergoes symmetric and, upon activation, asymmetric division (Fig. 1b). In the latter case, the division results in two daughter cells expressing either PAX7+/MYF5+ (progenitor cells capable of the differentiation process; Fig. 4b) and cells expressing PAX7 without MYF5 (PAX7+/MYF5−; Fig. 4a). PAX7+/MYF5- cells retain undifferentiated stem cell properties and are responsible for renewing the population of QSCs [124].
Epigenetic regulation of the quiescent state of SCs
SCs maintain and regenerate the damaged skeletal muscle tissue. Initially, it was thought that sustaining SCs at rest was the result of a lack of nutrients or extracellular signals that could induce cell proliferation. This dogma has changed radically as it was noted that quiescence is an active and reversible state, controlled by specific cellular epigenetic mechanisms [125]. The chromatin permissive and repressive epigenetic processes acting in SCs have been defined in the context of the expression (or lack of it) of specific transcription factors, some of which include myogenic regulatory factors (MRFs). MRFs are not expressed in QSCs but occur in an orderly and coordinated manner during the myogenesis process in ASCs, proliferating myoblasts and in mature muscle cells that fuse to form regenerated skeletal muscle fibers [126].
DNA methylation
DNA methylation has classically been postulated as one of the major repressive systems acting on the muscle gene loci. A recently published dataset of the whole transcriptome from QSCs and proliferating SCs showed downregulation of Dnmt3a (de novo DNMT) during activation of SCs, while the expression of another methyltransferase, Dnmt1 (maintenance DNMT), was increased [127]. These observations suggest that specific DNMTs play some role in initiating the MRF transcriptional program or in regulating the transition from SC quiescence and proliferation to differentiation. However, the precise mechanisms regulating methylation/demethylation are still elusive.
Histone modifications
In QSCs, the transcription factor PAX7 is expressed due to TrxG activity composed of MLL1/2, ASH2L, WDR5, and RBBP5 subunits [128], while modulators of cell cycle progression and other transcription factors responsible for myogenic differentiation, such as myogenic differentiation 1 (MYOD) and myogenin (MYOG), remain silenced (Fig. 4a) [129]. However, it was concluded that QSCs are not in a dormant state but rather are primed for activation and thus differentiation in response to external stimuli [130], such as muscle damage observed in DMD patients. At the chromatin level, it was shown that in QSCs permissive H3K4me3 marks can be found not only in actively transcribed but also in inactive genes, to be transcribed at a later time [130]. Particularly, a large number of genes, including MYF5, MYOD, and serum response factor (SRF, a negative transcription regulator of SC differentiation), were found to have opposing H3K4me3 and H3K27me3 marks at the transcription start sites due to the respective TrxG and Ying-Yang 1 (YY1), which recruits the EZH2 subunit (putatively the entire PRC2 complex), activities (Fig. 4a) [130, 131]. It is important to note that not all factors are regulated in the same manner as, e.g., muscle-specific regulatory factor 4 (Mrf4) promoter is devoid of active H3K4me3 or repressive H3K27me3 marks [132]. Moreover, somewhat undermining the role of epigenetic regulation, others noted Myf5 transcripts already in QSCs; however, sequestered inside the cytoplasmic messenger ribonucleoprotein granules as a result of miR-31 expression and phosphorylation of eIF2 [133, 134]. Following SC activation, the granules are disassembled, leading to Myf5 protein synthesis [133]. Similarly, MyoD transcripts were shown to be blocked in QSCs by Staufen 1, a regulator of mRNA localization, stability, and translation [135]. Loss of this repression enables MyoD translation and its accumulation that triggers the myogenic program [135].
MYOG expression in QSCs is halted by the PRC2 and YY1 complex and HDACs (Fig. 4a), which interact with chromatin through association with transcription factors and methyl MBD proteins [131, 136,137,138,139]. In contrast, MYF5 repression and MYOD repression occur not only through the PRC2 complex and removal of acetylation marks carried by HDACs, but it is also induced by the SUV39H1-driven addition of H3K9me2/3. The expression of MYOD is furthermore negatively regulated by a complex array of proteins that contains: an inhibitor of differentiation (ID), the NAD-dependent histone deacetylase activity of Sirtuin 2 (Sir2) [140], a histone acetyltransferase PCAF, as well as the myocyte enhancer factor-2 (MEF2), which also plays a role in recruiting HDACs, and Suv4-20h1/h2 (Fig. 4a) [141, 142]. Importantly, SCs deficient in Suv4-20h1/h2 and its associated H4K20me2/3, have a strong reduction of heterochromatin, which leads to the abnormal regulation of Myod expression, premature activation of SCs, and impaired long-term skeletal muscle regeneration [143].
Interestingly, the total levels of PRC2, EZH1, and EZH2 subunits differ during myogenesis [137, 144]. Studies in mice have shown that inactivation of the Ezh2 subunit in SCs results in a lower number of these cells and diminished regenerative potential following muscle-induced injury, attributed to the failure of SCs to proliferate and self-renew [136, 145]. In contrast to the well-defined functions of EZH2, the role of EZH1 is still unclear in QSCs [137].
Noncoding RNAs
miRNAs also maintain the specific epigenetic state of chromatin, necessary to keep SCs in quiescence. Castel et al. [145] reported a massive downregulation of miRNA expression during activation of QSCs isolated from mice. Similarly, others indicated that miRNAs have higher expression in QSCs compared to ASCs. This indicates that the quiescent state of SCs is actively suppressed by miRNAs [145]. Specifically, miR-195/497 and miR-489 as well as miR-27b, miR-489, miR-31, and miR-195/197 were identified as key regulators of the SC transition in these two phases [146]. Interestingly, the above observations based on experiments in mice are contradictory to the conclusion of Koning et al. [147], who observed that in human QSCs all miRNAs are downregulated and therefore have minimal regulatory activity. Other miRNAs that were indicated in regulating SC quiescence in adult resting muscles are miR-127 and miR-379. It was shown that their robust expression corresponds to an increase in Pax7 expression and reduced commitment towards differentiation [145].
A recent increase in interest in the field of lncRNA yielded a few reports that indicate the involvement of these molecules in QSCs. Particularly, high expression of lncRNA H19 was noted, indicative of its involvement in the maintenance of the QSC pool [148]. Furthermore, lncRNA, named Uc.283+ A, might be another key regulator of quiescence, as it can block the formation of miR-195 [149], which, as mentioned above, is needed to maintain the undifferentiated state of SCs.
Epigenetic control of SC activation and myoblast proliferation
In response to damage, QSCs undergo activation and divide. Symmetric divisions lead to self-renewal of PAX7+/MYF5-/MYOD-/MYOG- QSCs (Fig. 1b) [150]. In contrast, the asymmetric division results in one cell that returns to the quiescent state (PAX7+/MYF5-/MYOD-/MYOG-), while the other (PAX7+/MYF5+/MYOD+/MYOG-; myoblast) proliferates, differentiates, and fuses to form multinuclear myotubes [150]. In this context, myoblasts produce regulatory proteins, including MRFs, such as MYF5, MYOD (required for myoblast specification) (Fig. 4b), as well as MYOG and MRF4 expressed in the early and subsequent stages of differentiation, respectively (Fig. 5a, b). It is worth noting that the proper expression of MYOD and MYOG depends on the phosphorylation state of MEF2 (Figs. 4 and 5a) [151]. Ultimately, during terminal differentiation, proteins characteristic for mature skeletal muscle such as myosin heavy chain (MYHC), creatine kinase M-type (CKM), and α-actinin 1 (ACTN1) are generated (Fig. 5c) [152,153,154].
During symmetric and asymmetric divisions of QSCs, the PAX7 promoter is active, holding transcription permissive chromatin marks by the action of the TrxG complex (Fig. 4a) [155]. The study by von Maltzahn et al. [156] has revealed that PAX7 is a crucial player in the transcriptional regulation of SCs, as in the Pax7-deficient mice the population of SCs was completely absent, leading to muscle atrophy and premature death. This phenotype might partially stem from the fact that PAX7 is involved in the repressive regulation of MYOD by increasing expression of Id in cells undergoing self-renewal following asymmetric division [157]. Interestingly, PAX7 also triggers the synthesis of MYF5 in cells committed to myogenesis. Specifically, upon CARM1-mediated methylation of multiple arginine residues in the N terminus, PAX7 recruits TrxG to regulatory enhancers and the proximal promoter of MYF5 through direct interaction with MLL1/2 HMT (Fig. 4b) [158,159,160]. This activation occurs via PAX7 binding to different sites in the MYF5 promoter in a two-step manner. First, PAX7 binds to a site located in the enhancer marked by H3K4me2 [161], and then a strong H3K4me3 is induced, establishing a transcriptionally active domain. In addition to PAX7 and MYF5, proliferating myoblasts express MYOD, which inhibits the cell cycle and induces myoblast differentiation. In this context, MYOD expression starts with the detachment of HDACs and the PRC2 complex, and through the involvement of TrxG (H3K4me3), which is recruited by phosphorylated MEF2, as well as SRF, which binds to the serum response element (SRE) [162, 163]. As a consequence, the PCAF-p300/CBP and SWI/SNF chromatin remodeling complexes bind to the MYOD promoter and initiate transcription (Fig. 4b). Additionally, noncoding miRNAs are also involved in myoblast proliferation. For instance, miR133a enhances the proliferation of SCs by repressing the SRF (Fig. 7a) [164].
Epigenetic control of myoblast differentiation
The ability of myoblasts to differentiate into functional multinucleated myofibrils requires coordinated changes in the expression of muscle-specific genes [165]. The MYOD levels are highest at the end of myoblast proliferation (Fig. 4b) and in the initial stage of differentiation (Fig. 5a) as a result of PAX7 repression via H3K27me3 catalyzed by PRC2 (YY1-EZH2 complex) (Fig. 5a) [157, 166, 167]. Simultaneously, the high content of MYOD contributes to an increase in the production of MYOG (Fig. 5a), which in return inhibits MYF5 expression by repressive H3K27me3 marks (YY1-EZH2 complex and other unknown factors) (Fig. 5a) [168]. Furthermore, the combined action of MYOD and MYOG leads to the expression of genes characteristic for late differentiation, such as MRF4 (Fig. 5b), which allows for the formation of muscle fibers [169], and then, the MYOD and MYOG levels decline (Fig. 5b) [170]. In mature myofibers, the level of MRF4 remains high [171], and proteins characteristic for mature skeletal muscle such as MYHC, CKM, and ACNT1 are generated (Fig. 5c) [152,153,154].
Removal of repressive epigenetic marks
During differentiation, HDACs leave the promoters of muscle-specific genes, e.g., Myog, to enable the recruitment of transcription factors such as phosphorylated Mef2 and Myod, which in turn recruit the SWI/SNF chromatin remodeling complex and HATs leading to active transcription (Fig. 5a) [172]. Several mechanisms are known that allow HDACs to leave the gene promoters, including the reduction of their expression [172]. More specifically, upon myoblast differentiation, the disruption of the MYOD-HDAC I complex is observed [173] and the nuclear-to-cytoplasmic translocation of HDAC II occurs, thereby releasing the inhibitory constraints of MEF2, which activates the expression of muscle-specific genes [141, 174]. Additionally, upon reception of the differentiation-promoting signals, the NAD+/NADH ratio decreases, and as a consequence, inhibition of HDAC III (SIRT) and an increase in PCAF-p300/CBP complex activity occurs, which induces acetylation of histones in genes, such as MYOD (Figs. 4b, 5a) and MEF2 [175].
Activation of MYOG is also dependent on decreases in the PRC2 HKMT activity [176]. The total levels of PRC2 decrease significantly as myogenesis progresses, and correspondingly, the H3K27me3 mark is lifted from the MYOG promoter [176]. Additionally, it was also noted that PRC2-EZH1 replaces PRC2-EZH2 on the MYOG promoter (Fig. 5a), a process that is necessary to guarantee its activation in post-mitotic myotubes signified by the fact that the depletion of EZH1 negatively affects muscle differentiation and the ability of MYOD to regulate MYOG [177]. The Ezh2 to Ezh1 switch has been attributed to the mitogen- and stress-activated protein kinase 1 (MSK1)-mediated phosphorylation of histone H3 at serine 28 (H3S28ph) on, among others, the Myog regulatory region promoter [177].
Moreover, the loss of repressive histone marks from MYOG is also due to the removal of trimethylation of H3K27 by ubiquitously transcribed X chromosome tetratricopeptide repeat protein (UTX) activity (Fig. 5a), which belongs to the family of JmjC HDMs [178]. UTX acts in complex with several proteins, including mixed-lineage leukemia 4 (MLL4), the HMT subunit of TrxG [179, 180]. In turn, the removal of the repressive H3K9me2/3 marks from the Myog promoter [181] is catalyzed by HDM—JHDM2A (KDM4A) that also belongs to the JmjC family (Fig. 5a) [182] as underlined by the study revealing that knockdown of this enzyme leads to the significantly decreased levels of Myog expression [181]. Summarizing, the coordinated PRC2-EZH2 and PRC2-EZH1 switches as well as the activity of specific enzymes work together to remove inhibitory marks from the promoters of muscle differentiation-specific genes.
Addition of permissive epigenetic marks
MYOG activation is initiated upon MEF2 phosphorylation and its association with the ASH2L and MLL2 subunits of TrxG. This complex then binds to the MYOG promoter, catalyzing the H3K4me3 mark (Fig. 5a). Tri-methylation of H3K4 is also catalyzed by another HMT, SET7 (Fig. 5a), as underlined by siRNA knock-down experiments [183]. Apart from the acquisition of permissive histone marks, transcription initiation also requires the concerted recruitment of the chromatin remodeling complex SWI/SNF (Fig. 5a). The complex facilitates the binding and formation of the RNA polymerase II preinitiation complex and transcriptional elongation [184, 185] through recognition of acetylated lysines on histone tails by ATPase subunits BRG1 or BRM [186]. Interestingly, MYOD physically associates with the SWI/SNF complex on regulatory elements of MYOD-target genes, including MYOG, ensuring their active transcription (Fig. 5a) [187].
The combined action of MYOD and MYOG leads to the expression of genes characteristic for late differentiation, such as MRF4 (Fig. 5b), which allows for the formation of muscle fibers [169]. In this context, the permissive MRF4 marks are catalyzed by TrxG (H3K4me3) and by the PCAF-p300/CBP complex (acetylation). In the late differentiation, the levels of MYOD and MYOG decrease in response to the G9a HKMT-mediated H3K9me2 mark [188], the H3K27me3 addition by PRC2 (YY1-EZH2 complex) and probably due to other undiscovered factors (Fig. 5b). Conversely, the level of MRF4 is maintained at a high level [189] and proteins characteristic for mature skeletal muscle such as MYHC, CKM, and ACTN1 are generated (Fig. 5c) [152,153,154].
Noncoding RNA
miRNAs, including muscle-specific miRNAs (myomiRs), are one of the most important players orchestrating the processes of myoblast proliferation and myogenic differentiation [190]. In particular, myomiRs miR-1 and miR-206 target a subunit of DNA polymerase alpha complex, thus promoting differentiation by DNA synthesis inhibition. Other miRNAs involved in myogenesis involve, e.g., miR-29, a negative regulator of YY1 [191], or miR-214 [192] and miR-26a [193] that play crucial roles in the repression of PRC2 by targeting the Ezh2 mRNA. Moreover, miR-206 and miR-29 negatively regulate HDAC4 expression [194] and, hence, reduce the total levels of HDACs and in turn MYF5 as well as MYOG [195]. On the contrary, some of the described miRNAs are regulated by myogenic transcription factors that play pivotal functions in myogenesis. Specifically, MYOD overexpression translates into an increased concentration of miR-206 and enhanced myoblast differentiation capacity due to the MYOD direct binding to the miR-206 promoter [196]. Also, increasing amounts of MYOG and MYOD were found in regions upstream of miR-133, miR-1, and miR-206 [197], which suggests the involvement of MYOG and MYOD in the regulation of these miRNAs.
LncRNAs have been also shown to regulate myogenic differentiation. For instance, lncRNAs myogenic differentiation 1 (lncMyoD) is activated by MyoD [198]. Upon myoblast differentiation, lncMyoD accumulates and interacts with Igf2 mRNA-binding proteins (IMPs) to inhibit genes promoting myoblast proliferation, such as cyclin G1 (Ccng1) or c-Myc. As a result, myoblasts can exit the cell cycle and differentiate [198]. Another example is the long intergenic noncoding RNA activator of myogenesis (lincRAM), whose expression is also regulated by MYOD [198]. However, in this case, lincRAM directly interacts with MYOD and enhances its activity by promoting the assembly of the MYOD-BAF60c-BRG1, SWI/SNF complex which, in turn, remodels the chromatin of MYOD-target genes (e.g. MYOG), enabling their subsequent transcription [198]. Furthermore, recent data indicate that lncRNA Myoparr is an essential positive regulator of the myogenic process. It was shown that this MYOG promoter-associated lncRNA interacts with the transcriptional coactivator of MyoD, DEAD-Box Helicase 17 (Ddx17), and regulates binding of the latter to PCAF, activating MYOG transcription [199]. Additionally, lncRNA Irm is upregulated during myogenesis, promoting myogenic differentiation, while its inhibition has the opposite effect. Interestingly, lncRNA Irm blocks regeneration following cardiotoxin-induced muscle damage in mice and regulates the expression of myogenic genes through direct binding to MEF2, which in turn mediates MYOD/MEF2 interaction with target genes [200]. Another lncRNA implicated in myogenic differentiation involves a long intergenic non-protein coding RNA, muscle differentiation 1 (lncRNA lincMD1), which is localized in the cytoplasm and acts as a natural decoy for miR-133 and miR-135 [192]. Likewise, metastasis-associated lung adenocarcinoma transcript 1 (Malat1) lncRNA also acts as a sponge for miR-133. Particularly, its presence has been associated with the inhibition of SRF expression that allows for myoblast terminal differentiation [164].
Cell signaling control of epigenetic changes during muscle differentiation
The dynamic changes in the genome expression landscape of SCs during their activation and division as well as myoblast proliferation and differentiation are coordinated by extracellular signals that manage multicellular processes in response to microenvironmental requirements. These signals not only regulate genome expression through modulating the levels of transcription factors but also by influencing DNA structural alterations, e.g., by recruiting chromatin modifier enzymes (Fig. 6) [28, 201]. The following signaling pathways have been identified as epigenome regulators in the context of muscle differentiation: p38 MAPK, IGF1/Pi3K/AKT, Wnt, Ca2+/calmodulin-dependent protein kinase (CaMK), TNFα, and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB). Nevertheless, it is important to note that despite the growing number of studies, our knowledge is still limited and requires further exploration. Furthermore, while cell signaling influences epigenetic modifications, it can itself be tightly controlled by epigenetic events. As an example, the Notch pathway is implicated in the quiescence of SCs, the proliferation of myoblasts, and the transient inhibition of terminal differentiation of myoblasts into mature myofibers. Such a wide range of controlled processes the Notch pathway owes to a variety of Notch receptors and ligands required to activate downstream signaling [202]. Expression of Notch receptors and ligands has to be precisely regulated in a time- and space-restricted manner. Gene expression profiling and epigenetics studies performed by the Terragni et al. [202] revealed significant hypomethylation and very high enrichment of 5-hydroxymethylcytosine in myoblasts, myotubes, and skeletal muscle at intragenic or intergenic regions of some Notch receptors and ligand genes. Their results suggest that hypomethylation and/or hydroxymethylation of the Notch pathway genes is the mechanism of epigenetic regulation of Notch signaling activity.

Cell signaling pathways in the epigenetic regulation of myogenesis. p38α/β MAPK, p38γ MAPK, IGF1/Pi3K/AKT, CaMK, Wnt/β-catenin, and nNOS signaling either inactivates (marked red) or activates (marked green) downstream targets, dependent on the stage of the muscle formation. The consequences following particular signaling events and the final impact on myogenesis (in italics) are listed below the name of each signaling pathway
Exercise or muscle injury activates SCs via Wnt signaling. Wnt/β-catenin pathway modifies chromatin at the promoter regions of Myod and Myf5, activating their expression and, as a consequence, enhancing SC proliferation. In fact, Wnt signaling, which is further involved in the regulation of different myogenesis stages (expansion, proliferation-to-differentiation transition, cell motility), is under the epigenetic control of p38α MAPK [203, 204], the most studied signaling pathway in the context of muscle formation. In mammals, the family of p38 MAPK consists of four kinases, p38α, p38β, p38γ, and p38δ, all phosphorylated and activated by MAPK kinases (MKK). p38α is the most abundant isoform that was found to take part in the epigenetic regulation at various stages of muscle development and via different downstream mechanisms [204]. Upon SC activation, p38α/β MAPK signaling promotes asymmetric divisions and myogenic commitment via promoting the accumulation of MyoD. This occurs in the following manner: p38α/β MAPK phosphorylates MAPKAP2, which in turn inactivates, via phosphorylation, tristetraprolin (TTP). TTP is the mRNA decay factor, which in the active state promotes MyoD mRNA decay, leading to the maintenance of SCs quiescence [205].
The quiescence of SCs and their self-renewal via symmetric divisions is also controlled by p38γ MAPK, which phosphorylates Carm1, a process that prevents its nuclear localization and methylation of Pax7. Otherwise, methylated Pax7 recruits TrxG MLL1/2 HMT complex to regulatory enhancers and promoter regions of Myf5, leading to an “open mark” H3K4 methylation and Myf5 expression that favors asymmetric cell division (Fig. 4b) [159]. In the later stage of myogenesis, Carm1 additionally recruits SWI/SNF complex to promoters of muscle-specific genes like desmin (DES) or CKM [206]. p38γ MAPK signaling was found to contribute to the maintenance of SCs quiescence also via phosphorylation of MyoD and establishing MyoD and Suv39h1 HKMT on MYOG promoter, followed by methylation of H3K9, which induces transcriptionally repressive chromatin and prevents premature myoblast differentiation (Fig. 4b) [207]. Furthermore, p38γ MAPK activity was noticed at the proliferation and differentiation stages of myogenesis. However, its exact mechanistic involvement in the temporal patterning of gene expression remains unknown in these processes [207].
p38α MAPK phosphorylates EZH2, which plays a crucial role at the stage of transition from myoblast proliferation to its differentiation. TNF-activated p38α MAPK promotes PRC2 and YY1 interaction via phosphorylation of PRC2 EZH2 enzymatic subunit, which effects in repressive chromatin in the Pax7 promoter region (Fig. 5a) [208]. At the onset of differentiation, p38 MAPK phosphorylates P18hamlet, a subunit of SNF2-related CBP activator protein (SRCAP). As a consequence, SWI/SNF transcription-activating complex localizes to the MYOG promoter, which is accompanied by H2A.Z histone accumulation and expression of muscle-specific genes [209]. However, the mechanisms of epigenetic control via the p38α MAPK pathway are much more complex. p38α MAPK influences the epigenetics of muscle cells via interaction with various transcription factors, especially MyoD and the binding partner for MRFs (E47) [210]. Phosphorylation of E47 initiates the dimerization of E47 with MyoD and further localization of this heterodimer at myogenic loci [211]. Additionally, MyoD binding to target genes is facilitated by p38α MAPK-dependent phosphorylation of Baf60c, which results in MyoD-Baf60c incorporation into SWI/SNF chromatin remodeling complex and expression of MyoD-controlled genes [187]. In differentiating myoblasts, p38α/β activates Msk1, which in turn phosphorylates histone H4S28, implicated in Ezh2-containing PRC2 complex displacement of MYOG and CKM genes. Meanwhile, PRC2-Ezh2 is replaced by the PRC2-Ezh1 complex, precisely activating their expression [177, 203]. The p38α signaling pathway also regulates the EZH2 levels at the early stages of muscle differentiation, leading to its degradation through the proteasome, more specifically, by the E3 ubiquitin ligase Praja1 (PJA1) [212]. Transcription of MYOG or CKM in proliferating myoblasts is also activated via recruitment of Ash2L/MLL2-containing TrxG HMT complex by phosphorylated Mef2, a downstream target of the p38α/β MAPK pathway [213, 214]. This complex catalyzes tri-methylation of H3K4, a permissive epigenetic mark. Such a variety of actions allows for the speculation that p38α/β plays a bidirectional role in the proliferation-to-differentiation transition, both silencing genes responsible for proliferation and promoting the expression of prodifferentiation genes [204].
p38 MAPK signaling control of epigenetics is convergently accompanied by IGF1/Pi3K/AKT pathway. IGF1-activated AKT1 and AKT2 phosphorylates the acetyltransferase p300, which promotes its connection with MyoD and PCAF acetyltransferase. This results in hyperacetylation of muscle gene promoters and chromatin remodeling by the p38 MAPK-recruited SWI/SNF complex [215]. This is consistent with studies showing that a combination of both p300 and PCAF acetyltransferases acts as a strong activator of transcription, unlike PCAF alone that without the presence of p300 is just a moderate inducer [216]. Recruitment of HATs to muscle-specific gene promoters is also induced by CaMK signaling. At the onset of differentiation, CaMK directly phosphorylates members of the class II HDACs, HDAC4 and HDAC5, and forces them to move from the nucleus and release MEF2 from repressing interactions. In turn, MEF2 is potent to associate with HATs and promote the expression of muscle-specific genes [215, 217]. Another histone deacetylase, HDAC2 that belongs to the class I HDACs, loses affinity to chromatin and is released from specific miRNA promoters that enables their expression after NO-induced S-nitrosylation [218, 219]. Moreover, miR-133 enhances proliferation (via interaction with Akt pathway) [220], while miR-29 (downregulating Akt signaling) together with miR-1 and miR-30 promotes differentiation of myoblasts [220,221,222,223].
Epigenetic regulation of gene expression in Duchenne muscular dystrophy
Besides the well-established function as a mechanical anchor between the cytoskeleton and the ECM of myofibers, the DGC is now considered as a scaffold for signaling molecules in various cell types, including muscle fibers and SCs. Particularly, the absence of dystrophin in SCs of DMD patients has been associated with the signal transmission loss between the plasma membrane and the nucleus, leading to the SC aberrant epigenetic transcriptional activation and impaired regenerative ability [224,225,226].
Dystrophin deficiency in muscle regeneration
Dystrophin deficiency in ASCs, primed to divide asymmetrically, reduces the levels of the DGC and induces aberrant polarization of structural and signaling proteins. This leads to impaired signal transduction and transcriptional activity in the newly generated cells [227]. The defects in polarization, centrosome amplification, and prolonged cell divisions of dystrophic ASCs have been attributed to the loss of dystrophin interaction with MARK2 [228,229,230,231] as well as downregulation and mislocalization of another DGC component, β-syntrophin. Generally, β-syntrophin interacts with p38γ, modulating CARM1-mediated activation of MYF5 in the opposite cell that undergoes myogenic differentiation [232], while dystrophin deficiency leads to impaired polarization of p38γ, enhanced phosphorylation of CARM1, and reduced ability of MYF5 to be activated by PAX7 [233]. Furthermore, in DMD, elevated levels of TNFα and NFκB were found to diminish the regenerative potential of SCs and this is connected to epigenetic silencing of Notch-1 via hypermethylation of its promoter region [234]. What is more, the lack of dystrophin causes loss of nNOS binding sites and, as a consequence, reduces nNOS sarcolemmal localization [235]. This results in diminished NO signaling and, in turn, a decrease in NO-dependent S-nitrosylation of HDAC2. Importantly, restoration of NO-signaling-dependent inhibition of HDAC2 shows beneficial effects in dystrophic mice [236].
A recent study uncovered a crosstalk between fibro-adipogenic progenitors (FAPs) and the myogenic lineage, which sheds more light on adipocyte and myofibroblast accumulation in dystrophic skeletal muscle. The data indicate that soluble molecules released by myogenic progenitors activate the PI3K/Akt pathway in FAPs, stimulating their proliferation, while myotubes induce their differentiation through the secretion of pro-fibrogenic and anti-adipogenic factors [237]. As in DMD patients proliferation and differentiation of myogenic cells are disrupted, this results in excessive FAP proliferation [237] and their transformation into fibroadipocytes. Altogether, these processes mediate fat deposition and fibrosis in skeletal muscle [238]. A few signaling pathways associated with these pathological alterations have been described. Namely, Notch-mediated modulation of FAP adipogenesis was found compromised in FAPs from the mdx mouse, an animal model of DMD, supporting a model whereby the synergistic cooperation of Notch with other anti-adipogenic signals plays essential roles in the regulation of FAP adipogenesis in both healthy and dystrophic muscle [239]. Additionally, a recent report by Mázala et al. [240] indicated that muscle damage in mdx mice contributes to an increase in TGF-β activity accompanied by increased accumulation of FAPs, which leads to muscle fibrosis. Nevertheless, it is important to mention that although inhibition of TGF-β signaling blocked the accumulation of FAPs, it did not induce muscle regeneration [240, 241]. Communication between myogenic cells and FAPs expresses through appropriate epigenetic modifications, and FAPs are an important intervention target aimed at restoring the balance between skeletal muscle regeneration and degeneration in DMD. Specifically, histone deacetylase inhibitors (HDACi) are used to promote muscle gene expression and skeletal myogenesis. In this context, HDACi appears to selectively upregulate genes that are enriched in permissive H3K4me3 marks or marked by bivalent domains (H3K4me3/H3K27me3) [242].
At the epigenetic level, HDACi induce upregulation of MYOD and BAF60C (a subunit of SWI/SNF complex) and lead to the upregulation of miR-1, miR-133, and miR-206. These in turn target the alternative BAF60 variants, BAF60A and BAF60B, ultimately leading to promyogenic differentiation and simultaneous suppression of the fibro-adipogenic phenotype [243]. Interestingly, Saccone et al. [243] demonstrated that HDACi delivery by intraperitoneal injection into young mdx mice promotes myogenic differentiation of FAPs, while such an effect was not observed in wild-type or old mdx mice. This indicates that FAPs may support the activity of SCs or promote fibro-adipogenic degeneration and that the latent FAP myogenic phenotype may be induced in response to regenerative signals.
Concomitantly, fibrosis can be stimulated not only by FAPs but also by miRNAs, e.g., miR-21 and miR-29 that play opposing roles in DMD muscle fibrosis [244]. MiR-21 is involved in the pro-fibrotic effects induced by TGF-β treatment [245], and conversely, miR-29 downregulates the expression of ECM components such as collagen and elastin [246]. In particular, miR-29 is downregulated in mdx mice, a process that has been linked to fibrosis and impaired muscle regeneration [246]. At the epigenetic level, miR-29 promotes myogenesis by direct inhibition of a negative regulator of muscle genes, YY1 [191], as also shown by Zanotti et al. [244] (Fig. 7). The other miRNA involved in the pathogenesis of DMD is miR-206, which targets utrophin mRNA (a paralog of dystrophin) and whose appropriate concentration in skeletal muscle could inhibit the development of DMD [247, 248].

Dystrophin-nNOS signaling in epigenetic control of muscle differentiation. a The dystrophin-nNOS signaling regulates the epigenetic profile of myogenesis via S-nitrosylation of HDAC2 (class I HDAC), which affects gene expression through changes in histone acetylation. miR-221/222 are involved in the inhibition of cell cycle proteins and miR-222 targets β-syntrophin, while miR-31 temporarily targets dystrophin. Transcription of miR-1 and miR-133 is controlled by the HDAC2 S-nitrosylation state, regulated by nNOS activity. Also, miR-133 targets SRF during proliferation, which in a self-regulating manner promotes miR-133 expression, and miR-29 is coregulated by the HDAC2 S-nitrosylation state and YY1, while miR-206 is regulated by MRFs and the HDAC1 activity, and supports cell cycle inhibition by targeting PAX7. b In DMD, the epigenetic differentiation network is disturbed due to the absence of dystrophin-α/β-syntrophin-nNOS signaling and interrupted HDAC2 S-nitrosylation. As a consequence, a decrease in the levels of miR-133/1 and miR-29, followed by inhibition of muscle differentiating genes and ROS generation is observed. Also, diminished amounts of miR-29 levels correlate with an increase in collagen and fibrotic tissue. The miR-206 level is higher, resulting in an imbalance between proliferative and differentiated states. Additionally, miR-144 and miR-223 are also observed following an increase in inflammation and muscle degeneration. miRNA and protein names marked in bold and grey indicate their upregulation and downregulation, respectively; black arrows mark activation while red and grey blunt lines inhibition and reduction in inhibition, respectively. HDACi targeting HDACs and the corresponding processes are marked in yellow boxes
Epigenetic therapies in Duchenne muscular dystrophy
Myogenesis is coordinated by a complex set of epigenetic mechanisms that include DNA methylation, histone modifications, and rncRNA expression, and as such, targeting epigenetic modifiers is a promising pharmacological approach, opening new therapeutic avenues in muscle diseases. Particularly in DMD, one could envision changing the epigenetic status of SCs or myocytes to increase their regenerative potential.
Altered cell signaling poses a wide range of potential therapeutic strategies for DMD. Some pharmacological therapies are based on modulation of cell signaling, e.g., NO administration, stimulation of IGF-1, inhibition of TGFβ, or modulation of NFκB and TNFα pathways. However, changes in signaling pathways entail a risk of undesired side effects as distinct signaling pathways are differently activated depending on the muscle type [249] and differentiation stage. For this reason, epigenetic drugs aiming to modulate targets of signaling pathways seem to be a safer therapeutic approach [250]. Pharmacological inhibition of HDACs by HDACi enhances histone acetylation in the promoters of muscle-specific genes leading to their increased expression (Fig. 7b) [251]. However, it should be noted that HDACi acts systemically, affecting acetylation, and thus increases the expression of many genes. Indeed, HDACi has been associated with side effects such as nausea, neutropenia, thrombocytopenia, or ventricular arrhythmia [252].
Iezzi et al. [253] reported enhanced histone acetylation in the regulatory elements of Myod in wild-type myoblasts following HDACi treatment [253]. Moreover, HDACi delivery to myoblasts before the onset of differentiation upregulated Myf6 and Myog [254]. Interestingly, HDACi also increased follistatin expression [255], which downregulates myostatin, a major inhibitor of skeletal muscle regeneration [256, 257], and increased myoblast fusion [258]. In all, the results obtained in wild-type myoblasts highlight the potential of HDACi as a treatment having a positive effect on muscle regeneration in DMD.
HDACi ITF2357 (Givinostat) is the first epigenetic drug tested in preclinical studies in mdx mice [255, 259] and clinical trials [260]. Preclinical studies revealed that after 3.5 months of Givinostat treatment, mdx mice exhibited increased myofiber mass and size as well as restored muscle force to the levels observed in wild-type mice. At the same time, a decrease in the cellular inflammatory infiltrate, reduction in the formation of fibrosis, and accumulation of fat tissue were observed [259]. In another preclinical study, Givinostat alleviated the morphological and functional phenotypic consequences of dystrophin deficiency in mdx mice [255]. The success of these studies paved the way for I/II clinical trials with children affected by DMD (ClinicalTrials.gov Identifier: NCT01761292) [260]. Phase I and phase II clinical trials were conducted on 20 boys aged 7 to < 11 years. The current results indicate that long-term (over 1 year) treatment of Givinostat results in an increased fraction of muscle tissue as well as a decreased amount of fibrotic tissue and also reduced necrosis and fatty replacement [260] compared to untreated boys aged 7 to 10 years [261, 262]. Summarizing, Givnostat is the first pharmacological treatment shown to produce beneficial histological effects in muscle samples from DMD patients [260]. Based on these promising results, the clinical trial has been extended to the III phase (ClinicalTrials.gov Identifier: NCT03373968). Besides, one more clinical trial is currently underway to evaluate the efficacy and safety of Givinostat in ambulant DMD patients (6 to 17 years) (ClinicalTrials.gov Identifier: NCT02851797).
Trichostatin A (TSA) is another promising HDACi used to enhance myogenic regeneration. For example, in wild-type myoblasts (murine and human origin), TSA increased their fusion and favored myogenic differentiation but without leading to hypertrophy (fibrosis caused by an increased amount of fat components) of preformed myotubes [253, 254]. Besides, intraperitoneal TSA delivery to mdx mice increased utrophin expression and improved the structure and function of skeletal muscles [263]. TSA also ameliorated pathological alterations in a zebrafish model [264], which is an outstanding model for screening and evaluating novel drug therapies, e.g., in DMD [265]. In a recently published study, the authors performed a pilot screen of the commercially available Cayman Chemical Epigenetics Screening Library to identify epigenetic molecules that could improve muscle phenotype in the DMD zebrafish model. Interestingly, they proved that a novel combination of HDACi drugs, oxamflatin, and salermide significantly rescued muscle degeneration [266]. In particular, oxamflatin is an HDACi that inhibits HDACs classes I and II and is chemically similar to TSA. On the contrary, salermide belongs to a class III HDACi, which inhibits the NAD+-dependent deacetylases SIRT1 and SIRT2 [267] and represents a new class of HDACi in DMD treatment.
Conclusion
DMD is a quickly progressing and devastating genetic disorder. A medication that would alleviate the primary symptoms of the disease, i.e., the proper functioning of skeletal muscles and their regeneration, is of utmost need. In this review, we discussed the interplay of various factors that define the specific state of epigenetic homeostasis and contribute to the progression of DMD in skeletal muscles. The described studies show that myogenesis is strictly controlled by interdependent epigenetic pathways; however, it is also characterized by high cellular plasticity, amenable for therapeutic approaches altering the epigenetic status of chromatin. Particularly, HDACi delivery has proven to be an exceptionally effective strategy for restoring the regenerative ability of dystrophic muscles. Although further research is needed in this field, the outcome of the most recent therapeutic advances gives patients hope for a treatment that would significantly alleviate their condition.
Availability of data and materials
Not applicable.
Abbreviations
- ACTN1:
-
α-Actinin 1
- ASCs:
-
Activated satellite cells
- BAFs:
-
BRG1-associated factors
- BRG1 :
-
Brahma-related gene 1
- BRM :
-
Brahma
- CARM1:
-
Coactivator-associated arginine methyltransferase
- CBP:
-
CREB binding protein
- Ccng1:
-
Cyclin G1
- circ-BNC2:
-
Circular basonucling 2
- circ-QKI:
-
Circular QKI
- circRNAs:
-
Circular RNAs
- CKM:
-
Creatine kinase M-type
- CpG:
-
Cytosine-guanine adjacent sites
- CTX:
-
Cardiotoxin
- Ddx17:
-
DEAD-Box Helicase 17
- DGC:
-
Dystrophin–glycoprotein complex
- DMD:
-
Duchenne muscular dystrophy
- DMD :
-
Dystrophin
- DNMT3L:
-
DNMT3-like
- DNMTs:
-
DNA methyltransferases
- ECM:
-
Extracellular matrix
- FAPs:
-
Fibro-adipogenic progenitors
- HATs:
-
Histone acetyltransferases
- HDACi:
-
Histone deacetylase inhibitor
- HDACs:
-
Histone deacetylases
- HMTs:
-
Histone methyltransferase
- IL-10:
-
Interleukin 10
- IL-1β:
-
Interleukin 1 beta
- IMPs:
-
Igf2 mRNA-binding proteins
- JmjC:
-
Jumonji C domain
- lincRAM:
-
LincRNA activator of myogenesis
- lncMyoD:
-
LncRNAs myogenic differentiation 1
- lncRNA lincMD1:
-
Long intergenic non-protein coding RNA, muscle differentiation 1
- lncRNAs:
-
Long noncoding RNAs
- LSD1:
-
Lysine-specific histone demethylase 1
- Malat1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- MARK2:
-
Serine–threonine protein kinase 2
- MBD:
-
Methyl CpG binding domain
- MEF2:
-
Myocyte enhancer factor 2
- miRNA:
-
MicroRNA
- MLL1/2:
-
Mixed-lineage leukemia 1/2
- MLL4:
-
Mixed-lineage leukemia 4
- MRF4:
-
Muscle-specific regulatory factor 4
- MRFs:
-
Myogenic regulatory factors
- MSK1:
-
Mitogen- and stress-activated protein kinase 1
- MYF5 :
-
Myogenic factor 5
- MYF6 :
-
Myogenic factor 6
- MYHC:
-
Myosin heavy chain
- MYOD :
-
Myogenic differentiation 1
- MYOG :
-
Myogenin
- myomiRs:
-
MiRNAs that are specifically expressed in muscle tissue
- ncRNAs:
-
Noncoding RNAs
- nNOS:
-
Nitric oxide synthase
- NO:
-
Nitric oxide
- PAX7 :
-
Paired box 7
- PCAF:
-
P300/CBP-associated factor
- PHD:
-
Plant homeodomain
- PI3Kinase:
-
Phosphoinositide 3-kinase
- piRNAs:
-
PiwiRNAs
- PJA1 :
-
Praja1
- PRC2:
-
Polycomb repressive complex 2
- PRMTs:
-
Protein arginine methyltransferases
- QSCs:
-
Quiescent satellite cells
- RITS:
-
RNA-induced transcriptional silencing
- rncRNAs:
-
Regulatory noncoding RNAs
- ROS:
-
Reactive oxygen species
- SAM:
-
S-adenosyl-L-methionine
- SCs:
-
Satellite cells
- SET:
-
Su(var)3–9, enhancer of zeste, and trithorax catalytic domain
- SIRT2:
-
Sirtuin 2
- siRNAs:
-
Short interfering
- sncRNAs:
-
SMALL noncoding RNA
- SRF:
-
Serum response factor
- SUV39H1:
-
Suppressor variegation 3–9 homolog 1
- SUV4-20:
-
Suppressor of variegation 4–20
- SWI/SNF:
-
SWItch/sucrose nonfermentable
- TNF-α:
-
Tumor necrosis factor alfa
- TRDMT1:
-
TRNA aspartic acid methyltransferase 1
- TrxG:
-
Trithorax group
- TSS:
-
Transcriptional start sites
- UHRF1:
-
UBIQUITIN-like with PHD and ring finger domains 1
- YY1:
-
Yin Yang 1
References
An J, Xie Z, Jia F, Wang Z, Yuan Y, Zhang J, et al. Quantitative coordination evaluation for screening children with Duchenne muscular dystrophy. Chaos. 2020;30(2):023116.
Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–28.
Hendriksen RGF, Schipper S, Hoogland G, Schijns OEMG, Dings JTA, Aalbers MW, et al. Dystrophin distribution and expression in human and experimental temporal lobe epilepsy. Front Cell Neurosci. 2016;10:174.
Holder E, Maeda M, Bies RD. Expression and regulation of the dystrophin Purkinje promoter in human skeletal muscle, heart, and brain. Hum Genet. 1996;97(2):232–9.
Ganote CE, Armstrong SC. Dystrophin-associated protein complex and heart failure. Lancet. 2002;359:905–6.
Doorenweerd N, Mahfouz A, van Putten M, Kaliyaperumal R, T' Hoen P, Hendriksen J, Aartsma-Rus AM, et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep. 2017;7(1):12575. Erratum in: Sci Rep. 2018;8(1):4058.
Waite A, Tinsley CL, Locke M, Blake DJ. The neurobiology of the dystrophin-associated glycoprotein complex. Ann Med. 2009;41(5):344–59.
Morris GE, Simmons C, Man NT. Apo-dystrophins (DP140 and DP71) and dystrophin-splicing isoforms in developing brain. Biochem Biophys Res Commun. 1995;215:361–7.
Lidov HGW, Selig S, Kunkel LM. Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet. 1995;4(3):329–35.
Lidov HGW. Dystrophin in the nervous system. Brain Pathol. 1996;6(1):63–77.
Hoogland G, Hendriksen RGF, Slegers RJ, Hendriks MPH, Schijns OEMG, Aalbers MW, et al. The expression of the distal dystrophin isoforms Dp140 and Dp71 in the human epileptic hippocampus in relation to cognitive functioning. Hippocampus. 2019;29(2):102–10.
Emery AEH. Population frequencies of inherited neuromuscular diseases-A world survey. Neuromuscul Disord. 1991;1(1):19–29.
Mendell JR, Shilling C, Leslie ND, Flanigan KM, Al-Dahhak R, Gastier-Foster J, et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann Neurol. 2012;71(3):304–13.
Mendell JR, Lloyd-Puryear M. Report Of MDA Muscle Disease Symposium On Newborn Screening For Duchenne Muscular Dystrophy. Muscle Nerve. 2013;48(1):21–6.
Nozoe KT, Akamine RT, Mazzotti DR, Polesel DN, Grossklauss LF, Tufik S, et al. Phenotypic contrasts of duchenne muscular dystrophy in women: two case reports. Sleep Sci. 2016;9(3):129–33.
Song TJ, Lee KA, Kang SW, Cho H, Choi YC. Three cases of manifesting female carriers in patients with duchenne muscular dystrophy. Yonsei Med J. 2011;52(1):192–5.
Hibaoui Y, Reutenauer-Patte J, Patthey-Vuadens O, Ruegg UT, Dorchies OM. Melatonin improves muscle function of the dystrophic mdx mouse, a model for duchenne muscular dystrophy. J Pineal Res. 2011;51:163–71.
Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2(12):731–40.
Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018;17(4):347–61.
Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381(9869):845–60.
Yamamoto T, Awano H, Zhang Z, Sakuma M, Kitaaki S, Matsumoto M, et al. Cardiac dysfunction in duchenne muscular dystrophy is less frequent in patients with mutations in the dystrophin Dp116 coding region than in other regions. Circ Genom Precis Med. 2018;11(1):e001782.
Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. J Paediatr Child Health. 2015;51(8):759–64.
de Souza GT, Zanette Rde S, do Amaral DL, et al. Satellite cells: regenerative mechanisms and applicability in muscular dystrophy. Stem Cells Int. 2015;2015:487467.
Lepper C, Partridge TA, Fan CM. An absolute requirement for pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138(17):3639–46.
Sambasivan R, Yao R, Kissenpfennig A, Wittenberghe L Van, Paldi A, Gayraud-morel B, et al. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138(17):3647–56. Erratum in: Development. 2011;138(19):4333.
Dumont NA, Wang YX, Rudnicki MA. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development. 2015;142(9):1572–81.
Szcześniak KA, Ciecierska A, Ostaszewski P, Sadkowski T. Characterisation of equine satellite cell transcriptomic profile response to β-hydroxy-β-methylbutyrate (HMB). Br J Nutr. 2016;116(8):1315–25.
Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. Satellite cells and skeletal muscle regeneration. Compr Physiol. 2015;5(3):1027–59.
Schaaf GJ, Canibano-Fraile R, van Gestel TJM, van der Ploeg AT, Pijnappel WWMP. Restoring the regenerative balance in neuromuscular disorders: satellite cell activation as therapeutic target in Pompe disease. Ann Transl Med. 2019;7(13):280–280.
Dumont NA, Wang YX, von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, et al. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med. 2015;21(12):1455–63.
Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P, et al. Short telemeres and stem cell exhaustion model in mdx mice. Cell. 2010;143(7):1059–71.
Del Rocío C-G, Rodríguez-Cruz M, Cedillo REE. Systemic inflammation in Duchenne muscular dystrophy: Association with muscle function and nutritional status. Biomed Res Int. 2015;2015:891972.
Rosenberg AS, Puig M, Nagaraju K, Hoffman EP, Villalta SA, Rao VA, et al. Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med. 2015;7(299):299rv4.
Gao QQ, McNally EM. The dystrophin complex: structure, function, and implications for therapy. Compr Physiol. 2015;5(3):1223–39.
Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the Sarcolemma. Circ Res. 2004;94(8):1023–31.
Hoshino S, Ohkoshi N, Ishii A, Shoji S. The expression of α-dystrobrevin and dystrophin during skeletal muscle regeneration. J Muscle Res Cell Motil. 2002;23:131–8.
Bunnell TM, Jaeger MA, Fitzsimons DP, Prins KW, Ervasti JM. Destabilization of the dystrophin-glycoprotein complex without functional deficits in α-dystrobrevin null muscle. PLoS ONE. 2008;3(7):e2604.
Pilgram GSK, Potikanond S, Baines RA, Fradkin LG, Noordermeer JN. The roles of the dystrophin-associated glycoprotein complex at the synapse. Mol Neurobiol. 2010;41(1):1–21.
Shiao T, Fond A, Deng B, Wehling-Henricks M, Adams ME, Froehner SC, et al. Defects in neuromuscular junction structure in dystrophic muscle are corrected by expression of a NOS transgene in dystrophin-deficient muscles, but not in muscles lacking α- and β1-syntrophins. Hum Mol Genet. 2004;13(17):1873–84.
Petrillo S, Pelosi L, Piemonte F, Travaglini L, Forcina L, Catteruccia M, et al. Oxidative stress in Duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum Mol Genet. 2017;26(14):2781–90.
Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41(1):10–3.
Waterland RA. Epigenetic mechanisms and gastrointestinal development. J Pediatr. 2006;149(5 Suppl):S137–42.
Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10(3):192–206.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–54.
Wang Y, Liu C, Zhang X, Yang W, Wu F, Zou G, et al. Gene specific-loci quantitative and single-base resolution analysis of 5-formylcytosine by compound-mediated polymerase chain reaction. Chem Sci. 2018;9:3723–8.
Healy S, Khan P, He S, Davie JR. Histone H3 phosphorylation, immediate-early gene expression, and the nucleosomal response: A historical perspective. Biochem Cell Biol. 2012;90(1):39–54.
Sawicka A, Seiser C. Histone H3 phosphorylation—a versatile chromatin modification for different occasions. Biochimie Elsevier. 2012;94:2193–201.
Rossetto D, Avvakumov N, Côté J. Histone phosphorylation. Epigenetics. 2012;7(10):1098–108.
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95.
Mohammed SA, Ambrosini S, Lüscher T, Paneni F, Costantino S. Epigenetic control of mitochondrial function in the vasculature. Front Cardiovasc Med. 2020;7:28.
Zhu Q, Wani AA. Histone modifications: crucial elements for damage response and chromatin restoration. J Cell Physiol. 2010;223(2):283–8.
Kimura H. Histone modifications for human epigenome analysis. J Hum Genet. 2013;58(7):439–45.
Fauquier L, Azzag K, Parra MAM, Quillien A, Boulet M, Diouf S, et al. CBP and P300 regulate distinct gene networks required for human primary myoblast differentiation and muscle integrity. Sci Rep. 2018;8(1):1–16.
Dekker FJ, van den Bosch T, Martin NI. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov Today. 2014;19(5):654–60.
Barnes CE, English DM, Cowley SM. Acetylation and Co: an expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019;63(1):97–107.
Vaca HR, Celentano AM, Macchiaroli N, Kamenetzky L, Camicia F, Rosenzvit MC. Histone deacetylase enzymes as potential drug targets of Neglected Tropical Diseases caused by cestodes. Int J Parasitol Drugs Drug Resist. 2019;9:120–32.
McIntyre RL, Daniels EG, Molenaars M, Houtkooper RH, Janssens GE. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol Med. 2019;11(9):1–11.
Ververis K, Karagiannis TC. Overview of the classical histone deacetylase enzymes and histone deacetylase inhibitors. ISRN Cell Biol. 2012;2012:1–12.
Kong Y, Jung M, Wang K, Grindrod S, Velena A, Lee SA, et al. Histone deacetylase cytoplasmic trapping by a novel fluorescent HDAC inhibitor. Mol Cancer Ther. 2011;10(9):1591–9.
Lombard DB, Alt FW, Cheng H-L, Bunkenborg J, Streeper RS, Mostoslavsky R, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27(24):8807–14.
Zeng H, Xu W. Enzymatic assays of histone methyltransferase enzymes. In: Zheng YG, editor. Epigenetic technological applications. San Diego: Academic Press; 2015. p. 333–61.
Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284(5423):2174–7.
Kang MK, Mehrazarin S, Park NH, Wang CY. Epigenetic gene regulation by histone demethylases: emerging role in oncogenesis and inflammation. Oral Dis. 2017;6:709–20.
Licciardello MP, Kubicek S. Targeting histone methylation: the development of selective methyltransferase and demethylase inhibitors. In: Egger G, Arimondo P, editors. Drug discovery in cancer epigenetics. San Diego: Academic Press; 2016. p. 209–38.
Laugesen A, Højfeldt JW, Helin K. Molecular mechanisms directing PRC2 recruitment and H3K27 Methylation. Mol Cell. 2019;74(1):8–18.
Yu JR, Lee CH, Oksuz O, Stafford JM, Reinberg D. PRC2 is high maintenance. Genes Dev. 2019;33(15–16):903–35.
Nekrasov M, Wild B, Müller J. Nucleosome binding and histone methyltransferase activity of Drosophila PRC2. EMBO Rep. 2005;6(4):348–53.
Murzina NV, Pei XY, Zhang W, Sparkes M, Vicente-Garcia J, Pratap JV, et al. Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure. 2008;16(7):1077–85.
Boros J, Arnoult N, Stroobant V, Collet J-F, Decottignies A. Polycomb repressive complex 2 and H3K27me3 cooperate with H3K9 methylation to maintain heterochromatin protein 1 at chromatin. Mol Cell Biol. 2014;34(19):3662–74.
Canzio D, Chang EY, Shankar S, Kuchenbecker KM, Simon MD, Madhani HD, Narlikar GJ, Al-Sady B. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol Cell. 2011;41(1):67–81.
Wassef M, Margueron R. Regulation of cellular identity by polycomb and trithorax proteins. In: Göndör A, editor. Chromatin regulation and dynamics. Amsterdam: Elsevier; 2017. p. 165–89.
Becker PB, Hörz W. ATP-dependent nucleosome remodeling. Annu Rev Biochem. 2002;71:247–73.
Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304.
Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463(7280):474–84.
Workman JL. Nucleosome displacement in transcription. Genes Dev. 2006;20(15):2009–17.
Saladi SV, de la Serna IL. ATP dependent chromatin remodeling enzymes in embryonic stem cells. Stem Cell Rev Rep. 2010;6(1):62–73.
Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610.
Csankovszki G, Nagy A, Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol. 2001;153(4):773–83.
Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–5.
Razin A, Cedar H. DNA methylation and gene expression. Microbiol Rev. 1991;55(3):451–8.
Kandi V, Vadakedath S. Effect of DNA methylation in various diseases and the probable protective role of nutrition: a mini-review. Cureus. 2015;7(8):8–13.
Lee GE, Kim JH, Taylor M, Muller MT. DNA methyltransferase 1-associated protein (DMAP1) is a co-repressor that stimulates DNA methylation globally and locally at sites of double strand break repair. J Biol Chem. 2010;285(48):37630–40.
Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, Koch S, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. 2010;5(12):1–9.
Münzel M, Globisch D, Brückl T, Wagner M, Welzmiller V, Michalakis S, et al. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew Chem Int Ed. 2010;49(31):5375–7.
Schübeler D. Function and information content of DNA methylation. Nature. 2015;517:321–6.
Ichiyanagi T, Ichiyanagi K, Miyake M, Sasaki H. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res. 2013;41(2):738–45.
Swygert SG, Peterson CL. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Biophys Acta. 2014;1839(8):728–36.
Jorgensen BG, Berent RM, Ha SE, Horiguchi K, Sasse KC, Becker LS, et al. DNA methylation, through DNMT1, has an essential role in the development of gastrointestinal smooth muscle cells and disease. Cell Death Dis. 2018;9(5):2395–402.
Gujar H, Weisenberger DJ, Liang G. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes (Basel). 2019;10(2):172.
Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311(5759):395–8.
Jeltsch A, Ehrenhofer-Murray A, Jurkowski TP, Lyko F, Reuter G, Ankri S, Nellen W, et al. Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol. 2017;14(9):1108–23.
Norvil AB, Petell CJ, Alabdi L, Wu L, Rossie S, Gowher H. Dnmt3b methylates DNA by a noncooperative mechanism, and its activity is unaffected by manipulations at the predicted dimer interface. Biochemistry. 2018;57(29):4312–24.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57.
Fatemi M, Hermann A, Gowher H, Jeltsch A. Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur J Biochem. 2002;269(20):4981–4.
Zou X, Ma W, Solov’Yov IA, Chipot C, Schulten K. Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res. 2012;40(6):2747–58.
Liu K, Xu C, Lei M, Yang A, Loppnau P, Hughes TR, et al. Structural basis for the ability of MBD domains to bind methyl-CG and TG sites in DNA. J Biol Chem. 2018;293(19):7344–54.
Jarroux J, Morillon A, Pinskaya M. Long non coding RNA biology. Adv Exp Med Biol. 2017;1008:1–46.
Su Y, Wu H, Pavlosky A, Zou LL, Deng X, Zhang ZX, et al. Regulatory non-coding RNA: new instruments in the orchestration of cell death. Cell Death Dis. 2016;7:e2333.
Flynn RA, Chang HY. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell. 2014;14(6):752–61.
Garitano-trojaola A, Agirre X, Prósper F, Fortes P. Long non-coding RNAs in haematological malignancies. Int J Mol Sci. 2013;14(8):15386–422.
Erson-bensan AE. Noncoding RNAs and cancer noncoding RNAs and cancer. Turk J Biol. 2014;38:817–28.
Amin N, Mcgrath A, Chen YP. Evoluation of deep learning in non-coding RNA classification. Nat Mach Intell. 2019;1(5):246–56.
Friedman RC, Farh KKH, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105.
Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20(1):21–37.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74.
Yates LA, Norbury CJ, Gilbert RJ. The long and short of microRNA. Cell. 2013;153(3):516–9.
Huumonen K, Korkalainen M, Viluksela M, Lahtinen T, Naarala J, Juutilainen J. Role of microRNAs and DNA methyltransferases in transmitting induced genomic instability between cell generations. Front Public Health. 2014;2:139.
Shivram H, Le SV, Iyer VR. MicroRNAs reinforce repression of PRC2 transcriptional targets independently and through a feed-forward regulatory network. Genome Res. 2019;29(2):184–92.
Yao Q, Chen Y, Zhou X. The roles of microRNAs in epigenetic regulation. Curr Opin Chem Biol. 2019;51:11–7.
Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science. 2011;332(6031):848–52.
Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, et al. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303(5658):672–6.
Mattick JS, Rinn JL. Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol. 2015;22(1):5–7.
Elia L, Quintavalle M. Epigenetics and vascular diseases: influence of non-coding RNAs and their clinical implications. Front Cardiovasc Med. 2017;4:26.
Böhmdorfer G, Wierzbicki AT. Control of chromatin structure by long noncoding RNA. Trends Cell Biol. 2015;25(10):623–32.
Ng SY, Johnson R, Stanton LW. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012;31(3):522–33.
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322(5902):750–6.
Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143(1):46–58.
Nesmith AP, Wagner MA, Pasqualini FS, et al. A human in vitro model of Duchenne muscular dystrophy muscle formation and contractility. J Cell Biol. 2016;215(1):47–56.
Chal J, Pourquié O. Making muscle: skeletal myogenesis in vivo and in vitro. Development. 2017;144(12):2104–22.
Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci. 2018;19(6):1801.
Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, and therapeutic aspects. Antioxid Redox Signal. 2013;18(17):2352–63.
Saini J, McPhee JS, Al-Dabbagh S, Stewart CE, Al-Shanti N. Regenerative function of immune system: Modulation of muscle stem cells. Ageing Res Rev. 2016;27:67–76.
Schultz E, Jaryszak DL, Valliere CR. Response of satellite cells to focal skeletal muscle injury. Muscle Nerve. 1985;8(3):217–22.
Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129(5):999–1010.
Davegårdh C, Broholm C, Perfilyev A, Henriksen T, García-Calzón S, Peijs L, et al. Abnormal epigenetic changes during differentiation of human skeletal muscle stem cells from obese subjects. BMC Med. 2017;15:39.
Otto A, Collins-Hooper H, Patel K. The origin, molecular regulation and therapeutic potential of myogenic stem cell populations. J Anat. 2009;215(5):477–97.
Naito M, Mori M, Inagawa M, Miyata K, Hashimoto N, Tanaka S, et al. Dnmt3a regulates proliferation of muscle satellite cells via p57Kip2. PLoS Genet. 2016;12(7):e1006167.
McKinnell IW, Ishibashi J, Le Grand F, Punch VGJ, Addicks GC, Greenblatt JF, et al. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat Cell Biol. 2008;10(1):77–84.
Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93:23–67.
Liu L, Cheung TH, Charville GW, Hurgo BM, Leavitt T, Shih J, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4(1):189–204.
Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004;18(21):2627–38.
Handy DE, Castro R, Loscalzo J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation. 2011;123(19):2145–56.
Crist CG, Montarras D, Buckingham M. Muscle satellite cells are primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell Stem Cell. 2012;11(1):118–26; Erratum in: Cell Stem Cell. 2012;11(2):279.
Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, et al. Phosphorylation of eIF2α Is a translational control mechanism regulating muscle stem cell quiescence and self-renewal. Cell Stem Cell. 2016;18(1):79–90.
de Morrée A, van Velthoven CTJ, Gan Q, et al. Staufen1 inhibits MyoD translation to actively maintain muscle stem cell quiescence. Proc Natl Acad Sci USA. 2017;114(43):E8996–9005.
Juan AH, Derfoul A, Feng X, Ryall JG, Dell’Orso S, Pasut A, et al. Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev. 2011;25(8):789–94.
Luo SW, Zhang C, Zhang B, Kim CH, Qiu YZ, Du QS, et al. Regulation of heterochromatin remodelling and myogenin expression during muscle differentiation by FAK interaction with MBD2. EMBO J. 2009;28(17):2568–82.
Boonsanay V, Zhang T, Georgieva A, Kostin S, Qi H, Yuan X, Zhou Y, Braun T. Regulation of skeletal muscle stem cell quiescence by Suv4-20h1-dependent facultative heterochromatin formation. Cell Stem Cell. 2016;18(2):229–42.
Lande-Diner L, Zhang J, Ben-Porath I, Amariglio N, Keshet I, Hecht M, et al. Role of DNA methylation in stable gene repression. J Biol Chem. 2007;282(16):12194–200.
Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12(1):51–62.
Ma K, Chan JK, Zhu G, Wu Z. Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol Cell Biol. 2005;25(9):3575–82.
Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6(2):233–44.
Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callén E, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22(15):2048–61.
Woodhouse S, Pugazhendhi D, Brien P, Pell JM. Ezh2 maintains a key phase of muscle satellite cell expansion but does not regulate terminal differentiation. J Cell Sci. 2013;126(Pt2):565–79.
Castel D, Baghdadi MB, Mella S, Gayraud-Morel B, Marty V, Cavaillé J, et al. Small-RNA sequencing identifies dynamic microRNA deregulation during skeletal muscle lineage progression. Sci Rep. 2018;8(1):1–13.
Crist CG, Montarras D, Pallafacchina G, Rocancourt D, Cumano A, Conway SJ, et al. Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc Natl Acad Sci USA. 2009;106(32):13383–7.
Koning M, Werker PM, van Luyn MJ, Krenning G, Harmsen MC. A global downregulation of microRNAs occurs in human quiescent satellite cells during myogenesis. Differentiation. 2012;84(4):314–21.
Martinet C, Monnier P, Louault Y, Benard M, Gabory A, Dandolo L. H19 controls reactivation of the imprinted gene network during muscle regeneration. Development. 2016;143(6):962–71.
Ling H, Michlewski G, Calin GA, Liz J, Portela A, Soler M, et al. Short article regulation of pri-miRNA processing by a long noncoding RNA transcribed from an ultraconserved region. Mol Cell. 2014;55:138–47.
Liu W, Wen Y, Bi P, Lai X, Liu XS, Liu X, et al. Hypoxia promotes satellite cell self-renewal and enhances the efficiency of myoblast transplantation. Development. 2012;139(16):2857–65.
Hernández-Hernández JM, García-González EG, Brun CE, Rudnicki MA. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin Cell Dev Biol. 2017;72:10–8.
Muscat GE, Perry S, Prentice H, Kedes L. The human skeletal alpha-actin gene is regulated by a muscle-specific enhancer that binds three nuclear factors. Gene Expr. 1992;2(2):111–26.
Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, Reggiani C. Developmental myosins: expression patterns and functional significance. Skeletal Muscle. 2015;5(1):1–14.
Murphy ACH, Young PW. The actinin family of actin cross-linking proteins—a genetic perspective. Cell Biosci. 2015;5:49.
Buckingham M, Relaix F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin Cell Dev Biol. 2015;44:115–25.
Von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci USA. 2013;110:16474–9.
Olguin HC, Yang Z, Tapscott SJ, Olwin BB. Reciprocal inhibition between Pax7 and muscle regulatory factors modulates myogenic cell fate determination. J Cell Biol. 2007;177(5):769–79.
Wang YX, Bentzinger CF, Rudnicki MA. Molecular regulation of determination in asymmetrically dividing muscle stem cells. Cell Cycle. 2013;12(1):3–4.
Kawabe YI, Wang YX, McKinnell IW, Bedford MT, Rudnicki MA. Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell. 2012;11:333–45.
Olguín HC, Pisconti A. Marking the tempo for myogenesis: Pax7 and the regulation of muscle stem cell fate decisions. J Cell Mol Med. 2012;16(5):1013–25.
Soleimani VD, Punch VG, Kawabe Y, Jones AE, Palidwor GA, Porter CJ, et al. Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev Cell. 2012;22(6):1208–20.
L’honore A, Rana V, Arsic N, Franckhauser C, Lamb NJ, Fernandez A. Identification of a new hybrid serum response factor and myocyte enhancer factor 2-binding element in MyoD enhancer required for MyoD expression during myogenesis. Mol Biol Cell. 2007;18(6):1992–2001.
Li S, Czubryt MP, McAnally J, Bassel-Duby R, Richardson JA, Wiebel FF, et al. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc Natl Acad Sci USA. 2005;102(4):1082–7.
Han X, Yang F, Cao H, Liang Z. Malat1 regulates serum response factor through miR-133 as a competing endogenous RNA in myogenesis. FASEB J. 2015;29(7):3054–64.
Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435(7044):948–53.
MacQuarrie KL, Yao Z, Fong AP, Diede SJ, Rudzinski ER, Hawkins DS, et al. Comparison of genome-wide binding of MyoD in normal human myogenic cells and rhabdomyosarcomas identifies regional and local suppression of promyogenic transcription factors. Mol Cell Biol. 2013;33(4):773–84.
Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR. Muscle satellite cells adopt divergent fates: A mechanism for self-renewal? J Cell Biol. 2004;166(3):347–57.
Deato MDE, Marr MT, Sottero T, Inouye C, Hu P, Tjian R. MyoD targets TAF3/TRF3 to activate myogenin transcription. Mol Cell. 2008;32(1):96–105.
Tomczak KK, Marinescu VD, Ramoni MF, Sanoudou D, Montanaro F, Han M, Kunkel LM, Kohane IS, Beggs AH. Expression profiling and identification of novel genes involved in myogenic differentiation. FASEB J. 2004;18(2):403–5.
Michela P, Velia V, Aldo P, Ada P. Role of connexin 43 in cardiovascular diseases. Eur J Pharmacol. 2015;768:71–6.
Zammit PS. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol. 2017;72:19–32.
Cabane C, Coldefy AS, Yeow K, Dérijard B. The p38 pathway regulates Akt both at the protein and transcriptional activation levels during myogenesis. Cell Signal. 2004;16(12):1405–15.
Puri PL, Iezzi S, Stiegler P, Chen TT, Schiltz RL, Muscat GEO, et al. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol Cell. 2001;8(4):885–97.
McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci U S A. 2000;97(26):14400–5.
Sartorelli V, Caretti G. Mechanisms underlying the transcriptional regulation of skeletal myogenesis. Curr Opin Genet Dev. 2005;15(5):528–35.
Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20(9):1123–36.
Stojic L, Jasencakova Z, Prezioso C, Stützer A, Bodega B, Pasini D, Klingberg R, Mozzetta C, Margueron R, Puri PL, Schwarzer D, Helin K, Fischle W, Orlando V. Chromatin regulated interchange between polycomb repressive complex 2 (PRC2)-Ezh2 and PRC2-Ezh1 complexes controls myogenin activation in skeletal muscle cells. Epigenetics Chromatin. 2011;4:16.
Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449(7163):731–4.
Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA. 2007;104(47):18439–44.
Seenundun S, Rampalli S, Liu QC, Aziz A, Palii C, Hong S, Blais A, Brand M, Ge K, Dilworth FJ. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 2010;29(8):1401–11.
Verrier L, Escaffit F, Chailleux C, Trouche D, Vandromme M. A new isoform of the histone demethylase JMJD2A/KDM4A is required for skeletal muscle differentiation. PLoS Genet. 2011;7(6):e1001390.
Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125(3):483–95.
Tao Y, Neppl RL, Huang ZP, Chen J, Tang RH, Cao R, et al. The histone methyltransferase Set7/9 promotes myoblast differentiation and myofibril assembly. J Cell Biol. 2011;194(4):551–65.
Giacinti C, Bagella L, Puri PL, Giordano A, Simone C. MyoD recruits the cdk9/cyclin T2 complex on myogenic-genes regulatory regions. J Cell Physiol. 2006;206(3):807–13.
Ohkawa Y, Yoshimura S, Higashi C, Marfella CGA, Dacwag CS, Tachibana T, et al. Myogenin and the SWI/SNF ATPase Brg1 maintain myogenic gene expression at different stages of skeletal myogenesis. J Biol Chem. 2007;282(9):6564–70.
de la Serna IL, Ohkawa Y, Berkes CA, Bergstrom DA, Dacwag CS, Tapscott SJ, Imbalzano AN. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol Cell Biol. 2005;25(10):3997–4009.
Forcales SV, Albini S, Giordani L, Malecova B, Cignolo L, Chernov A, et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 2012;31(2):301–16.
Ling BMT, Gopinadhan S, Kok WK, Shankar SR, Gopal P, Bharathy N, et al. G9a mediates Sharp-1-dependent inhibition of skeletal muscle differentiation. Mol Biol Cell. 2012;23(24):4778–85.
Hinterberger TJ, Sassoon DA, Rhodes SJ, Konieczny SF. Expression of the muscle regulatory factor MRF4 during somite and skeletal myofiber development. Dev Biol. 1991;147(1):144–56.
Horak M, Novak J, Bienertova-Vasku J. Muscle-specific microRNAs in skeletal muscle development. Dev Biol Elsevier. 2016;410:1–13.
Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, et al. NF-κB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell. 2008;14:369–81.
Juan AH, Kumar RM, Marx JG, Young RA, Sartorelli V. Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell. 2009;36:61–74.
Chung FW, Tellam RL. MicroRNA-26a targets the histone methyltransferase enhancer of zeste homolog 2 during myogenesis. J Biol Chem. 2008;283(15):9836–43.
Winbanks CE, Wang B, Beyer C, Koh P, White L, Kantharidis P, et al. TGF-β regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J Biol Chem. 2011;286(16):13805–14.
Hassan N, Tchao J, Tobita K. Concise review: skeletal muscle stem cells and cardiac lineage: potential for heart repair. Stem Cells Transl Med. 2014;3(2):183–93.
Koutalianos D, Koutsoulidou A, Mastroyiannopoulos NP, Furling D, Phylactou LA. MyoD transcription factor induces myogenesis by inhibiting Twist-1 through miR-206. J Cell Sci. 2015;128(19):3631–45.
Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci USA. 2006;103(23):8721–6.
Yu X, Zhang Y, Li T, Ma Z, Jia H, Chen Q, et al. Long non-coding RNA Linc-RAM enhances myogenic differentiation by interacting with MyoD. Nat Commun. 2017;8:14016.
Hitachi K, Nakatani M, Takasaki A, Ouchi Y, Uezumi A, Ageta H, et al. Myogenin promoter-associated lncRNA Myoparr is essential for myogenic differentiation. EMBO Rep. 2019;20(3):e47468.
Sui Y, Han Y, Zhao X, Li D, Li G. Long non-coding RNA Irm enhances myogenic differentiation by interacting with MEF2D. Cell Death Dis. 2019;10:181.
Sincennes MC, Brun CE, Rudnicki MA. Concise review: epigenetic regulation of myogenesis in health and disease. Stem Cells Transl Med. 2016;5(3):282–90.
Terragni J, Zhang G, Sun Z, Pradhan S, Song L, Crawford GE, Lacey M, Ehrlich M. Notch signaling genes: myogenic DNA hypomethylation and 5-hydroxymethylcytosine. Epigenetics. 2014;9(6):842–50.
Segalés J, Perdiguero E, Muñoz-Cánoves P. Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front Cell Dev Biol. 2016;4:91.
Segalés J, Islam ABMMK, Kumar R, Liu Q, Sousa-victor P, Dilworth FJ, et al. Chromatin-wide and transcriptome profiling integration uncovers p38 α MAPK as a global regulator of skeletal muscle differentiation. Skelet Muscle. 2016;6:9.
Hausburg MA, Doles JD, Clement SL, Cadwallader AB, Hall MN, Blackshear PJ, et al. Post-transcriptional regulation of satellite cell quiescence by TTP-mediated mRNA decay. Elife. 2015;4:e03390.
Dacwag CS, Bedford MT, Sif S, Imbalzano AN. Distinct protein arginine methyltransferases promote ATP-dependent chromatin remodeling function at different stages of skeletal muscle differentiation. Mol Cell Biol. 2009;29(7):1909–21.
Gillespie MA, Le Grand F, Scimè A, Kuang S, von Maltzahn J, Seale V, et al. p38-{gamma}-dependent gene silencing restricts entry into the myogenic differentiation program. J Cell Biol. 2009;187(7):991–1005.
Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, et al. TNF/p38α /polycomb signalling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7(4):455–69.
Cuadrado A, Corrado N, Perdiguero E, Lafarga V, Muñoz-Canoves P, Nebreda AR. Essential role of p18Hamlet/SRCAP-mediated histone H2A.Z chromatin incorporation in muscle differentiation. EMBO J. 2010;29(12):2014–25.
Lluís F, Perdiguero E, Nebreda AR, Muñoz-Cánoves P. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol. 2006;16(1):36–44.
Lluís F, Ballestar E, Suelves M, Esteller M, Muñoz-Cánoves P. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J. 2005;24(5):974–84.
Consalvi S, Brancaccio A, Dall’agnese A, Puri PL, Palacios D. Praja1 E3 ubiquitin ligase promotes skeletal myogenesis through degradation of EZH2 upon p38α activation. Nat Commun. 2017;8:13956.
Penn BH, Bergstrom DA, Dilworth FJ, Bengal E, Tapscott SJ. A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 2004;18(19):2348–53.
Rampalli S, Li LF, Mak E, Ge K, Brand M, Tapscott SJ, et al. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat Struct Mol Biol. 2007;14(12):1150–6.
Serra C, Palacios D, Mozzetta C, et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol Cell. 2007;28(2):200–13.
Dilworth FJ, Seaver KJ, Fishburn AL, Htet SL, Tapscott SJ. In vitro transcription system delineates the distinct roles of the coactivators pCAF and p300 during MyoD/E47-dependent transactivation. Proc Natl Acad Sci USA. 2004;101(32):11593–8.
McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408(6808):106–11.
Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M, et al. MicroRNAs involved in molecular circuitries relevant for the duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 2010;12(4):341–51.
Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455(7211):411–5.
Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38(2):228–33.
Wei W, He H, Zhang W, Zhang H, Bai J, Liu H, et al. miR-29 targets Akt3 to reduce proliferation and facilitate differentiation of myoblasts in skeletal muscle development. Cell Death Dis. 2013;4(6):e668.
Guess MG, Barthel KK, Harrison BC, Leinwand LA. miR-30 Family microRNAs regulate myogenic differentiation and provide negative feedback on the microRNA pathway. PLoS ONE. 2015;10(2):e0118229.
Yang Q, Tang Y, Imbrogno K, Lu A, Proto JD, Chen A, et al. AAV-based shRNA silencing of NF-κB ameliorates muscle pathologies in mdx mice. Gene Ther. 2012;19(12):1196–204.
Dumont NA, Rudnicki MA. Targeting muscle stem cell intrinsic defects to treat Duchenne muscular dystrophy. NPJ Regen Med. 2016;1:16006.
Ribeiro AF, Souza LS, Almeida CF, Ishiba R, Fernandes SA, Guerrieri DA, et al. Muscle satellite cells and impaired late stage regeneration in different murine models for muscular dystrophies. Sci Rep. 2019;9:1–11.
Boldrin L, Zammit PS, Morgan JE. ScienceDirect Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. 2015;14:20–9.
Chang NC, Chevalier FP, Rudnicki MA. Satellite cells in muscular dystrophy—lost in polarity. Trends Mol Med. 2016;22(6):479–96.
Kottlors M, Kirschner J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010;340(3):541–8.
de Souza GT, de Zanette RSS, do Amaral DLAS, da Guia FC, Maranduba CP, de Souza CM, et al. Satellite cells: regenerative mechanisms and applicability in muscular dystrophy. Stem Cells Int. 2015;2015:487467.
Miyagoe-Suzuki Y, Fukada S, Take S. Muscle satellite cells and duchenne muscular dystrophy. In: Hegde M, editor. Muscular dystrophy. London: IntechOpen; 2012. p. 20112.
Duan D. Duchenne muscular dystrophy gene therapy: lost in translation? Res Rep Biol. 2011;2:31.
Troy A, Cadwallader AB, Fedorov Y, Tyner K, Tanaka KK, Olwin BB. Coordination of satellite cell activation and self-renewal by par-complex-dependent asymmetric activation of p38α/β MAPK. Cell Stem Cell. 2012;11:541–53.
Chang NC, Sincennes MC, Chevalier FP, Brun CE, Lacaria M, Segalés J, et al. The dystrophin glycoprotein complex regulates the epigenetic activation of muscle stem cell commitment. Cell Stem Cell. 2018;22(5):755-768.e6.
Acharyya S, Sharma SM, Cheng AS, Ladner KJ, He W, Wang H, et al. TNF inhibits notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression : implications in duchenne muscular dystrophy. PLoS ONE. 2010;5(8):e12479.
Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci USA. 2009;106(5):1679.
Consalvi S, Saccone V, Giordani L, Minetti G, Mozzetta C, Puri PL. Histone deacetylase inhibitors in the treatment of muscular dystrophies: epigenetic drugs for genetic diseases. Mol Med. 2011;17(5–6):457–65.
Moratal C, Arrighi N, Dechesne CA, Dani C. Control of muscle fibro-adipogenic progenitors by myogenic lineage is altered in aging and duchenne muscular dystrophy. Cell Physiol Biochem. 2019;53(6):1029–45.
Uezumi A, Fukada SI, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12:143–52.
Marinkovic M, Fuoco C, Sacco F, Perpetuini AC, Giuliani G, Micarelli E, et al. Fibro-adipogenic progenitors of dystrophic mice are insensitive to NOTCH regulation of adipogenesis. Life Sci Alliance. 2019;2(3):1–17.
Mázala DA, Novak JS, Hogarth MW, Nearing M, Adusumalli P, Tully CB, et al. TGF- β-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight. 2020;5(6):e135703.
Ismaeel A, Kim JS, Kirk JS, Smith RS, Bohannon WT, Koutakis P. Role of transforming growth factor-β in skeletal muscle fibrosis: A review. Int J Mol Sci. 2019;20(10):2446.
Karantzali E, Schulz H, Hummel O, Hubner N, Hatzopoulos A, Kretsovali A. Histone deacetylase inhibition accelerates the early events of stem cell differentiation: transcriptomic and epigenetic analysis. Genome Biol. 2008;9(4):R65.
Saccone V, Consalvi S, Giordani L, et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 2014;28(8):841–57.
Zanotti S, Gibertini S, Curcio M, Savadori P, Pasanisi B, Morandi L, et al. Opposing roles of miR-21 and miR-29 in the progression of fibrosis in Duchenne muscular dystrophy. Biochim Biophys Acta. 2015;1852:1451–64.
Bowen T, Jenkins RH, Fraser DJ. MicroRNAs, transforming growth factor beta-1, and tissue fibrosis. J Pathol. 2013;229(2):274–85.
Wang L, Zhou L, Jiang P, Lu L, Chen X, Lan H, et al. Loss of miR-29 in myoblasts contributes to dystrophic muscle pathogenesis. Mol Ther. 2012;20(6):1222–33.
Guiraud S, Edwards B, Babbs A, Squire SE, Berg A, Moir L, et al. The potential of utrophin and dystrophin combination therapies for duchenne muscular dystrophy. Hum Mol Genet. 2019;28(13):2189–200.
Kennedy TL, Moir L, Hemming S, Edwards B, Squire S, Davies K, et al. Utrophin influences mitochondrial pathology and oxidative stress in dystrophic muscle. Skelet Muscle. 2017;7(1):22.
Lang JM, Esser KA, Dupont-Versteegden EE. Altered activity of signaling pathways in diaphragm and tibialis anterior muscle of dystrophic mice. Exp Biol Med (Maywood). 2004;229(6):503–11.
Consalvi S, Sandoná M, Saccone V. Epigenetic reprogramming of muscle progenitors: inspiration for clinical therapies. Stem Cells Int. 2016;2016:6093601.
Nebbioso A, Manzo F, Miceli M, Conte M, Manente L, Baldi A, et al. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 2009;10(7):776–82.
Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol. 2018;8:92.
Iezzi S, Cossu G, Nervi C, Sartorelli V, Puri PL. Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases. Proc Natl Acad Sci USA. 2002;99(11):7757–62.
Iezzi S, Di Padova M, Serra C, Caretti G, Simone C, Maklan E, et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell. 2004;6(5):673–84.
Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12(10):1147–50.
Tsuchida K. Myostatin inhibition by a follistatin-derived peptide ameliorates the pathophysiology of muscular dystrophy model mice. Acta Myol. 2008;27(1):14–8.
Mendell JR, Sahenk Z, Malik V, Gomez AM, Flanigan KM, Lowes LP, et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol Ther. 2015;23(1):192–201.
Zhu J, Li Y, Lu A, Gharaibeh B, Ma J, Kobayashi T, et al. Follistatin improves skeletal muscle healing after injury and disease through an interaction with muscle regeneration, angiogenesis, and fibrosis. Am J Pathol. 2011;179(2):915–30.
Consalvi S, Mozzetta C, Bettica P, Germani M, Fiorentini F, Del Bene F, et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med. 2013;19(1):79–87.
Bettica P, Petrini S, D’Oria V, D’Amico A, Catteruccia M, Pane M, et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2016;26:643–9.
Desguerre I, Mayer M, Leturcq F, Barbet J, Gherardi RK, Christov C. Endomysial fibrosis in duchenne muscular dystrophy : a marker of poor outcome associated with macrophage alternative activation. J Neuropathol Exp Neurol. 2009;68(7):762–73.
Peverelli L, Testolin S, Villa L, D’Amico A, Petrini S, Favero C, et al. Histologic muscular history in steroid-treated and untreated patients with Duchenne dystrophy. Neurology. 2015;85(21):1886–93.
Loro E, Sengupta K, Bogdanovich S, Whig K, Schultz DC, Huryn DM, et al. High-throughput identification of post-transcriptional utrophin up-regulators for Duchenne muscle dystrophy (DMD) therapy. Sci Rep. 2020;10:2132.
Bajanca F, Vandel L. Epigenetic regulators modulate muscle damage in duchenne muscular dystrophy model. PLoS Curr. 2017;9.
Widrick JJ, Kawahara G, Alexander MS, Beggs AH, Kunkel LM. Discovery of novel therapeutics for muscular dystrophies using zebrafish phenotypic screens. J Neuromuscul Dis. 2019;6(3):271–87.
Farr GH III, Morris M, Gomez A, Pham T, Kilroy E, Parker EU, et al. A novel chemical-combination screen in zebrafish identifies epigenetic small molecule candidates for the treatment of Duchenne muscular dystrophy. Skelet Muscle. 2020;10:29.
Benedetti R, Conte M, Altucci L. Targeting histone deacetylases in diseases: where are we? Antioxid Redox Signal. 2015;23(1):99–126.
Acknowledgements
Not applicable.
Funding
This work was supported by the Polish National Science Centre [Grant No. 2018/30/E/NZ5/00065] (to P.K.).
Author information
Authors and Affiliations
Contributions
A.R., A.S. and P.K. wrote the manuscript and prepared the figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rugowska, A., Starosta, A. & Konieczny, P. Epigenetic modifications in muscle regeneration and progression of Duchenne muscular dystrophy. Clin Epigenet 13, 13 (2021). https://doi.org/10.1186/s13148-021-01001-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01001-z