
Abstract
The clinical, social and economic burden of cardiovascular disease (CVD) associated with diabetes underscores an urgency for understanding the disease aetiology. Evidence suggests that the hyperglycaemia associated with diabetes is, of itself, causal in the development of endothelial dysfunction (ED) which is recognised to be the critical determinant in the development of CVD. It is further recognised that epigenetic modifications associated with changes in gene expression are causal in both the initiation of ED and the progression to CVD. Understanding whether and how hyperglycaemia induces epigenetic modifications therefore seems crucial in the development of preventative treatments. A mechanistic link between energy metabolism and epigenetic regulation is increasingly becoming explored as key energy metabolites typically serve as substrates or co-factors for epigenetic modifying enzymes. Intriguing examples are the ten-eleven translocation and Jumonji C proteins which facilitate the demethylation of DNA and histones respectively. These are members of the 2-oxoglutarate-dependent dioxygenase superfamily which require the tricarboxylic acid metabolite, α-ketoglutarate and molecular oxygen (O2) as substrates and Fe (II) as a co-factor. An understanding of precisely how the biochemical effects of high glucose exposure impact upon cellular metabolism, O2 availability and cellular redox in endothelial cells (ECs) may therefore elucidate (in part) the mechanistic link between hyperglycaemia and epigenetic modifications causal in ED and CVD. It would also provide significant proof of concept that dysregulation of the epigenetic landscape may be causal rather than consequential in the development of pathology.
Similar content being viewed by others


Introduction
A startling increase in obesity driven by a sedentary lifestyle and high-calorie diet has resulted in a worldwide epidemic of type 2 diabetes (T2D) that is set to further rise dramatically [1, 2]. Thus, current estimates suggest that by 2045, 700 million people will be living with diabetes [2]. The hyperglycaemia characteristic of diabetes represents a major risk factor for the development of cardiovascular disease (CVD) which is ultimately the most common cause of mortality in affected individuals [3]. Further, the phenomena of hyperglycaemic memory, where phenotypic alterations persist despite the restoration of normal glycaemic control, renders CVD a life-long risk to individuals who have been affected by diabetes. This predisposition to CVD additionally affects the offspring of mothers who exhibit gestational diabetes (defined as glucose intolerance resulting in hyperglycaemia with onset or first discovery in pregnancy), cases of which have similarly burgeoned with the obesity crisis [4]. Such developmental priming has also been shown to exhibit inheritance to the second (F2) generation and therefore additionally compounds the current problems onto the future population [5, 6]. The economic burden associated with this is vast and in the current economic climate, unaffordable. It is therefore vital that better preventative and therapeutic clinical strategies are developed. The underlying mechanisms which predispose individuals who are, or have in the past been exposed to, hyperglycaemia must therefore be determined.
Hyperglycaemia, endothelial dysfunction and cardiovascular disease
Diabetes is associated with the development of complications affecting both the microvasculature (diabetic retinopathy, neuropathy and nephropathy) and macrovasculature (peripheral artery disease, cardiomyopathy, myocardial infarction and stroke) [7]. Macrovascular complications are primarily responsible for the reduced life expectancy of diabetic patients; T2D is associated with a two to sixfold greater risk of cardiovascular mortality than amongst non-diabetics [8, 9] and similarly high relative risks are reported for type 1 diabetics [10]. It is well established that the dysfunction of the vascular endothelium, which comprises the one cell thick, innermost layer of the vascular wall is both a hallmark of vascular disease and is critical in the development of both macrovascular and microvascular pathologies [11, 12]. Endothelial dysfunction (ED) is characterised by a proinflammatory, prothrombotic state with impaired vasodilation [11, 13, 14] and a reduced bioavailability of the critical vascular mediator, nitric oxide (NO) [11]. Given the direct contact between endothelial cells (ECs) and blood, it is not surprising that they are critically affected by metabolic changes in blood plasma such as hyperglycaemia. Indeed, a significant body of evidence from in vitro studies suggests that hyperglycaemia per se is causative in the development of ED [15,16,17,18,19,20,21,22,23,24,25,26,27]. Thus, it has been shown that even short-term exposure of ECs to high glucose is sufficient to induce monocyte adhesion [15], promote endothelial to mesenchymal transition [16], activate prothrombotic signalling [20], reduce eNOS activity [21] and increase apoptosis [22]. High-glucose culture conditions also enhanced the response of human umbilical vein endothelial cells (HUVECs) to the pro-inflammatory stimulus interleukin-1β, resulting in augmented endothelial ICAM-1 and VCAM-1 expression and promoting increased leukocyte adhesion [26], further demonstrating the effect of hyperglycaemia upon the inflammatory responses characteristic of ED.
Endothelial dysfunction and epigenetics
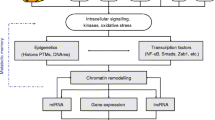
The mechanisms whereby chronic hyperglycaemia mediate ED have classically been attributed to involve the overproduction of mitochondrial-generated superoxide via PKG activation and upregulation of the hexosamine and polyol pathways leading to the production of advanced glycosylation end-products (AGEs) [28,29,30]. However, these mechanisms cannot account for the observation of hyperglycaemic memory, including developmental priming. It is now evident from epidemiological and clinical studies that the development of T2D and associated CVD is correlated with, and in part depends upon, environmentally influenced epigenetic changes which modulate gene expression (reviewed in [31]). The term epigenetics has come to mean heritable alterations in gene expression and phenotype that do not involve changes in the primary DNA sequence [32]. Epigenetic mechanisms thus involve stable cellular modifications, capable of governing functional changes in gene expression patterns in response to external environmental stimuli, or as part of normal development [33]. Epigenetic modifications include changes to histone post-translational modifications, non-coding RNA and DNA methylation patterns, which serve to activate or repress transcription of a gene [32]. In essence, epigenetic modifications govern remodelling of chromatin, placing it in a conformation which is ‘open’ (euchromatin) or ‘closed’ (heterochromatin) to transcription factors and other transcription machinery [32]. The epigenetic landscape of a cell is thus a major regulator of gene transcription and accounts for the vastly different phenotypes and functions of cells containing identical DNA, providing a link between phenotype and genotype.
Hyperglycaemia and epigenetic regulation
Although not documented here, the (ever-increasing) recognised associations between changes in the epigenome and T2D-related CVD are the subject of many excellent reviews [34,35,36,37]. Further, the study of the regulation of these epigenetic changes in the aetiology and progression of ED and CVD in diabetes is an emerging field. The influence of high glucose upon the EC epigenetic landscape and transcriptome has clearly been demonstrated in vitro. Thus in an important study, it was shown that the incubation of human aortic endothelial cells (HAECs) in high glucose (30 mM) for 2 days resulted in significant changes in the expression of genes associated with diabetes and vascular complications, many of which correlated with (histone and DNA) epigenetic changes at these loci [38]. Thus, hyperglycaemia per se can mediate epigenetic changes that associate with changes in transcription in ECs.
Of particular significance in this regard is the emerging intimate relationship between metabolic alterations and epigenetic modifications in both health and disease [39, 40]. The epigenome must be maintained by the balance of the actions of a variety of epigenetic modifiers, including the ‘writers’ and ‘erasures’ of epigenetic marks such as posttranslational histone modifications and DNA methylation. Crucially, many of these modifiers use key metabolites (including S-adenosyl methionine (SAM), ATP and acetylCoA) as obligate substrates or co-factors, suggesting a mechanistic link between metabolic changes and epigenetic (dys)regulation of gene transcription (reviewed in [40]). It is therefore tempting to speculate that metabolic changes induced by the hyperglycaemia characteristic of diabetes may affect the activities of these epigenetic modifiers by altering the availability of their substrates (and/or co-factors). In this regard, it is particularly intriguing that the enzymes which facilitate the demethylation of both DNA and histones are members of the superfamily of 2-oxoglutarate-dependent dioxygenases (2-OGDDs). These require the tricarboxylic acid (TCA) metabolite, α-ketoglutarate (2-OG) and molecular oxygen (O2) as substrates and Fe (II) as a co-factor.
DNA methylation
DNA methylation represents the best characterised epigenetic mark to date. It occurs via the transfer of a methyl group from the universal methyl donor; SAM by a DNA methyltransferase (DNMT) to the fifth position of a cytosine ring to form 5-methylcytosine (5mC) [41]. This modification predominantly occurs at CpG sites, which are often found clustered in ‘CpG islands’ of regulatory elements such as promoters and enhancers [41]. The pattern of DNA methylation is regulated by writers and erasers which add and remove methyl groups respectively. Thus, members of the DNMT family act as writers; DNMT3A/DNMT3B are involved in de novo methylation, whilst DNMT1 is involved in the maintenance of the methyl mark in subsequent mitotic divisions [42, 43]. DNMTs are essential for development [42, 43] and play an important role in maintaining genomic integrity [44]. DNA methylation is ‘read’ by various chromatin-binding proteins containing methyl-CpG-binding domains which can form repressive complexes to silence transcription [41].
Traditionally, DNA methylation was believed to be a permanent epigenetic modification which could only be lost passively during DNA replication [45]. However, relatively recently, this notion was challenged by the discovery of ten-eleven translocation (TET) enzymes: TET1, TET2 and TET3 [46] which act as erasers of this modification by active DNA demethylation mechanisms [47, 48]. TETs catalyse the successive oxidations of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (Fig. 1a) [47, 48]. These variants can be either lost passively during DNA replication or actively removed and replaced with unmethylated cytosine by base excision repair machinery including thymine DNA glycosylase (TDG) (Fig. 1a) [48]. Not only is the removal of 5mC important for reversing transcriptional repression, but it is now recognised that the oxidised intermediates formed by TETs may perform distinct regulatory functions in their own right [49]. Evidence for this comes from differential genomic positions of these oxidised derivatives [50, 51], the existence of distinct proteins which recognise and read the cytosine variants [52], and reported roles for 5fC and 5caC in fine-tuning the rate of transcription by RNA polymerase II [53]. As stated above, these TET proteins are 2-OGDDs which require 2-OG and molecular oxygen (O2) as substrates and Fe (II) as a co-factor (Fig. 1b).

Action of ten-eleven translocation (TET) and Jumonji C (JmjC) enzymes. a Cytosine is converted to 5-methylcytosine (5mC) by the addition of a methyl group by DNA methyltransferase enzymes (DNMTs). TET enzymes successively oxidise 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). Base excision repair (BER) machinery excise and replace the intermediates with unmethylated cytosine. b TET enzymes require 2-oxoglutarate (2-OG), Fe2+ and molecular oxygen (O2) for their DNA demethylase activity. c Histone methyltransferases (HMTs) can deposit either repressive or activating methyl marks depending on which site is methylated and to what extent (mono/di/trimethylation). This results in a ‘closed’ and ‘open’ conformation of chromatin, respectively. Hence, the removal of methyl groups by JmjCs can result in the activation or repression of a gene. d JmjC enzymes require 2-oxoglutarate (2-OG), Fe2+ and molecular oxygen (O2) for their histone lysine demethylase activity
Histone methylation
In chromatin, DNA is wrapped around histone proteins to form nucleosomes, which in turn form a ‘beads on a string’ structure that folds to form a 30 nm fibre [54]. Further condensation results in 250 nm supercoiled fibres which make up the chromatid of a chromosome [54]. The canonical nucleosome contains a tetramer of histone H3 and H4, interacting with two dimers of histone H2A-H2B [54]. The tails of these histones protrude from the nucleosome and can be post-translationally modified by methylation, acetylation, phosphorylation, ubiquitination, SUMOylation or O-GlcNAcylation to alter the interaction of DNA with the nucleosome to facilitate or repress gene transcription [55]. Of these, the most well studied are histone methylation and acetylation [41]. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) respectively comprise the writers and erasers of the histone acetylation mark, which occurs predominantly at lysine residues [56]. This neutralises the positive charge of lysine, causing disruption of electrostatic interactions between the histone and DNA, typically resulting in enhanced transcription [57]. The readers of histone acetylation are chromatin-binding proteins containing an acetyl-lysine reader domain, which participate in transcriptional activation [41].
Histone methylation at lysine or arginine residues is conferred by the transfer of a methyl group (again donated from SAM) by histone methyltransferases (HMTs) and (as in the case of DNA methylation) can be actively erased by histone demethylases (Fig. 1c) [56]. Compared to histone acetylation, histone methylation patterns are more complex and have more diverse effects depending on the position and extent of methylation [57]. Thus there are three degrees (mono-, di- and tri-) of methylation which occur on lysine residues, while arginine residues can be monomethylated and symmetrically or asymmetrically demethylated [58]. Further, methylation at specific amino acid residues within the histone tails recruit either activating or repressive complexes. For example, trimethylation of histone 3 lysine 4 (H3K4me3) is generally associated with active transcription (euchromatin), whilst trimethylation of lysine 9 or 27 (H3K9me3, H3K27me3) are associated with transcriptionally repressive state (constitutive and facultative heterochromatin respectively, Fig. 1c) [57]. The complexity of the histone methyl marks is reflected by the large number of HMTs and demethylases which observe a high degree of specificity for the specific lysine/arginine and degree of methylation being modified (reviewed in [59]). In the case of histone demethylases, there are two evolutionarily conserved families which employ distinct mechanisms, both of which involve oxidation of the methyl group. Lys-specific demethylases (LSDs) catalyse a flavin adenine dinucleotide (FAD)-dependent oxidation reaction [60]. By contrast, the Jumonji C demethylases (JmjCs) are, like the TET enzymes, members of the 2-OGDD superfamily which employ molecular oxygen and 2-OG as substrates (Fig. 1d) [61].
Hyperglycaemia and the regulation of TETs and JmjCs
The dependency of the epigenetic demethylases; TETs and JmjCs, upon O2, 2-OG and Fe2+ is particularly intriguing in the context of diabetes, as the availability of each of these factors is potentially affected by hyperglycaemia. We, here, review the current evidence of dysregulation of these epigenetic erasers by hyperglycaemia in the endothelium and explore the known biochemical mechanisms which might underlie this dysregulation.
Hyperglycaemia and availability of molecular oxygen
As stated above, molecular oxygen is an obligate substrate for all 2-OGDDs, of which the best characterised are the related hypoxia-inducible factor-1prolyl hydroxylases (HIF-PHD1–3). Under normoxic conditions, PHDs hydroxylate HIF1α, leading to its association with von Hippel-Lindau and subsequent degradation [62]. The inhibition of PHDs by relative hypoxia, due to their requirement for molecular oxygen, leads to HIF stabilisation and HIF-dependent activation of transcription [62]. Studies performed to determine the KM values for O2 of these enzymes suggest that their activities may be modulated by fluctuations in [O2] over physiological ranges, at least in some tissues, further substantiating their roles as bona fide O2 sensors [63].
Physiological normoxia within eukaryotic cells is maintained by the balance of its diffusional uptake and metabolic usage, of which oxidative phosphorylation (OxPhos) represents by far the largest consumer [64]. An upset in this metabolic flux will therefore impact upon the intracellular [O2] and potentially modulate the activities of enzymes which use O2 as a substrate, including the TET and JmjC demethylases. Despite being positioned in a typically highly oxygenated environment, the primary method of adenosine triphosphate (ATP) production in ECs is glycolysis, which is estimated to generate 85% of endothelial ATP [65, 66]. Thus, compared to other cell types, ECs are much less reliant on oxidative processes in the mitochondria [66] which account for less than 5% of the total cell volume (compared to, for instance, approximately 25% of the hepatocyte cell volume) [64]. Instead, glycolysis allows rapid and sufficient energy production for the supply of homeostatic processes even under hypoxic conditions, such as proliferation and migration required for angiogenesis [65].
To maintain physiological normoxia, the relative consumption of O2 through OxPhos (and glycolysis) is tightly regulated in response to different levels of oxygenation [67] and this balance can be dysregulated in a cell-specific manner under hyperglycaemic conditions. The metabolic response of ECs to increased glucose abundance is currently not fully defined. Glucose enters ECs by facilitated diffusion via GLUT 1[68] in a manner that is not regulated by insulin [69] and therefore intracellular levels increase in hyperglycaemia (Fig. 2). Glucose utilisation is controlled by its flux through glycolysis, the TCA cycle, OxPhos, and alternative metabolic pathways [69]. Under hyperglycaemic conditions, ECs have been shown to exhibit the ‘Crabtree effect’, where the O2 consumption rate of cells is decreased despite high glucose concentrations, potentially resulting in higher intracellular [O2] (Fig. 2a) [70]. This is thought to be due to a higher glycolytic flux resulting in significant ATP production and increased substrate-level phosphorylation, which in turn acts to decrease the requirement for ATP produced by OxPhos, thus reducing O2 consumption (Fig. 2a) [70]. An increase in anaerobic glucose metabolism is also evidenced by reports of elevated lactate production and resultant acidosis in ECs cultured under high glucose conditions and in peripheral tissues of diabetic patients [31, 70,71,72].

Potential effects of hyperglycaemia on endothelial oxygen consumption. a Endothelial cells (EC) have been reported to decrease oxygen consumption in hyperglycaemia (HG) (the Crabtree Effect), perhaps due to ATP requirements being met through glycolysis, resulting in a decreased requirement for ATP production by the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (ox-phos). b HG has also been reported to increase oxygen consumption and cause intracellular hypoxia perhaps in a reactive oxygen species (ROS)-dependent manner
By contrast, endothelial O2 consumption rate has, in some reports, also been reported to increase upon exposure to high glucose culture conditions (Fig. 2b) [73]. Using pimonidazole staining as an indicator of hypoxia, Sada et al. showed significantly elevated hypoxic staining in bovine aortic endothelial cells (BAECs) cultured in media containing 25 mM glucose compared to that containing 5 mM glucose [73]. This effect was diminished by blocking the mitochondrial electron transport chain or overexpressing manganese superoxide dismutase (SOD), indicating a potential involvement of mitochondrial ROS in this finding [73]. This study also reported intensified hypoxic staining in the glomeruli of diabetic mice, in vivo. Increased O2 consumption under hyperglycaemia leading to activation of HIF signalling and hypoxia-induced pathways was also demonstrated in isolated rat islets and pancreatic beta cells in vitro [74] and it is thought that this may contribute to apoptosis and oxidative stress-associated cellular damage in diabetic patients [75]. Furthermore, the pathological effects of hyperglycaemia and hypoxia in diabetes are thought to be additive. Thus, in retinas isolated from normal rats, high glucose and hypoxia were shown to increase cytosolic free NADH via distinct metabolic mechanisms to potentially fuel biochemical pathways associated with the progression of diabetic retinopathy [76]. Hyperglycaemia can also worsen tissue responses to hypoxia. For example, Capla et al. observed decreased mobilisation of endothelial progenitor cells from the bone marrow of diabetic mice following ischaemic injury, and demonstrated in vitro that these cells displayed impaired adhesion, migration and proliferation in response to hypoxic culture conditions [77].
The altered cellular availability of O2 in hyperglycaemia, whether increased or decreased, could, as stated above, potentially alter the activities of TETs and JmjCs in ECs, dependent upon the KMs of the proteins for O2 and the (patho)physiological ranges of O2 to which they are exposed. The physiologically normoxic intracellular environment of ECs has been determined to be 3.5-4 kPa [64]. Although not yet comprehensively completed, some studies have been performed to determine the KMs of individual purified TET and JmjC proteins which suggest that, (at least) in some cases, their activities may be modulated by moderate fluctuations in physiological levels of O2 [78, 79]. There is also increasing evidence from cellular studies that relative hypoxia can act to inhibit the activities of both TETs and JmjCs [80,81,82,83]. Further studies need to be carried out to determine whether the activities of these demethylases may be modulated by any changes in consumption and therefore availability of O2 resulting from hyperglycaemia in ECs.
It is also noteworthy that, perhaps paradoxically, the transcription of TET1, together with that of some JmjC demethylase family members; JMJD1A, JMJD2B and JARID1B, is under the positive control of HIF or hypoxia-driven pathways [84,85,86,87]. Thus, a reduction in intracellular [O2] may result in a decrease in enzymatic activity that is balanced by a (potentially later-onset) increase in expression, suggesting a complex mode of hypoxia-induced regulation.
Hyperglycaemia and metabolism
The dependence of 2-OGDDs, including TETs and JmjC demethylases on their co-substrate, 2-OG, positions them at the interface between cellular metabolism and epigenetic regulation. It may therefore be speculated that they could be involved in mediating changes in gene expression in response to an altered metabolic state induced by hyperglycaemia. Perhaps significantly in this regard, injection of mice with glucose, glutamine or glutamate rapidly increased TET activity, as assessed by an increase in hepatic 5hmC levels, concomitant with elevated hepatic 2-OG levels (Fig. 3) [88]. Similar increases in 5hmC were observed in kidneys and skeletal muscle [88]. Both TETs and JmjCs have conversely been shown to be strongly inhibited via steric competition of 2-OG by the downstream TCA metabolites, succinate and fumarate (Fig. 3) [89, 90]. Accordingly, accumulation of fumarate and succinate following knockdown of fumarate hydratase and succinate dehydrogenase enzymes in HEK293T cells and mouse liver resulted in altered DNA and histone methylation patterns consistent with altered TET and JmjC demethylase activity (Fig. 3) [89]. Changes in the flux through the TCA cycle resulting in altered availability of these metabolites could therefore have profound effects on these epigenetic modulators and hence on cell function. Indeed, dysregulation of the TCA cycle has been reported to be an early manifestation of endothelial dysfunction [91].

Dysregulation of metabolites by hyperglycaemia. Hyperglycaemia has been reported to increase levels of lactate and succinate, which have been reported to decrease 2-oxoglutarate dependent dioxygenase (2-OGDD) activity. Increased flux through the tricarboxylic acid cycle may occur during hyperglycaemia, which could alter intracellular levels of 2-oxoglutarate, fumarate and succinate. High levels of 2-hydroxyglutarate (2HG), formed by the action of lactate dehydrogenase (LDH)-A on glutamine-derived 2-OG, or by mutant isocitrate dehydrogenase (IDH), is also reported to inhibit 2-OGDD activity. Abbreviations: glutamate dehydrogenase (GDH), glutaminase (GLS)
Although ATP synthesis primarily occurs via glycolysis in ECs, the mitochondria remain active, performing vital roles in NO generation, maintaining calcium homeostasis and regulating ROS production, in addition to some ATP generation [92]. Additionally, as stated above, metabolites generated by the TCA cycle act as cofactors or inhibitors of many enzymes including epigenetic modifiers [89, 90]. Reports of the effects of hyperglycaemia upon O2 consumption, and hence flux through the TCA cycle in high glucose conditions are, as stated above, varied [70, 73]. Further, to our knowledge, studies of 2-OG, succinate and fumarate levels in response to high glucose have not been conducted in ECs. However, the culture of isolated rat pancreatic islets in high glucose conditions was demonstrated to increase islet succinate content by 40% [93]. Given their potential effects upon the epigenetic landscape, the effects of hyperglycaemia upon TCA flux and steady-state metabolite levels in ECs warrants further investigation.
Another intermediate metabolite which has been shown to function as a competitive inhibitor of 2-OGDDs including TETs and JmjCs is 2-hydroxyglutarate (2HG) [94]. This is a chiral molecule which exists as a D-or L-enantiomer, both of which have been shown to inhibit 2-OGDDs (Fig. 3) [94,95,96,97]. Normal cells generate small quantities of both enantiomers (via poorly-understood pathways), but intracellular levels are typically kept low by the actions of D- and L-2-hydroxyglutarate dehydrogenases [98]. Intriguingly, D-2HG, produced by mutant gain-of-function isocitrate dehydrogenase is considered an ‘oncometabolite’ [99]. High levels of D-2HG are thus associated with and contribute to cancer pathogenesis, via inhibition of 2-OGDDs, including JmjC and TET enzymes [100]. More recently, it has been shown that L-2HG is selectively generated by cells under hypoxia and that this increase is both necessary and sufficient for increased histone methylation marks, acting through inhibition of specific JmjC proteins [100]. Biochemically, the hypoxia-induced L-2HG was found to be generated from glutamine-derived 2-OG, via the action of lactate dehydrogenase (LDH)-A, demonstrating promiscuous substrate usage. It is noteworthy that the levels of LDH have been shown to increase in diabetic rabbits, and that mammalian LDH enzymes, other than LDH-A, are known to have the potential to produce L-2HG under more acidic environments [101], such as those resulting from the increased lactic acid production in hyperglycaemia (Fig. 3) [102]. In another report, levels of L-2HG were similarly shown to accumulate in response to hypoxia, via the action of malate dehydrogenase (MDH) on (increased levels of) mitochondrial 2-OG [103] resulting from TCA dysfunction and mitochondrial stress. In turn, the increased levels of L-2HG acted to inhibit electron transport and glycolysis. The relationship between hyperglycaemia, hypoxia, altered TCA flux and the regulation of epigenetic modifiers by L-2HG is intriguing and remains to be explored.
The observed increases in endothelial lactate production resulting from increased glycolytic flux during hyperglycaemia [70,71,72] also has the potential to influence 2-OGDD activity. Thus, it has been reported that that the uptake of high levels of exogenous lactate increased HIF-1α expression in ECs, leading to angiogenesis, independently of hypoxia [104]. Mechanistically, it was proposed that the conversion of lactate to pyruvate by LDH-B leads to competitive substrate inhibition of PHD2 by pyruvate, reducing the ability of PHD2 to trigger HIF-1α degradation [104]. Investigation of the regulation of other 2-OGDDs by glycolysis metabolites has not yet been conducted. However, this provides further support to the idea that 2-OGDDs may act as sensors of the metabolic status of ECs.
Hyperglycaemia, cellular redox and iron homeostasis
2-OGDDs require Fe2+ as a cofactor for their catalytic activity, so the bioavailability of iron in its reduced form is important in maintaining their function [105, 106]. In evidence of this, two groups identified that supplementation with ascorbic acid (vitamin C), which promotes the reduction of Fe3+ to Fe2+, enhanced TET-mediated oxidation of 5mC [105, 106]. The histone demethylation activity of JmjCs has similarly been demonstrated to increase with vitamin C supplementation [107,108,109].
The bioavailability of Fe2+ in vivo is determined by multiple mechanisms which regulate cellular redox and iron homeostasis (transport, storage and metabolism) which may all impact upon the activities of TETs and JmjCs [110]. In hyperglycaemia, the cellular redox balance becomes perturbed, favouring excessive production of reactive oxygen species (ROS) and overloading the antioxidant defences which usually counteract this (Fig. 4a) [111]. A major source of hyperglycaemia-induced ROS production in ECs is the mitochondrial electron transport chain [111]. This is evidenced by the finding that the use of mitochondrial complex II inhibitors or oxidative phosphorylation uncouplers prevents endothelial ROS production [30, 112] ROS production in hyperglycaemia has also been reported to be mediated by increased NADPH oxidase activity [113, 114], eNOS uncoupling [115] and xanthine oxidase activity [116]. Hyperglycaemia-induced formation of AGEs induces an inflammatory response which can also generate ROS [117]. The direct effects of ROS-generating systems upon the activities of the epigenetic modifiers has so-far been little investigated. However, in one study, the administration of 150 μM H2O2 to epithelial cells resulted in increased global levels of several methylation marks (purported to be due to attenuated JmjC activity) together with reduced (global) TET activity [118]. By contrast, an oxidant-dependent increase in TET activity, in response to high glucose exposure, has been reported in bovine retinal endothelial cells [119]. Thus, a glucose-induced activation of TET2 was observed, as evidenced by higher 5hmC levels specifically at the matrix metalloproteinase-9 (MMP-9) gene promoter, which could be ablated by the addition of a mitochondrial SOD mimetic [119]. The significance of this (locus-specific) effect of hyperglycaemia-induced ROS in respect of the global levels of 5hmC and TET activity are at present unclear.

Hyperglycaemia may affect ten-eleven translocation (TET) and Jumonji C (JmjC) activity by altering Fe2+ availability. a Hyperglycaemia increases flux through the polyol pathway, which consumes reduced nicotinamide adenine dinucleotide phosphate (NADPH). Reduced glutathione (GSH) and vitamin C levels may decrease as a result, decreasing the regeneration of Fe2+ which is required by TET and JmjC enzymes. Increased generation of reactive oxygen species (ROS) in hyperglycaemia may also affect Fe2+ regeneration. b An increase in cyclic adenosine monophosphate (cAMP) levels in hyperglycaemia may increase the intracellular labile Fe2+ pool and thus increase TET and JmjC activity. Abbreviations: glucose-6-phosphate (G6P), fructose-6-phosphate (F6P), Fructose-1,6-bisphosphate (F1,6P2), glyceraldehyde-3-phosphate (G3P), 1,3-bisphosphoglycerate (1,3 BPG)
Steady-state cellular redox is often measured by ratios of oxidised and reduced forms of NAD and glutathione (NADH/NAD+ and GSH/GSSG respectively). The delicate balance of these agents determines the ability of a cell to undergo oxidation and reduction reactions which are crucial for many cellular processes including iron metabolism [120]. When ECs are exposed to hyperglycaemia, excess glucose is utilised by the polyol pathway, which consumes NADPH and generates NADH, resulting in an abnormal NADH/NAD+ balance [121]. Furthermore, this impairs generation of the reduced form of glutathione by glutathione reductase, which requires NADPH for its activity [121]. Glutathione is a major cellular redox buffer which, amongst many other functions, is responsible for regenerating the reduced form of vitamin C, necessary for optimal 2-OGDD activity (Fig. 4a) [122].
Interestingly, cyclic AMP (cAMP) has been shown to increase 5hmC levels by increasing the labile Fe2+ pool of primary Schwann cells, HEK293 and mouse embryonic fibroblasts [123]. Accordingly, TET activity is augmented by the increased availability of the Fe2+ cofactor, leading to increased detection of 5hmC [123]. Whether cAMP-dependent signalling acts to increase intracellular labile Fe2+ via increased iron uptake, storage, or conversion of Fe3+ to Fe2+, is not currently understood, but acidification of endosomes has been suggested to play a role [123]. Although changes in cAMP levels in hyperglycaemic conditions have not been quantified in ECs, high glucose exposure has been shown to increase cAMP levels in mesangial cells, which uptake glucose in a similar fashion to ECs, via GLUT-1 [124]. Therefore, hyperglycaemia-induced disturbances in cellular redox and iron homeostasis may be factors which potentially could mediate the activities of both TETs and JmjCs, due to their dependence on Fe2+ (Fig. 4b).
Hyperglycaemia and non-catalytic roles of 2-OGDD demethylases
There is an increasing body of research to suggest that TETs perform some epigenetic regulatory functions, which are distinct from their catalytic activity, by serving a scaffolding role via binding to DNA and recruiting other proteins or complexes [125,126,127,128,129]. These highlights the potential cross-talk between specific TET proteins and the regulation of histone methylation. Thus, physical and/or functional interactions between TET1 and TET2 and components of the Polycomb repressive complex 2 (PRC2), which mediates the methylation of H3K27 have been demonstrated [126, 130, 131]. Further, all 3 TET proteins have been shown to physically interact with O-GlcNAc Transferase (OGT) [127,128,129, 132]. A proven specific regulatory target of O-GlcNAcylation by (at least some) TET-OGT complexes is host cell factor 1 (HCF1), which is an integral element of the histone H3K4 methyltransferase SET1/COMPASS complex [127]. The substrate for both N- and O-linked glycosylation of proteins is UDP-GlcNAc, which is generated by the hexosamine biosynthesis pathway [133]. This is noteworthy with respect to hyperglycaemia because flux through the hexosamine pathway is increased by high glucose concentrations [133], likely resulting in higher UDP-GlcNAc production and subsequent GlcNAcylation of proteins (Fig. 5). Accordingly, O-GlcNac levels have been shown to be significantly increased in the aortae of obese, insulin-resistant mice compared to lean mice [134] and in rat aortic smooth muscle upon culture in high glucose media [135]. Thus, increased O-GlcNAcylation of SET1/COMPASS, resulting in elevated methylation of H3K4 may represent another mechanism whereby hyperglycaemia may influence epigenetic modifications, in a TET-dependent fashion.

Hyperglycaemia may influence ten-eleven translocation (TET)-dependent O-GlcNAcylation of the histone methyltransferase SET1/COMPASS complex. TET2 and TET3 interact with O-GlcNAc Transferase (OGT), promoting its O-GlcNAcylation of the SET1/COMPASS complex which mediates trimethylation of histone H3 lysine 4 (H3K4me3). Hyperglycaemia increases hexosamine pathway flux so increases production of the substrate UDP-GlcNAc. Abbreviations: glucose-6-phosphate (G6P), fructose-6-phosphate (F6P), fructose-1,6-bisphosphate (F1,6P2), glyceraldehyde-3-phosphate (G3P), 1,3-bisphosphoglycerate (1,3 BPG)
Transcriptional and post-translational regulation of TETs and JmjCs by hyperglycaemia
The activities and functions of TET and JmjC demethylases are not only regulated by the availability of their cofactors and substrates. Thus, like many proteins, their levels of activity within a cell are subject to both changes in their expression and post-translational modifications. Using a zebrafish model of diabetes, induced by streptozotocin injection, Dhliwayo et al. observed that hyperglycaemia induced a 5-10 fold increase in RNA expression of TET1, TET2 and TET3 in fin samples within 1 week, which persisted to 3 weeks after induction of hyperglycaemia [136]. This was supported by a significant global accumulation of 5hmC [136]. Administration of poly(ADP-ribose) polymerase (PARP) inhibitors rescued the hyperglycaemia-induced impairment of fin regeneration and prevented the increase in 5hmC, suggesting a role for PARPs in regulating TETs [136]. The authors suggest that this is due to suppression of TET expression, as PARP1 activity has previously been reported to be required for TET1 transcriptional upregulation in developing mouse primordial germ cells [137].
Post-translational modifications of 2-OGDD demethylases have not thus far been extensively studied. However, in a significant recent study, Wu et al. found that TET2 is phosphorylated at serine 99 by 5'AMP activated protein kinase (AMPK) in peripheral blood mononuclear cells (PBMCs) (Fig. 6) [138]. High glucose levels disrupted AMPK-mediated phosphorylation, leading to decreased stability of the TET2 protein (Fig. 6) [138]. This is likely to account for their finding that global levels of 5hmC are lower in PBMCs from type 2 diabetic patients compared to healthy donors (Fig. 6) [138]. This is perhaps the strongest evidence, to date, of high glucose levels affecting the activity of a 2-OGDD, and it would be interesting to investigate whether this finding extends to other cell types including ECs, and whether other 2-OGDDs including JmjCs are similarly affected.

Hyperglycaemia impairs adenosine monophosphate-activated protein kinase (AMPK)-mediated stabilisation of ten-eleven translocation (TET)2. a In peripheral blood mononuclear cells (PBMCs) from healthy controls, AMPK phosphorylates TET2 at serine 99, thereby stabilising it and increasing levels of its product, 5-hydroxymethylcytosine (5hmC). b Hyperglycaemia impairs AMPK mediated phosphorylation of TET2 in PBMCs from diabetic patients, leading to destabilisation and reduced activity of TET2
Several JmjC family members have been reported to possess phosphorylated peptides [139] including lysine demethylase-3A and -7C (KDM3A and KDM7C) which have been shown to be specifically phosphorylated by mitogen- and stress-activated protein kinase 1 (PK1) and PKA respectively [140, 141]. In the latter case, KDM7C is reportedly inactive unless phosphorylated by PKA, which enables it to associate with the DNA binding protein, AT-Rich Interaction Domain 5B (ARID5B) and demethylate H3K9me2 [141]. This highlights the potential for phosphorylation to activate 2OGDDs either directly or by promoting the formation of active complexes. Investigating whether hyperglycaemia affects the phosphorylation of 2-OGDD members other than TET2, and the effects of this on endothelial cell function, could be a valuable area of study.
Conclusions and future perspectives
The challenge to develop better preventative treatments to alleviate the escalating problem of CVD associated with T2D necessitates a detailed mechanistic understanding of the disease aetiology. The causal links between hyperglycaemia, altered epigenetic patterns and endothelial dysfunction now appear irrefutable. It is therefore now critical to understand how epigenetic patterns are normally regulated in the endothelium and how hyperglycaemia might dysregulate this. In addition, such a biochemical understanding of the relationship between high glucose exposure and epigenetic regulation would serve as powerful proof of the concept of the epigenetic basis of disease.
It is noteworthy that ED may, in part, be initiated or exacerbated by changes in other cell types, such as leukocytes participating in the inflammatory response which is strongly associated with cardiovascular disease [142]. In addition, dysfunctional vascular smooth muscle cells can contribute to the CVD associated with diabetes [143]. Therefore, although the focus here has been ECs, hyperglycaemia-mediated epigenetic changes in immune cells and other vascular cells are also highly relevant. Further studies on these cell populations are important to understand not only generalised mechanisms of 2OGDD regulation but also the involvement of these other cell types in the development of CVD in T2D.
The fact that many epigenetic modifiers use energy metabolites has initiated a large field of study aimed at elucidating the link between epigenetic adaption to the environment and the progression of non-communicable diseases, most notably cancer [39, 144, 145]. The clear associations between altered epigenetic methylation patterns and diabetes-associated CVD suggest that the dysregulation of the methylome is of critical importance in its aetiology [34,35,36,37,38]. In addition, altered methylation patterns are capable of inducing lasting gene expression changes, which could contribute to developmental priming in offspring of diabetic mothers, which perpetuates the disease burden throughout successive generations [5, 6]. Therefore, understanding the epigenetic mechanisms at play could have a far-reaching impact.
The substrate and co-factor requirements of the 2-OGDD family of epigenetic demethylases suggest they may act as sensors and integrators of not only energy metabolic fluxes (for instance, glycolysis versus OxPhos) but also O2 availability and cellular redox. Their activities may further be impacted by post-translational modifications, perhaps also modulated by hyperglycaemia. However, the relationship between glucose exposure, the bioavailability of the obligate substrates/cofactors in endothelial cells and the catalytic activities of these enzymes remains poorly understood and the available data is sometimes contradictory. Investigating this may aid our understanding of hyperglycaemia-induced ED, and potentially inform significant improvements in the future prevention and management of CVD in diabetes.
Intriguingly, somatic mutations in 2-OGDD enzymes have increasingly been found to associate with other pathologies, most notably certain cancers. Thus, mutations in TET enzymes, and in particular TET2, have been identified in haematological cancers including myelodysplastic syndromes [146], myeloproliferative neoplasms [147], mixed lineage leukaemia [148], T cell lymphomas [149] and B cell non-Hodgkin lymphoma [150]. A recent study also revealed that TET2 is one of the most commonly mutated genes in peripheral blood cells of individuals with clonal haematopoiesis (benign clonal expansion of blood cells), which associated with a higher incidence of atherosclerosis (assessed as coronary artery calcification), myocardial infarction and coronary heart disease [151]. Thus, (dysregulated) TETs are associated with haematological and (non-diabetes-associated) cardiovascular diseases [146,147,148,149,150,151]. Mutations in JmjCs have also been associated with cancers, developmental and inflammatory diseases, which have been reviewed elsewhere [152]. The study of the potential epigenetic basis of these pathologies is therefore a field of active investigation.
It is imperative also to gain a full understanding of the epigenetic mechanisms underlying the cell-type-specific transcriptional regulation of the genome. The recent rise in single-cell epigenomic analyses, coupled with transcriptomics, shows great potential in understanding subtle molecular differences in heterogeneous cell populations [153]. Further, the development of CRISPR-Cas technologies facilitating RNA-guided epigenetic regulators to specifically edit the epigenetic code will further elucidate the functional consequences of specific epigenetic marks [154]. The application of these technologies to ECs challenged with stressors, such as high glucose, could provide unprecedented insight into the epigenetic basis of gene regulation and endothelial (dys)function.
Given that modulating DNA methylation status or histone post-translational modifications can co-ordinate transcriptional silencing or activation of many genes, the regulators involved in this process, including TETs and JmjCs, may be attractive targets for epigenetic-based therapies. These could potentially co-ordinate the broad transcriptional changes that would be required to maintain or restore normal cell function in unfavourable conditions (such as hyperglycaemia). Epigenetic modifiers, such as inhibitors of HDACs and DNA methylation, are already undergoing clinical trials for some cancers, and a number of these have gained approval from the Food and Drug Administration (reviewed in [155, 156]). It is likely that the number of epigenetic modifying drugs available and the number of conditions for which they are used will expand, informed by further research.
Availability of data and materials
Not applicable; no new data are presented.
Abbreviations
- CVD:
-
Cardiovascular disease
- ED:
-
Endothelial dysfunction
- TET:
-
Ten-eleven translocation
- JmjC:
-
JumonjiC
- T2D:
-
Type 2 diabetes
- EC:
-
Endothelial cell
- HUVEC:
-
Human umbilical vein endothelial cell
- AGE:
-
Advanced glycosylation end-products
- HAEC:
-
Human aortic endothelial cell
- SAM:
-
S-adenosyl methionine
- 2-OGDD:
-
2-oxoglutarate-dependent dioxygenase
- 2-OG:
-
α-ketoglutarate
- DNMT:
-
DNA methyltransferase
- 5mC:
-
5-methylcytosine
- 5hmC:
-
5-hydroxymethylcytosine
- 5fC:
-
5-formylcytosine
- 5caC:
-
5-carboxylcytosine
- TDG:
-
Thymine DNA glycosylase
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- HMT:
-
Histone methyltransferase
- LSD:
-
Lys-specific demethylase
- HIF:
-
Hypoxia-inducible factor
- PHD:
-
Prolyl hydroxylase
- OxPhos:
-
Oxidative phosphorylation
- TCA:
-
Tricarboxylic acid
- BAEC:
-
Bovine aortic endothelial cells
- LDH:
-
Lactate dehydrogenase
- MDH:
-
Malate dehydrogenase
- 2HG:
-
2-hydroxyglutarate
- ROS:
-
Reactive oxygen species
- MMP-9:
-
Matrix metalloproteinase-9
- SOD:
-
Superoxide dismutase
- PRC2:
-
Polycomb repressive complex 2
- OGT:
-
O-GlcNAc transferase
- HCF1:
-
Host cell factor 1
- PBMC:
-
Peripheral blood mononuclear cell
- KDM:
-
Lysine demethylase
- ARID5B:
-
AT-rich interaction domain 5B
- AMPK:
-
5'AMP activated protein kinase
- PARP:
-
Poly(ADP-ribose) polymerase
References
Zimmet P, Alberti KG, Magliano DJ, Bennett PH. Diabetes mellitus statistics on prevalence and mortality: facts and fallacies. Nat Rev Endocrinol. 2016;12(10):616–22.
Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843.
Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc Diabetol. 2018;17(1):83.
Sallam NA, Palmgren VAC, Singh RD, John CM, Thompson JA. Programming of vascular dysfunction in the intrauterine milieu of diabetic pregnancies. Int J Mol Sci. 2018;19(11):3665.
Leybovitz-Haleluya N, Wainstock T, Landau D, Sheiner E. Maternal gestational diabetes mellitus and the risk of subsequent pediatric cardiovascular diseases of the offspring: a population-based cohort study with up to 18 years of follow up. Acta Diabetol. 2018;55(10):1037–42.
Moore TR. Fetal exposure to gestational diabetes contributes to subsequent adult metabolic syndrome. Am J Obstet Gynecol. 2010;202(6):643–9.
Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88–98.
Gaede P, Vedel P, Larsen N, Jensen GV, Parving HHPO. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. J Cardpulm Rehabil. 2003;348(5):383–93.
Kannel WB, McGee DL. Diabetes and cardiovascular disease: the Framingham study. JAMA J Am Med Assoc. 1979;241(19):2035–8.
De Ferranti SD, De Boer IH, Fonseca V, Fox CS, Golden SH, Lavie CJ, et al. Type 1 diabetes mellitus and cardiovascular disease a scientific statement from the american heart association andamerican diabetes association. Circulation. 2014;130(13):1110–30.
Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23(2):168–75.
Sitia S, Tomasoni L, Atzeni F, Ambrosio G, Cordiano C, Catapano A, et al. From endothelial dysfunction to atherosclerosis. Autoimmun Rev. 2010;9(12):830–4.
Schalkwijk CG, Stehouwer CDA. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci. 2005;109(2):143–59.
Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–4.
Piga R, Naito Y, Kokura S, Handa O, Yoshikawa T. Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis. 2007;193(2):328–34.
Yu CH, Suriguga GM, Liu WJ, Cui NX, Wang Y, et al. High glucose induced endothelial to mesenchymal transition in human umbilical vein endothelial cell. Exp Mol Pathol. 2017;102(3):377–83.
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications. N Engl J Med. 1993;329(14):977–86.
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837–53.
Bethel MA, Neil HAW, Paul SK, Matthews DR, Holman RR. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359(15):1577–89.
Iwasaki H, Okamoto R, Kato S, Konishi K, Mizutani H, Yamada N, et al. High glucose induces plasminogen activator inhibitor-1 expression through rho/rho-kinase-mediated NF-κB activation in bovine aortic endothelial cells. Atherosclerosis. 2008;196(1):22–8.
Srinivasan S, Hatley ME, Bolick DT, Palmer LA, Edelstein D, Brownlee M, et al. Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia. 2004;47(10):1727–34.
Ho FM, Liu SH, Liau CS, Huang PJ, Lin-Shiau SY. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-JUN NH2-terminal kinase and caspase-3. Circulation. 2000;101(22):2618–24.
Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36(7):808–12.
Kowluru RA. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes. 2003;52(3):818–23.
Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, et al. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation. 2002;106(16):2067–72.
Azcutia V, Abu-Taha M, Romacho T, Vázquez-Bella M, Matesanz N, Luscinskas FW, et al. Inflammation determines the pro-adhesive properties of high extracellular D-glucose in human endothelial cells in vitro and rat microvessels in vivo. PLoS One. 2010;5(4):10091.
Zoungas S, Arima H, Gerstein HC, Holman RR, Woodward M, Reaven P, et al. Effects of intensive glucose control on microvascular outcomes in patients with type 2 diabetes: a meta-analysis of individual participant data from randomised controlled trials. Lancet Diabetes Endocrinol. 2017;5(6):431–7.
Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–25.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–20.
Nishikawa T, Edelstein D, Du XL, Yamagishi SI, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–90.
Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58(12):2718–25.
Zarzour A, Kim HW, Weintraub NL. Epigenetic regulation of vascular diseases. Arterioscler Thromb Vasc Biol. 2019;39(6):984–90.
Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol. 2010;6(12):665–75.
Pasquier J, Hoarau-Véchot J, Fakhro K, Rafii A, Abi KC. Epigenetics and cardiovascular disease in diabetes. Curr Diab Rep. 2015;118(11):1706–22.
Othman SH. The epigenetics of diabetes, obesity, overweight and cardiovascular disease. AIMS Genet. 2019;6(3):36–45.
Rodriguez-Araujo G, Nakagami H. Pathophysiology of cardiovascular disease in diabetes mellitus. Cardiovasc Endocrinol Metab. 2018;7(1):4–9.
De Rosa S, Arcidiacono B, Chiefari E, Brunetti A, Indolfi C, Foti DP. Type 2 diabetes mellitus and cardiovascular disease: genetic and epigenetic links. Front Endocrinol (Lausanne). 2018;9:2.
Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011;21(10):1601–15.
Johnson C, Warmoes MO, Shen X, Locasale JW. Epigenetics and cancer metabolism. Cancer Lett. 2015;356(2):309–14.
Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol. 2012;227(9):3169–77.
Stratton MS, Farina FM, Elia L. Epigenetics and vascular diseases. J Mol Cell Cardiol. 2019;133:148–63.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57.
Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–26.
Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014;4:80.
Siegfried Z, Cedar H. DNA methylation: a molecular lock. Curr Biol. 1997;7(5):305–7.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (80- ). 2009;324(5929):930–5.
Sun M, Song MM, Wei B, Gao Q, Li L, Yao B, et al. 5-Hydroxymethylcytosine-mediated alteration of transposon activity associated with the exposure to adverse in utero environments in human. Hum Mol Genet. 2016;25(11):2208–19.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (80- ). 2011;333(6047):1300–3.
Song CX, He C. Potential functional roles of DNA demethylation intermediates. Trends Biochem Sci. 2013;38(10):480–4.
Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153(3):678–91.
Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149(6):1368–80.
Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PWTC, Bauer C, et al. Dynamic readers for 5-(Hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152(5):1146–59.
Kellinger MW, Song CX, Chong J, Lu XY, He C, Wang D. 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nat Struct Mol Biol. 2012;19(8):831–3.
Hübner MR, Eckersley-Maslin MA, Spector DL. Chromatin organization and transcriptional regulation. Curr Opin Genet Dev. 2013;23(2):89–95.
Schiattarella GG, Madonna R, Van Linthout S, Thum T, Schulz R, Ferdinandy P, et al. Epigenetic modulation of vascular diseases: assessing the evidence and exploring the opportunities. Vasc Pharmacol. 2018;107:43–52.
Saksouk N, Simboeck E, Déjardin J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics and Chromatin. 2015;8:3.
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95.
Youn HD. Methylation and demethylation of DNA and histones in chromatin: the most complicated epigenetic marker. Exp Mol Med. 2017;49(4):321.
Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48(4):491–507.
Anand R, Marmorstein R. Structure and mechanism of lysine-specific demethylase enzymes. J Biol Chem. 2007;282(49):35425–9.
Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715–27.
Pollard P, Yang M, Su H, Soga T, Kranc K. Prolyl hydroxylase domain enzymes: important regulators of cancer metabolism. Hypoxia. 2014;2:127–42.
Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–54.
Keeley TP, Mann GE. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol Rev. 2019;99(1):161–234.
Bierhansl L, Conradi L-C, Treps L, Dewerchin M, Carmeliet P. Central role of metabolism in endothelial cell function and vascular disease. Physiology. 2017;32(2):126–40.
Pircher A, Treps L, Bodrug N, Carmeliet P. Endothelial cell metabolism: a novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis. 2016;253:247–57.
Taylor CT. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J. 2008;409(1):19–26.
Watson RT, Pessin JE. Intracellular organization of insulin signaling and GLUT4 translocation. Recent Prog Horm Res. 2001;56:175–93.
Artwohl M, Brunmair B, Fürnsinn C, Hölzenbein T, Rainer G, Freudenthaler A, et al. Insulin does not regulate glucose transport and metabolism in human endothelium. Eur J Clin Investig. 2007;37(8):643–50.
Dobrina A, Rossi F. Metabolic properties of freshly isolated bovine endothelial cells. Biochim Biophys Acta. 1983;762(2):295–301.
La Selva M, Beltramo E, Pagnozzi F, Bena E, Molinatti PA, Molinatti GM, et al. Thiamine corrects delayed replication and decreases production of lactate and advanced glycation end-products in bovine retinal and human umbilical vein endothelial cells cultured under high glucose conditions. Diabetologia. 1996;39(11):1263–8.
Ido Y, Carling D, Ruderman N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells. Diabetes. 2002;51(1):159–67.
Sada K, Nishikawa T, Kukidome D, Yoshinaga T, Kajihara N, Sonoda K, et al. Hyperglycemia induces cellular hypoxia through production of mitochondrial ROS followed by suppression of aquaporin-1. PLoS One. 2016;11(7):0158619.
Bensellam M, Duvillié B, Rybachuk G, Laybutt DR, Magnan C, Guiot Y, et al. Glucose-induced O 2 consumption activates hypoxia inducible factors 1 and 2 in rat insulin-secreting pancreatic beta-cells. PLoS One. 2012;7(1):29807.
Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxidants Redox Signal. 2017;26(10):501–18.
Nyengaard JR, Ido Y, Kilo C, Williamson JR. Interactions between hyperglycemia and hypoxia: implications for diabetic retinopathy. Diabetes. 2004;53(11):2931–8.
Capla JM, Grogan RH, Callaghan MJ, Galiano RD, Tepper OM, Ceradini DJ, et al. Diabetes impairs endothelial progenitor cell-mediated blood vessel formation in response to hypoxia. Plast Reconstr Surg. 2007;119(1):59–70.
Hancock RL, Masson N, Dunne K, Flashman E, Kawamura A. The activity of JmjC histone lysine demethylase KDM4A is highly sensitive to oxygen concentrations. ACS Chem Biol. 2017;12(4):1011–9.
Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016;537(7618):63–8.
Sánchez-Fernández EM, Tarhonskaya H, Al-Qahtani K, Hopkinson RJ, Mccullagh JSO, Schofield CJ, et al. Investigations on the oxygen dependence of a 2-oxoglutarate histone demethylase. Biochem J. 2013;449(2):491–6.
Burr S, Caldwell A, Chong M, Beretta M, Metcalf S, Hancock M, et al. Oxygen gradients can determine epigenetic asymmetry and cellular differentiation viadifferential regulation of Tet activity in embryonic stem cells. Nucleic Acids Res. 2018;46(3):1210–26.
Dobrynin G, McAllister TE, Leszczynska KB, Ramachandran S, Krieg AJ, Kawamura A, et al. KDM4A regulates HIF-1 levels through H3K9me3. Sci Rep. 2017;7(1):11094.
Batie M, Frost J, Frost M, Wilson JW, Schofield P, Rocha S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science (80- ). 2019;363(6432):1222–6.
Melvin A, Rocha S. Chromatin as an oxygen sensor and active player in the hypoxia response. Cell Signal. 2012;24(1):35–43.
Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, et al. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014;15(12):513.
Muecke C, Dabral S, Seeger W, Pullamsetti SS. Role and regulation of Jumonji C domain-containing histone demethylases 1A and 2B in pulmonary hypertension. Eur Respir J. 2016;48(60):5092.
Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 enhances hypoxic gene expression and tumor growth. Mol Cell Biol. 2010;30(1):344–53.
Yang H, Lin H, Xu H, Zhang L, Cheng L, Wen B, et al. TET-catalyzed 5-methylcytosine hydroxylation is dynamically regulated by metabolites. Cell Res. 2014;24(8):1017–20.
Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–38.
Lu X, Zhao BS, He C. TET family proteins: oxidation activity, interacting molecules, and functions in diseases. Chem Rev. 2015;115(6):2225–39.
Addabbo F, Ratliff B, Park HC, Kuo MC, Ungvari Z, Ciszar A, et al. The Krebs cycle and mitochondrial mass are early victims of endothelial dysfunction: proteomic approach. Am J Pathol. 2009;174(1):34–43.
Koziel A, Woyda-Ploszczyca A, Kicinska A, Jarmuszkiewicz W. The influence of high glucose on the aerobic metabolism of endothelial EA.hy926 cells. Pflugers Arch Eur J Physiol. 2012;464(6):657–69.
Alarcon C, Wicksteed B, Prentki M, Corkey BE, Rhodes CJ. Succinate is a preferential metabolic stimulus-coupling signal for glucose-induced proinsulin biosynthesis translation. Diabetes. 2002;51(8):2496–504.
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30.
Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–9.
Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–8.
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–8.
Linster CL, Van Schaftingen E, Hanson AD. Metabolite damage and its repair or pre-emption. Nat Chem Biol. 2013;9(2):72–80.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–34.
Intlekofer AM, DeMatteo RG, Venneti S, Finley LWS, Lu C, Judkins AR, et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015;22(2):304–11.
Teng X, Emmett MJ, Lazar MA, Goldberg E, Rabinowitz JD. Lactate dehydrogenase C produces S-2-hydroxyglutarate in mouse testis. ACS Chem Biol. 2016;11(9):2420–7.
Gillies RJ, Pilot C, Marunaka Y, Fais S. Targeting acidity in cancer and diabetes. Biochim Biophys Acta - Rev Cancer. 2019;1871(2):273–80.
Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-mediated increases in l-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015;22(2):291–303.
Sonveaux P, Copetti T, de Saedeleer CJ, Végran F, Verrax J, Kennedy KM, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One. 2012;7(3):33418.
Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, et al. Ascorbic acid enhances tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013;135(28):10396–403.
Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288(19):13669–74.
Ebata KT, Mesh K, Liu S, Bilenky M, Fekete A, Acker MG, et al. Vitamin C induces specific demethylation of H3K9me2 in mouse embryonic stem cells via Kdm3a/b. Epigenetics and Chromatin. 2017;10:36.
Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442(7100):312–6.
Tsukada YI, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–6.
Lamadema N, Burr S, Brewer AC. Dynamic regulation of epigenetic demethylation by oxygen availability and cellular redox. Free Radic Biol Med. 2019;131:282–98.
Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS? Int J Mol Sci. 2019;131:282–98.
Stefano GB, Challenger S, Kream RM. Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disorders. Eur J Nutr. 2016;55(8):2339–45.
Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, et al. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105(14):1656–62.
Miele C, Paturzo F, Teperino R, Sakane F, Fiory F, Oriente F, et al. Glucose regulates diacylglycerol intracellular levels and protein kinase C activity by modulating diacylglycerol kinase subcellular localization. J Biol Chem. 2007;282:31835–43.
Sasaki N, Yamashita T, Takaya T, Shinohara M, Shiraki R, Takeda M, et al. Augmentation of vascular remodeling by uncoupled endothelial nitric oxide synthase in a mouse model of diabetes mellitus. Arterioscler Thromb Vasc Biol. 2008;28(6):1068–76.
Matsumoto S, Koshiishi I, Inoguchi T, Nawata H, Utsumi H. Confirmation of superoxide generation via xanthine oxidase in streptozotocin-induced diabetic mice. Free Radic Res. 2003;37(7):767–72.
Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114(6):597–605.
Niu Y, Desmarais TL, Tong Z, Yao Y, Costa M. Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med. 2015;82:22–8.
Kowluru RA, Shan Y. Role of oxidative stress in epigenetic modification of MMP-9 promoter in the development of diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2017;255(5):955–62.
Kumar C, Igbaria A, D’Autreaux B, Planson AG, Junot C, Godat E, et al. Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011;30(10):2044–56.
Zheng H, Wu J, Jin Z, Yan L-J. Protein modifications as manifestations of hyperglycemic glucotoxicity in diabetes and its complications. Biochem Insights. 2016;9:1–9.
Montecinos V, Guzmán P, Barra V, Villagrán M, Muñoz-Montesino C, Sotomayor K, et al. Vitamin C is an essential antioxidant that enhances survival of oxidatively stressed human vascular endothelial cells in the presence of a vast molar excess of glutathione. J Biol Chem. 2007;282(21):15506–15.
Camarena V, Sant DW, Huff TC, Mustafi S, Muir RK, Aron AT, et al. CAMP signaling regulates DNA hydroxymethylation by augmenting the intracellular labile ferrous iron pool. Elife. 2017;6:29750.
Deb DK, Bao R, Li YC. Critical role of the cAMP-PKA pathway in hyperglycemia-induced epigenetic activation of fibrogenic program in the kidney. FASEB J. 2017;31(5):2065–75.
Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525(7569):389–93.
Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–93.
Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32(5):645–55.
Shi FT, Kim H, Lu W, He Q, Liu D, Goodell MA, et al. Ten-eleven translocation 1 (Tet1) is regulated by o-linked n-acetylglucosamine transferase (ogt) for target gene repression in mouse embryonic stem cells. J Biol Chem. 2013;288(29):20776–84.
Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493(7433):561–4.
Hasegawa N, Oshima M, Sashida G, Matsui H, Koide S, Saraya A, et al. Impact of combinatorial dysfunctions of Tet2 and Ezh2 on the epigenome in the pathogenesis of myelodysplastic syndrome. Leukemia. 2017;31(4):861–71.
Neri F, Incarnato D, Krepelova A, Rapelli S, Pagnani A, Zecchina R, et al. Genome-wide analysis identifies a functional association of Tet1 and Polycomb repressive complex 2 in mouse embryonic stem cells. Genome Biol. 2013;14(8):91.
Vella P, Scelfo A, Jammula SG, Chiacchiera F, Williams K, Cuomo A, et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell. 2013;49(4):645–56.
Issad T, Kuo MS. O-GlcNAc modification of transcription factors, glucose sensing and glucotoxicity. Trends Endocrinol Metab. 2008;19(10):380–9.
Luo B, Soesanto Y, McClain DA. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting Akt activity in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28(4):651–7.
Akimoto Y, Kreppel LK, Hirano H, Hart GW. Hyperglycemia and the O-GlcNAc transferase in rat aortic smooth muscle cells: elevated expression and altered patterns of O-GlcNAcylation. Arch Biochem Biophys. 2001;389(2):166–75.
Dhliwayo N, Sarras MP, Luczkowski E, Mason SM, Intine RV. Parp inhibition prevents ten-eleven translocase enzyme activation and hyperglycemia-induced DNA demethylation. Diabetes. 2014;63(9):3069–76.
Ciccarone F, Klinger FG, Catizone A, Calabrese R, Zampieri M, Bacalini MG, et al. Poly(ADP-ribosyl)ation acts in the DNA demethylation of mouse primordial germ cells also with DNA damage-independent roles. PLoS One. 2012;7(10):46927.
Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature. 2018;559(7715):637–41.
Brauchle M, Yao Z, Arora R, Thigale S, Clay I, Inverardi B, et al. Protein complex interactor analysis and differential activity of KDM3 subfamily members towards H3K9 methylation. PLoS One. 2013;8(4):60549.
Cheng MB, Zhang Y, Cao CY, Zhang WL, Zhang Y, Shen YF. Specific phosphorylation of histone demethylase KDM3A determines target gene expression in response to heat shock. PLoS Biol. 2014;12(12):1002026.
Baba A, Ohtake F, Okuno Y, Yokota K, Okada M, Imai Y, et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat Cell Biol. 2011;13(6):668–75.
Lopez-Candales A, Hernández Burgos PM, Hernandez-Suarez DF, Harris D. Linking chronic inflammation with cardiovascular disease: from normal aging to the metabolic syndrome. J Nat Sci. 2017;3(4):341.
Montero D, Walther G, Pérez-Martin A, Vicente-Salar N, Roche E, Vinet A. Vascular smooth muscle function in type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetologia. 2013;56(10):2122–33.
Miranda-Gonçalves V, Lameirinhas A, Henrique R, Jerónimo C. Metabolism and epigenetic interplay in cancer: regulation and putative therapeutic targets. Front Genet. 2018;9:427.
Ferrari A, Longo R, Silva R, Mitro N, Caruso D, De Fabiani E, et al. Epigenome modifiers and metabolic rewiring: new frontiers in therapeutics. Pharmacol Ther. 2019;193:178–93.
Langemeijer SMC, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–42.
Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–10.
Huang H, Jiang X, Li Z, Li Y, Song CX, He C, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2013;110(29):11994–9.
Couronné L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366(1):95–6.
Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, et al. Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica. 2013;98(12):1912–20.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–21.
Johansson C, Tumber A, Che KH, Cain P, Nowak R, Gileadi C, et al. The roles of Jumonji-type oxygenases in human disease. Epigenomics. 2014;6(1):89–120.
Bheda P, Schneider R. Epigenetics reloaded: the single-cell revolution. Trends Cell Biol. 2014;24(11):712–23.
Vora S, Tuttle M, Cheng J, Church G. Next stop for the CRISPR revolution: RNA-guided epigenetic regulators. FEBS J. 2016;283(17):3181–93.
Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 2005;97(20):1498–506.
Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1(3):117–36.
Acknowledgements
Not applicable
Funding
This work was supported by a BHF 4 year PhD Studentship FS/18/60/34181 to Hannah Green.
Author information
Authors and Affiliations
Contributions
Both authors contributed to the writing of the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Both authors have reviewed the manuscript and given consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Green, H.L.H., Brewer, A.C. Dysregulation of 2-oxoglutarate-dependent dioxygenases by hyperglycaemia: does this link diabetes and vascular disease?. Clin Epigenet 12, 59 (2020). https://doi.org/10.1186/s13148-020-00848-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-020-00848-y