
Abstract
Background
Alzheimer’s disease results from a neurodegenerative process that starts well before the diagnosis can be made. New prognostic or diagnostic markers enabling early intervention into the disease process would be highly valuable. Environmental and lifestyle factors largely modulate the disease risk and may influence the pathogenesis through epigenetic mechanisms, such as DNA methylation. As environmental and lifestyle factors may affect multiple tissues of the body, we hypothesized that the disease-associated DNA methylation signatures are detectable in the peripheral blood of discordant twin pairs.
Results
Comparison of 23 disease discordant Finnish twin pairs with reduced representation bisulfite sequencing revealed peripheral blood DNA methylation differences in 11 genomic regions with at least 15.0% median methylation difference and FDR adjusted p value ≤ 0.05. Several of the affected genes are primarily associated with neuronal functions and pathologies and do not display disease-associated differences in gene expression in blood. The DNA methylation mark in ADARB2 gene was found to be differentially methylated also in the anterior hippocampus, including entorhinal cortex, of non-twin cases and controls. Targeted bisulfite pyrosequencing of the DNA methylation mark in ADARB2 gene in 62 Finnish and Swedish twin pairs revealed that, in addition to the disease status, DNA methylation of this region is influenced by gender, age, zygosity, APOE genotype, and smoking. Further analysis of 120 Swedish twin pairs indicated that this specific DNA methylation mark is not predictive for Alzheimer’s disease and becomes differentially methylated after disease onset.
Conclusions
DNA methylation differences can be detected in the peripheral blood of twin pairs discordant for Alzheimer’s disease. These DNA methylation signatures may have value as disease markers and provide insights into the molecular mechanisms of pathogenesis. We found no evidence that the DNA methylation marks would be associated with gene expression in blood. Further studies are needed to elucidate the potential importance of the associated genes in neuronal functions and to validate the prognostic or diagnostic value of the individual marks or marker panels.
Similar content being viewed by others
Introduction
Alzheimer’s disease (AD) is an aging-associated neurodegenerative disorder and the most common cause of dementia. The molecular mechanisms of AD are not known in detail. The disease is characterized by accumulation of beta-amyloid plaques and hyperphosphorylated tau protein in the brain tissue. The pathological changes can start decades before the first clinical symptoms appear [1,2,3]. In the early-onset form of AD, which accounts for approximately 2–10% of the cases, the symptoms can start already before age of 30 years. In 5–10% of the early onset cases, the disease is caused by autosomal dominant mutations in the genes APP, PSEN1, and/or PSEN2 [1, 4]. The majority of AD cases are of the late-onset form, which typically manifests after 65 years of age [1, 4]. Based on twin studies, the estimated heritability of the disease exceeds 50% [5]. Genetic association with apolipoprotein E (APOE) epsilon 4 (ε4) allele is common in both early- and late-onset form. Furthermore, genome-wide association studies have identified over 20 additional variants each contributing less than 1% to the heritability of liability. Together with APOE ε4, these known variants explain approximately 29% of the heritability [4, 6,7,8] leaving a large fraction unexplained.
In addition to genetic factors, the morbidity of AD is influenced by modifiable lifestyle factors, such as physical activity and cognitive ability, nutrition, alcohol use, and smoking [1, 9]. These factors may influence the disease process through epigenetic mechanisms, such as DNA methylation, by causing changes detectable in various tissues. Such changes may provide insights into the molecular mechanisms of the disease and have potential as informative biomarkers. DNA methylation marks associated with AD have been discovered in several brain regions [10,11,12,13,14]. Whether similar marks exist in peripheral blood is still unclear [15]. The question is complicated by the fact that methylation levels at CpG sites associated with the disease may be driven by genetic variants. Within-pair comparison of disease-discordant monozygotic (MZ) twin pairs with, in practice, identical genomes, the same age and sex, and who share many intrauterine and early-life environmental factors provides a powerful approach for detection of the disease-associated methylation sites [16]. Dizygotic (DZ) disease-discordant twin pairs also provide improved sensitivity to the analyses as they share on average 50% of their genome and are of the same age and matched for prenatal and shared rearing environmental factors.
Modern technologies, such as positron emission tomography (PET), enable monitoring of the disease status in the brains of living subjects and are already utilized in diagnostics together with clinical examination and biomarkers to identify the individuals with first symptoms [9, 17, 18]. However, novel high-throughput methods enabling early detection and large scale monitoring of the disease status would be highly valuable. The aim of this study was to identify epigenetic marks associated with late-onset AD in peripheral blood by comparing DNA methylation profiles of the disease-discordant twin pairs and to examine overlap in brain tissue. To evaluate potential as prognostic or diagnostic marker, one of the most interesting loci was selected for validation in extended twin cohorts from Finland and Sweden.
Results
Twin pairs discordant for Alzheimer’s disease have similar genetic risk load for the disease
Before identification of the epigenetic marks, the genetic risk load of the twin pairs for Alzheimer’s disease was characterized. To measure the genetic risk load for AD in the Finnish study subjects, we analyzed the APOE genotypes and 21 loci previously associated with AD as a risk or protective variants (Table 2, Additional file 1) [4, 6]. In this analysis, we included 9 full MZ and 12 full DZ AD-discordant twin pairs, 9 unrelated controls, and 18 unrelated cases (Table 1). Monozygotic co-twins were confirmed to have identical single nucleotide polymorphism (SNP) profiles. All dizygotic co-twins were identical for the DSG2, CASS4, and SORL1 alleles but carried differences in 2–10 other variants. Most of the twin pairs (17 of 23 pairs) had APOE ε3ε3 genotype. Standardized GRS were calculated for the study groups based on the 21 loci previously associated with AD (Table 2). According to the generalized linear regression analysis, the GRS was not associated with the disease status (Wald test z value < 1, Pr(>[z]) > 0.5).
Blood DNA methylation marks associated with Alzheimer’s disease in the Finnish twin cohort
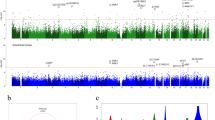
Our first research question was whether DNA methylation marks associated with AD can be detected in the peripheral blood of disease discordant twin pairs. The null hypothesis was that peripheral blood DNA methylation is not associated with the disease status. To address this hypothesis, we first identified the within-pair methylation differences associated with AD in 11 MZ twin pairs with reduced representation bisulfite sequencing (RRBS) as described in the “Methods” section (Table 1). This comparison revealed 427 CpG sites with increased and 474 sites with decreased methylation in AD cases when using cut off values of at least 15.0% median methylation difference and Benjamin-Hochberg false discovery rate (B-H FDR) adjusted p value ≤ 0.05 (Fig. 1a, Additional file 2a). Similarly, CpG methylome differences were then identified in 12 DZ twin pairs discordant for AD. This comparison revealed 1337 sites with increased and 764 sites with decreased methylation in AD cases (Fig. 1b, Additional file 2b). We then examined the overlap of differentially methylated CpG sites detected in both MZ and DZ within-pair comparisons. This revealed 11 common sites with consistent methylation difference present in both zygosity groups (adjusted p value ≤ 0.05), (Fig. 1c, d, Additional file 2c and Additional file 3). The genes nearest or overlapping with these regions included DEFA1 (intron), TSNARE1 (intron), DEAF1 (intron), ARAP2 (− 2,197,329 bp from TSS), CNPY1 (+ 764 bp from TSS), ADARB2 (exon), ARHGAP8 (intron), GTF3C2 (− 1157 bp from TSS), ACTA1 (+ 29543 bp from TSS), SEMA5A (+ 694751 bp from TSS), and CLIP2 (+ 121538 bp from TSS). No methylation differences were detected in the mitochondrial DNA or X chromosome with the chosen filtering criteria. In summary, DNA methylation changes in 11 genomic regions were found to be associated with AD in peripheral blood of AD-discordant Finnish twin pairs. These regions are henceforth referred to as AD-associated loci in blood. Based on these results, we could reject the first null hypothesis for the 11 CpG sites.

Peripheral blood CpG methylation differences in Finnish twin pairs discordant for Alzheimer’s disease. Peripheral blood CpG methylomes of 11 monozygotic (MZ) and 12 dizygotic (DZ) Finnish twin pairs discordant for Alzheimer’s disease (AD) were profiled with reduced representation bisulfite sequencing. Differentially methylated sites associated with AD in autosomes were identified using RADMeth algorithm and ± 15.0% average methylation difference and B-H FDR adjusted p value cut off 0.05 in a MZ and b DZ twin pairs separately. c Differentially methylated sites detected in both MZ and DZ twin pairs were extracted to identify overlaps between the AD-associated sites detected in both groups (Additional file 2). d Combined median methylation difference and adjusted p value (adj. pval) distribution of these 11 regions associated with AD in both MZ and DZ twin pairs is illustrated. The size of the dot indicates the percentage of the twin pairs, which had the required ≥ 10× coverage in the region. The closest or overlapping gene is indicated for the selected differentially methylated regions
Overlap of DNA methylation marks associated with Alzheimer’s disease in the anterior hippocampus and peripheral blood
Our second research question was whether the DNA methylation marks associated with AD in peripheral blood are also detectable in brain tissue. Our null hypothesis was that DNA methylation marks associated with AD in the anterior hippocampus, including the entorhinal cortex, do not overlap with those detected in peripheral blood. Anterior hippocampus, including entorhinal cortex, was selected for analysis, as these brain regions are crucial for memory formation and learning. Both regions were analyzed together as they were not available separately from the biobank. To test the hypothesis, the DNA methylation was profiled with the RRBS from postmortem tissue sections collected from six cases with AD and six controls (Table 1). Two cases were excluded from the final analysis as one was identified to be an outlier in the principal component analysis, and according to the neuropathological examination, one sample was from amygdala. Comparison of cases and controls with MethylKit R package revealed increased methylation at 114 sites and decreased methylation at 87 genomic sites among AD cases (at least 15.0% methylation difference, SLIM adjusted q value ≤ 0.05), overlapping or closest to the 176 gene identifiers, in the anterior hippocampus (Fig. 2a). These sites are henceforth referred to as AD-associated loci in the brain. Comparison of the gene identifiers closest to the AD-associated loci in the brain to those detected in blood revealed one overlapping gene ADARB2 (Fig. 2b). In the gene ADARB2, the differentially methylated sites in blood and hippocampus were localized within the same region chr10:1404752-1405717 in exon 3, in the proximity of the exon 3-intron junction. Therefore, the second null hypothesis was rejected.

Differentially methylated sites associated with Alzheimer’s disease in the anterior hippocampus. DNA methylation profiles of anterior hippocampus samples of 10 non-twin subjects, including six references and four with Alzheimer’s disease (AD), were examined with reduced representation bisulfite sequencing. The samples were from NIH NeuroBioBank. a Differentially methylated sites (201 CpGs) were identified with MethylKit package in R by using a minimum of 15% methylation difference and q value below 0.05 as filtering cutoff (Additional file 2d). The 201 differentially methylated CpG sites overlapped or were closest to 176 genes (AD-associated genes in the brain). Examples of the genes are shown. b Overlap of genes with AD-associated DNA methylation marks in both peripheral blood and anterior hippocampus of the brain
We also examined the correlation of AD-associated loci in blood with the brain tissue in the publicly available Illumina 450K data by using the online tool https://epigenetics.essex.ac.uk/bloodbrain/. Only two of the AD-associated loci in blood identified in this study are detected by Illumina 450K arrays, including probe cg26376030 in DEAF1 gene and cg22909083 in DEFA1B gene locus. The peripheral blood DNA methylation of probe cg22909083 had high correlation (> 0.95, p < 0.05) with all the four regions of brain available in the database, including prefrontal cortex, entorhinal cortex, superior temporal gyrus, and cerebellum. The probe cg26376030 had modest correlation (> 0.26, p < 0.05) with prefrontal cortex, entorhinal cortex, and superior temporal gyrus (Additional file 4).
Gene expression levels in whole blood and individual lymphocytes
Our third research question was whether the genes closest to DNA methylation marks in the blood are also associated with differences in gene expression in peripheral blood from AD cases compared to controls. Our null hypothesis was that the expression of the genes closest to the AD-associated loci in blood is not associated with AD. To address this hypothesis, expression of the 11 genes overlapping or closest to the AD-associated loci (Fig. 1d, Additional file 2c) were examined in the data from whole blood (GSE63061, Europeans) of 122 controls and 121 cases with AD [19] available at the GEO database. Expression of 9 genes was detected; however, there were no differences between AD cases and controls (Additional file 5). Expression of these genes was further examined in individual peripheral blood mononuclear cells (PBMCs) of two twin pairs discordant for episodic memory, one of which were also discordant for mild AD. The proportions of blood cell types expressing these genes were similar between the co-twins and no major differences were detected in the expression of the genes within the twin pairs (Additional files 6 and 7). In conclusion, analysis of blood did not provide evidence that the genes would display major disease associated functional significance in the PBMC populations. Based on our data, we could not reject the third null hypothesis.
Targeted validation of disease-associated methylation changes in ADARB2 gene in Finnish and Swedish twin pairs discordant for AD
Our fourth research question was whether the DNA methylation mark in the ADARB2 gene can be validated with targeted bisulfite pyrosequencing in an extended cohort including an independent twin cohort from Sweden. The null hypothesis was that the DNA methylation mark in ADARB2 is not associated with AD in an extended cohort. To address this hypothesis, the CpG methylation of the region (chr10:1,405,405-366) in ADARB2 gene was analyzed with targeted bisulfite pyrosequencing in 62 of AD discordant twin pairs including 33 Finnish and 29 Swedish twin pairs. Association of the CpG methylation with AD was analyzed using linear mixed effects model (lme) [20]. Initially, several models were compared with ANOVA including or excluding zygosity, age, and gender as fixed effects and twin pair identification number nested with genomic position and country of origin as random effects. The model with lowest AIC value was selected as the final model: methylation level (%) ~ disease status + zygosity + age + gender + diseases status × zygosity + disease status × age + disease status × gender + (1 | twin pair identification number/genomic position). Influence of APOE genotype (ε34/ε44 and ε33) was examined separately, including disease status and gender as fixed effects, to ensure sufficient number of observations in each subgroup. The influence of APOE genotype was examined because it is a well-known risk factor for AD and can influence the progression and severity of the disease, which in turn may be reflected in the state of the DNA methylation marks associated with the disease. Increased CpG methylation level of the region chr10:1,405,405-366 was validated to be associated with AD (Wald t value 4.68) and was influenced by interaction with gender, zygosity, and age. Separate examination by sex revealed that the CpG methylation level was increased in male cases (Wald t value 6.10) (Fig. 3a, Additional file 8). The methylation difference of the region was higher in DZ than MZ twin pairs (estimate 7.67%, standard error (SE) 1.44, Wald t value 5.34) (Fig. 3b), indicating that the association is influenced by genetic factors. In addition, in males the CpG methylation difference between discordant twin pairs increased with age (estimate 1.00% per year, SE 0.15, t value 6.43) (Fig. 3c). Interestingly, in females, the CpG methylation was increased in cases with APOE ε34/ε44 genotype (Wald t value 3.44); however, it was not increased in cases with the ε33 genotype (Wald t value − 1.67) (Fig. 3d). In male cases, the CpG methylation level was higher among cases than controls in both APOE genotype groups (Wald t value > 3.83), and in male cases, the level in APOE ε34/ε44 group was higher in comparison to the male cases with ε33 genotype (Wald t value 4.59). Of note, visual examination revealed that also the male cases with ε33 genotype had polarized into high and low/intermediate CpG methylation level groups (Fig. 3e).

The DNA methylation status of the ADARB2 gene is associated with Alzheimer’s disease. The DNA methylation level of region chr10:1,405,405-366 in exon 3 of ADARB2 gene was measured with targeted pyrosequencing in blood DNA samples collected from Finnish (33 pairs) and Swedish (29 pairs) twin pairs discordant for Alzheimer’s disease (AD). The association of the disease status with DNA methylation was examined with linear mixed effects model including gender, zygosity, age, and interaction terms as fixed effects and twin pair information nested with genomic position as a random effect. The country of origin was not associated with the CpG methylation level and was excluded from the final models. a The CpG methylation level in male and female controls (ctrl) and cases. Wald t value for disease association including both genders is 4.68, for males 6.10, and for females 1.17. b Influence of zygosity of CpG methylation difference between twin pairs discordant for AD. c Influence of age on the CpG methylation difference between AD discordant twin pairs. Influence of APOE genotype on the CpG methylation level was examined separately including only gender as a covariate. Only group ε33 and combined groups ε34/44 were included in the statistical analysis. The CpG methylation levels for d female and e male ctrls and cases in different APOE groups. f CpG methylation difference in AD discordant twin pairs before onset of the diseases (not predictive for disease outcome based on Cox mixed effects model). In b–f, the representative data for CpG site 6 in the region is shown, and in a, c, and f, the lines are the smoothed conditional means with 0.95 confidence intervals area
Next, we determined whether the association of the methylation mark with AD is attenuated and if also smoking history is included as a fix effect and interaction term in the linear mixed effects model. Smoking data was available for 60 twin pairs, including 49 current or past smokers and 15 smoking discordant pairs. The association of the methylation mark with the disease status was not attenuated when smoking was included in the analysis (estimate 64.51%, SE 9.23, and Wald t value 6.99). However, the smoking did interact with the disease (estimate 8.10%, SE 1.43, and Wald t value 5.68). The median methylation level of the mark was higher in those cases who were current or past smokers in comparison to controls and cases who had never smoked or controls who were current or past smokers (Additional file 9). The association with the disease remained significant also in the model with adjustment for APOE genotype (estimate 35.41%, SE 9.41, and Wald t = 3.76).
In conclusion, our findings from targeted analysis confirm the association of CpG methylation in region chr10:1,405,405-366 with AD and the methylation is influenced by gender, age, zygosity, APOE genotype, and smoking. Thus, based on our results, the fourth null hypothesis was rejected.
CpG methylation status of region chr10:1,405,405-366 in ADARB2 gene does not predict the disease outcome
Our fifth and last research question was whether the DNA methylation mark in ADARB2 is predictive for AD. The null hypothesis was that the DNA methylation mark in ADARB2 gene is not associated with incident AD. To address this hypothesis, the methylation level in region chr10:1,405,405-366 was analyzed with targeted bisulfite sequencing and Cox mixed effects model was used to examine the association of CpG methylation with disease outcome in samples collected 0.5–18.5 years before disease onset from 120 discordant twin pairs from Sweden. The gender and age at blood draw were included as fixed effects in the analysis and relatedness nested with genomic position as a random effect. According to the results, the disease outcome was not associated with the DNA methylation level of the region before disease onset (hazard ratio = 1.00, 95% confidence interval = 1.00–1.00, p = 0.63). Thus, the fifth null hypothesis was not rejected (Fig. 3f).
Discussion
Our results show that differences in DNA methylation associated with AD can be detected in the peripheral blood in at least 11 genomic regions. Although the functional importance of the affected regions in transcriptional regulation requires further studies, our results suggest that the DNA methylation marks are not associated with gene expression in peripheral blood. Several of these genes have been previously linked to neurological functions or pathologies. Among the most interesting findings was the AD-associated methylation signature in the exon 3 of ADARB2 gene, which was detected in both peripheral blood and anterior hippocampus in brain tissue. Based on previous studies, expression of ADARB2 is brain specific; however, its function has not been fully characterized. ADARB2 may have a function in the inhibition of RNA editing by ADAR1 and ADAR2 proteins [21,22,23]. Importantly, a recent study showed that mice lacking the corresponding exon 3 of ADARB2 gene display impaired hippocampus-dependent memory formation, learning, and regulation of genes implicated in synaptic functions [24]. Consistently, a variant in ADARB2 gene has been associated with accelerated cognitive decline after conversion from mild cognitive impairment (MCI) to AD [25]. With targeted validation including independent twin pairs discordant for AD from both Finnish and Swedish cohorts, we further found that the methylation level in ADARB2 was associated with APOE genotype, age, gender, and smoking which all also influence risk of AD. Although these results together suggest potential importance of ADARB2 in the molecular pathology of AD, the high inter-individual variance and relatively small within-pair differences in blood suggest that methylation at this region alone is unlikely to have value as a diagnostic marker. Furthermore, CpG methylation in ADARB2 was not predictive for AD when measured before disease onset.
Similarly to ADARB2, several of the differentially methylated regions in blood overlapped or were closest to genes that are highly expressed in the brain or have been previously linked to neuronal functions or pathologies, such as neurogenesis, synaptic function, cognitive decline, intellectual disability, social interaction, schizophrenia, and Parkinson’s and Alzheimer’s diseases [24, 26,27,28,29,30,31]. For example, increased levels of alpha defensins (encoded by DEFA1) have been reported in the CSF of patients with AD [29], whereas mutations in DEAF1 gene can cause severe intellectual disability and behavioral problems. Conditional knockout of Deaf1 gene in mouse model leads to a phenotype with impaired memory and increased anxiety [26]. TSNARE1 gene has been previously associated with schizophrenia and rate of cognitive decline in late MCI [32,33,34].
Why DNA methylation marks associated with neurodegenerative disease can be detected in the blood is not fully clear. One potential explanation is that these marks are caused by environmental or lifestyle factors, which affect the methylation status of DNA also in peripheral blood; however, the influence on the disease process manifests only in the specific tissues where the affected genes are functionally active or important. Nonetheless, potential importance of these genes in the neuronal functions and disease process remains to be elucidated. Further studies are also needed to elucidate whether combinations of differentially methylated regions in multiple AD-associated loci would increase sensitivity in distinguishing AD cases from cognitively preserved controls and to evaluate the value of these changes as predictive disease markers.
Previously, two independent large-scale studies on different regions of the brain have found DNA methylation marks in the ANK1 and RHBDF2 genes to be associated with AD [11, 12]. Both studies utilized Illumina 450K arrays. These regions were not captured by the RRBS method utilized in our study. Vice versa, the differentially methylated region in ADARB2 gene found in this study is not covered by the probes present in 450K array.
A limitation in our study is that no adjustment for smoking was performed in the RRBS analysis due to the small number of samples for this variable available in the discovery cohort. To our knowledge, the genes nearest to the differentially methylated sites in peripheral blood have not been previously associated with smoking, suggesting that the results were not confounded by this factor. Another limitation is that the analysis was carried out by using whole blood and cell type heterogeneity can potentially confound the results. The adjustment of the RRBS results with the Bacon method is expected to correct for potential bias caused by cell type heterogeneity.
Conclusions
Our results show that DNA methylation differences can be detected in the peripheral blood of twin pairs discordant for Alzheimer’s disease. These DNA methylation signatures may have value as disease marker candidates and may provide insights into the molecular mechanisms of pathogenesis. Our results suggest that the DNA methylation marks do not associate with gene expression in blood. Further studies are needed to elucidate the functional importance of the affected genes and to validate the prognostic or diagnostic value of the individual marks or marker panels.
Methods
Study participants and samples
A total of 395 peripheral blood samples from Finnish and Swedish cohorts and 12 brain samples from NIH NeuroBioBank were analyzed in this study (Table 1). In more detail, the older Finnish twin cohort [35, 36] of 2483 individuals, born 1922–1937, had been previously screened for cognitive functions by phone interview [37], and the discordant twin pairs were invited to neuropsychological testing, brain magnetic resonance imaging, PET amyloid imaging [18, 38,39,40,41], and collection of EDTA blood samples. Based on the overall evaluation, a diagnosis of AD was made (29 pairs). The controls were not required to have negative PET amyloid imaging. In addition to the original cohort, four twin pairs born in 1915–1950 were included in the study. Furthermore, BD Vacutainer® CPT™ samples were collected from two MZ twin pairs (age 76 years) discordant for episodic memory, where one pair was also discordant for mild AD. These two twin pairs were from a younger Finnish twin cohort (born in 1938–1944). The blood DNA samples from Swedish twin pairs (149 pairs), born 1907–1953, were from the Swedish Twin Registry [42] and had been collected before (120 pairs) or after (29 pairs) the onset of the disease. The participants originated from three sub-studies of aging within the Swedish Twin Registry [42], namely the Swedish Adoption/Twin Study of Aging (SATSA) [43], the Study of Dementia in Swedish Twins (HARMONY) [44], and TwinGene [42]. Disease information was available through linkage to nationwide health registers for all Swedish twins, and in addition, AD was clinically evaluated as part of the SATSA and HARMONY study [45]. Furthermore, our study material included fresh frozen post-mortem brain tissue from anterior hippocampus, including dentate gyrus and entorhinal cortex. The samples had been collected from 12 adults (57–90 years), including six patients with AD and six non-demented controls. Five of the individuals were white and seven had unknown ethnic origin.
Nucleic acid extractions
The DNA was extracted from EDTA blood with Qiagen QIAamp DNA Blood kit and from fresh frozen tissue with QIAamp DNA Micro kit. Qubit 3.0 dsDNA HS Assay, Thermo Scientific NanoDrop 2000, and Fragment Analyzer HS Genomic DNA assay (Advanced Analytical) were used for the quality analysis and quantitation.
Targeted genotyping and variant data analysis
Genotyping of APOE alleles and 21 SNPs previously associated with AD [6] (Table 2) was performed with Illumina TruSeq Custom Amplicon assay and sequencing (500 cycles) with Illumina MiSeq. Illumina BaseSpace was used for variant analysis. Genetic risk scores were calculated based on 21 genetic variants by summing up the number of risk or protective alleles, weighted by the effect size from the stage 1 and 2 meta-analyses in a genome-wide association study of AD [6]. Influence of the GRS on the disease outcome was examined with generalized linear model in R version 3.4.3 [46]. The influence of sex, age, and APOE genotype on the model was examined separately due to small number of samples. Packages lmtest [47], multiwayvcov [48], and fmsb [49] were utilized in the model and in calculation of Nagelkerke’s R square.
Reduced representation bisulfite sequencing and data analysis
The libraries for RRBS were prepared as previously described [50, 51] and were sequenced (1 × 50bp) with Illumina HiSeq2500/3000. The trimmed reads (Trim Galore v0.4.1 [52] were mapped to hg19 (blood) or hg38 (brain) reference genome using Bismark v0.14.5/0.15.0 [53]. The differentially methylated sites in blood were identified using a paired comparison with RADMeth [54] using CpG sites with coverage ≥ 10× in both twins in a pair, and at least four pairs per study group, as an input. The genomic inflation factors and bias caused by unobserved factors were calculated and corrected with BACON v1.10.1 package [55]. The genomic inflation factor was 1.07 (bias 0.30) for MZ and 1.14 (bias 0.50) for DZ twins. We expect this step to correct for any bias caused by cell type heterogeneity. The differentially methylated regions were filtered according to Benjamin-Hochberg false discovery rate adjusted p value combined with RADMeth algorithm (< 0.05) followed by absolute average within-pair methylation difference (≥ 15.0%) and central interval (70% interval < 0 or 70% interval > 0) for these sites. Due to small cohort size, the analysis was not adjusted for smoking. Information of the smoking history of the twin pairs is provided in the Additional file 9. The effect sizes of the top-ranked differentially methylated sites from the monozygotic twin pairs (“discovery set”) were examined for reproducibility in the dizygotic twin pairs (“validation set”) by comparing the corresponding values in a scatterplot. The effect size was measured as methylation difference (% estimate by RADMeth) and the standardized effect size was measured as estimated methylation difference over standard deviation of pairwise methylation. Also, for brain samples, the CpG sites with coverage ≥ 10× and less than 99.9th percentile of coverage in each sample were included. Methylkit v.1.1.7 [56] was used to detect differentially methylated CpG sites with a minimum and consistent difference of 15% and q value ≤ 0.05 as a cutoff. MethylKit uses the SLIM method to adjust p values to q values. The bisulfite conversion efficiency of all the samples was above 99% according to the lambda spike-in control.
Targeted bisulfite pyrosequencing analysis
The PCR primers 5′-gtaatttagtggtgttgttgaat-3′, 5′-biotin-cctaacccccaaccaacttcttactac-3′ and the sequencing primer 5′-gggttgagttaagtgtgtttggtaga-3′ for the region chr10:1405336-1405409 (hg19) were designed with Qiagen PyroMark AssayDesign SW 2.0. The samples were prepared with Qiagen EpiTect Fast Bisulfite Conversion and Qiagen PyroMark PCR kits and sequenced with Qiagen PyroMark Q24 Advanced system. The amplicons were analyzed with PyroMark Q24 Advanced software version 3.0.0. The statistical analysis was carried in R v3.4.3 [46] using packages lme4 v1.1-15 [20], car v2.1-6 [57], survival 2.42-4 [58], coxme 2.2-10 [59], and ggplot2 v2.2.1 [60]. Association of AD with CpG methylation was examined with lme4 package, with modeling methylation level as a function of disease status. Influence of covariates and their interaction with AD in the model was examined, including gender, age, zygosity, APOE genotype, and smoking (never, ever) as fixed effects and twin pair nested with genomic position and country of origin as random effects as specified in the results. The model with the lowest AIC was chosen. The Cox mixed effects model with coxme package was utilized to examine CpG methylation level as a prognostic marker of disease outcome. In this model, only age and gender were included as fixed effects and twin pair information nested with genomic position as a random effect.
Single-cell transcriptome analysis
PBMCs were isolated from the BD Vacutainer® CPT™ Cell Preparation tubes. Single-cell RNA sequencing (scRNA-seq) libraries were prepared using ChromiumTM controller, Single Cell 3′ Reagents kit (10x Genomics), targeting at the recovery of 3000 cells per sample, and were sequenced with Hiseq2500. The data was preprocessed using Cell Ranger v. 1.2.0 (10x Genomics) and the GRCh38 genome reference. The filtered gene-barcode unique molecular identifier count matrix of the aggregated sample (Cell Ranger aggr tool) was normalized using a global-scaling normalization from the Seurat R package v. 1.4.0.9 [61]. Finally, the full Seurat scRNA-seq analysis was performed for each sample individually. Cells with unique gene count over 1750 or proportion of mitochondrial genes over 10% were first filtered out. Ten most significant principal components were selected for the graph-based clustering, the different PBMC cell types were identified using canonical markers, and the cell type frequencies were estimated. Paired t test was used to test differential expression between sample groups.
Public data used
The data from GSE63061 [19] available at NCBI GEO database was analyzed with GEO2R using default settings [62, 63].
Abbreviations
- AD:
-
Alzheimer’s disease
- ADD:
-
Alzheimer’s disease-discordant
- APOE:
-
Apolipoprotein E (APOE)
- DZ:
-
Dizygotic
- GRS:
-
Genetic risk score
- MCI:
-
Mild cognitive impairment
- MZ:
-
Monozygotic
- PBMC:
-
Peripheral blood mononuclear cells
- PET:
-
Positron emission tomography
- RRBS:
-
Reduced representation bisulfite sequencing
- scRNA-seq:
-
Single-cell RNA sequencing
- SE:
-
Standard error
- SNP:
-
Single nucleotide polymorphism
References
Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12(4):459–509.
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–44.
Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12(3):292–323.
Cuyvers E, Sleegers K. Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. Lancet Neurol. 2016;15(8):857–68.
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–74.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–13.
Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD, et al. GWAS on family history of Alzheimer’s disease. Transl Psychiatry. 2018;8(1):99–018-0150-6.
Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137–52.
Roubroeks JAY, Smith RG, van den Hove DLA, Lunnon K. Epigenetics and DNA methylomic profiling in Alzheimer’s disease and other neurodegenerative diseases. J Neurochem. 2017;143(2):158–70.
De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014 Sep;17(9):1156–63.
Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014 Sep;17(9):1164–70.
Watson CT, Roussos P, Garg P, Ho DJ, Azam N, Katsel PL, et al. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016;8(1):5–015-0258-8.
Zhao J, Zhu Y, Yang J, Li L, Wu H, De Jager PL, et al. A genome-wide profiling of brain DNA hydroxymethylation in Alzheimer’s disease. Alzheimers Dement. 2017;13(6):674–88.
Fransquet PD, Lacaze P, Saffery R, McNeil J, Woods R, Ryan J. Blood DNA methylation as a potential biomarker of dementia: a systematic review. Alzheimers Dement. 2018;14(1):81–103.
Tsai PC, Bell JT. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. Int J Epidemiol. 2015;44(4):1429–41.
Nordberg A, Rinne JO, Kadir A, Langstrom B. The use of PET in Alzheimer disease. Nat Rev Neurol. 2010;6(2):78–87.
Scheinin NM, Scheinin M, Rinne JO. Amyloid imaging as a surrogate marker in clinical trials in Alzheimer’s disease. Q J Nucl Med Mol Imaging. 2011;55(3):265–79.
Sood S, Gallagher IJ, Lunnon K, Rullman E, Keohane A, Crossland H, et al. A novel multi-tissue RNA diagnostic of healthy ageing relates to cognitive health status. Genome Biol. 2015;16:185-015-0750-x.
Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48.
Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, et al. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550(7675):249–54.
Oakes E, Anderson A, Cohen-Gadol A, Hundley HA. Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J Biol Chem. 2017;292(10):4326–35.
Gaisler-Salomon I, Kravitz E, Feiler Y, Safran M, Biegon A, Amariglio N, et al. Hippocampus-specific deficiency in RNA editing of GluA2 in Alzheimer’s disease. Neurobiol Aging. 2014;35(8):1785–91.
Mladenova D, Barry G, Konen LM, Pineda SS, Guennewig B, Avesson L, et al. Adar3 Is involved in learning and memory in mice. Front Neurosci. 2018;12:243.
Lee E, Giovanello KS, Saykin AJ, Xie F, Kong D, Wang Y, et al. Single-nucleotide polymorphisms are associated with cognitive decline at Alzheimer’s disease conversion within mild cognitive impairment patients. Alzheimers Dement (Amst). 2017;8:86–95.
Vulto-van Silfhout AT, Rajamanickam S, Jensik PJ, Vergult S, de Rocker N, Newhall KJ, et al. Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am J Hum Genet. 2014;94(5):649–61.
Bartelt-Kirbach B, Langer-Fischer K, Golenhofen N. Different regulation of N-cadherin and cadherin-11 in rat hippocampus. Cell Commun Adhes. 2010;17(4-6):75–82.
Duan Y, Wang SH, Song J, Mironova Y, Ming GL, Kolodkin AL, et al. Semaphorin 5A inhibits synaptogenesis in early postnatal- and adult-born hippocampal dentate granule cells. Elife. 2014;3. https://doi.org/10.7554/eLife.04390.
Szekeres M, Ivitz E, Datki Z, Kalman J, Pakaski M, Varhelyi ZP, et al. Relevance of defensin beta-2 and alpha defensins (HNP1-3) in Alzheimer’s disease. Psychiatry Res. 2016;239:342–5.
Zhu B, Chen C, Xue G, Moyzis RK, Dong Q, Chen C, et al. The SEMA5A gene is associated with hippocampal volume, and their interaction is associated with performance on Raven’s Progressive Matrices. Neuroimage. 2014;88:181–7.
Vandeweyer G, Van der Aa N, Reyniers E, Kooy RF. The contribution of CLIP2 haploinsufficiency to the clinical manifestations of the Williams-Beuren syndrome. Am J Hum Genet. 2012;90(6):1071–8.
Li QS, Parrado AR, Samtani MN, Narayan VA. Alzheimer’s Disease Neuroimaging Initiative. Variations in the FRA10AC1 fragile site and 15q21 are associated with cerebrospinal fluid Abeta1-42 level. PLoS One. 2015;10(8):e0134000.
Gu LZ, Jiang T, Cheng ZH, Zhang YC, Ou MM, Chen MC, et al. TSNARE1 polymorphisms are associated with schizophrenia susceptibility in Han Chinese. J Neural Transm (Vienna). 2015;122(6):929–32.
Sleiman P, Wang D, Glessner J, Hadley D, Gur RE, Cohen N, et al. GWAS meta analysis identifies TSNARE1 as a novel Schizophrenia / Bipolar susceptibility locus. Sci Rep. 2013;3:3075.
Kaprio J, Koskenvuo M. Genetic and environmental factors in complex diseases: the older Finnish Twin Cohort. Twin Res. 2002 Oct;5(5):358–65.
Kaprio J. The Finnish Twin Cohort Study: an update. Twin Res Hum Genet. 2013;16(1):157–62.
Jarvenpaa T, Rinne JO, Raiha I, Koskenvuo M, Lopponen M, Hinkka S, et al. Characteristics of two telephone screens for cognitive impairment. Dement Geriatr Cogn Disord. 2002;13(3):149–55.
Jarvenpaa T, Raiha I, Kaprio J, Koskenvuo M, Laine M, Kurki T, et al. Regional cerebral glucose metabolism in monozygotic twins discordant for Alzheimer’s disease. Dement Geriatr Cogn Disord. 2003;16(4):245–52.
Jarvenpaa T, Laakso MP, Rossi R, Koskenvuo M, Kaprio J, Raiha I, et al. Hippocampal MRI volumetry in cognitively discordant monozygotic twin pairs. J Neurol Neurosurg Psychiatry. 2004;75(1):116–20.
Scheinin NM, Aalto S, Kaprio J, Koskenvuo M, Raiha I, Rokka J, et al. Early detection of Alzheimer disease: (1)(1)C-PiB PET in twins discordant for cognitive impairment. Neurology. 2011;77(5):453–60.
Virta JJ, Karrasch M, Kaprio J, Koskenvuo M, Raiha I, Viljanen T, et al. Cerebral glucose metabolism in dizygotic twin pairs discordant for Alzheimer’s disease. Dement Geriatr Cogn Disord. 2008;25(1):9–16.
Magnusson PK, Almqvist C, Rahman I, Ganna A, Viktorin A, Walum H, et al. The Swedish Twin Registry: establishment of a biobank and other recent developments. Twin Res Hum Genet. 2013;16(1):317–29.
Finkel D, Reynolds CA, McArdle JJ, Pedersen NL. The longitudinal relationship between processing speed and cognitive ability: genetic and environmental influences. Behav Genet. 2005;35(5):535–49.
Gatz M, Fratiglioni L, Johansson B, Berg S, Mortimer JA, Reynolds CA, et al. Complete ascertainment of dementia in the Swedish Twin Registry: the HARMONY study. Neurobiol Aging. 2005;26(4):439–47.
Gatz M, Pedersen NL, Berg S, Johansson B, Johansson K, Mortimer JA, et al. Heritability for Alzheimer’s disease: the study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci. 1997;52(2):M117–25.
R Core Team. R: A language and environment for statistical computing. 2017; Available at: https://www.R-project.org/, 2017-2018.
Zeileis A, Hothorn T. Diagnostic checking in regression relationships. R News. 2002;2:7–10.
Graham N, Arai M, Hagströmer B. multiwayvcov: Multi-Way Standard Error Clustering version 1.2.3. 2018; Available at: http://sites.google.com/site/npgraham1/research/code. Accessed 02, 2018.
Nakazawa M. fmsb: Functions for Medical Statistics Book with some Demographic Data version 0.6.1 . 2017; Available at: http://minato.sip21c.org/msb/. Accessed 02, 2018.
Boyle P, Clement K, Gu H, Smith ZD, Ziller M, Fostel JL, et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 2012;13(10):R92.
Konki M, Pasumarthy K, Malonzo M, Sainio A, Valensisi C, Soderstrom M, et al. epigenetic silencing of the key antioxidant enzyme catalase in karyotypically abnormal human pluripotent stem cells. Sci Rep. 2016;6:22190.
Krueger F. Trim Galore! 2015; Available at: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/. Accessed 2015, 2015.
Krueger F, Andrews S. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–2.
Dolzhenko E, Smith AD. Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments. BMC Bioinformatics. 2014;15:215-2105-15-215.
van Iterson M, van Zwet EW, BIOS Consortium, Heijmans BT. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 2017;18(1):19-016-1131-9.
Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87–2012-13-10-r87.
Fox J, Weisberg S. An R companion to applied regression. 2nd ed. Thousand Oaks: Sage; 2011.
Therneau T. A Package for Survival Analysis in S. version 2.38. 2018; Available at: https://CRAN.R-project.org/package=survival. Accessed 07/30, 2018.
Therneau T. coxme: Mixed Effects Cox Models. 2018; Available at: https://CRAN.R-project.org/package=coxme. Accessed 30 July 2018.
Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2009.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20.
Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–5.
Acknowledgements
We acknowledge Finnish Functional Genomics Centre, Biocenter Finland, University of Turku, and Åbo Akademi University for the infrastructure support, NIH NeuroBioBank for the brain samples, and Sinikka Collanus for the H&E staining of the brain samples.
Availability of the data and materials
The data from RRBS analysis will be available in the NCBI SRA database. The data from GSE63061 [19] available at NCBI GEO database was analyzed with GEO2R using default settings [62, 63].
Funding
This study was supported by State Research Funding, Hospital District of Southwest Finland, Finnish Cultural, Yrjö Jahnsson and Wihuri foundations, and by Tirkkonen family. JK has been supported by the Academy of Finland grants # 265240, 263278, 308248, 312073, and MO by # 297908. JOR has been supported by the Sigrid Juselius Foundation, by the Academy of Finland grant # 2310962 and by State funding for university-level health research (VTR).
Author information
Authors and Affiliations
Contributions
JR, JK, NS, and NL were responsible for the characterization and recruitment of the Finnish twin pairs and provided the study samples. NP and IK provided the samples from Swedish Twin cohorts. MO and JK provided additional Finnish Twin cohort samples and data. MR performed the neuropathological examination of the brain tissue samples, and MK the sample preparation for RRBS under supervision of RLu. MM performed the bioinformatics analysis of the blood RRBS data under supervision of HL and RLu. The sample preparation for scRNA-seq was carried out by MK and TL. The bioinformatics analysis of the scRNA-seq data was performed by JS under supervision of AL and LE. The bioinformatics analysis of the brain RRBS data was carried out by BG under supervision of AL, LE, and RLu. The genotyping and pyrosequencing analysis were performed by MK, IK, and RLu. Nearly all the authors participated in the processing of the preparation of the manuscript and interpretation of the data. All authors revised and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The studies on the living human subjects have been approved by the Ethics Committee Hospital District of Southwest Finland, Turku, Finland (ETMK: 77/1801/2013) and Regional Ethics Board at Karolinska Institutet, Stockholm, Sweden. Written informed consent was obtained from all the participants. The study was conducted by following good clinical and scientific practices and in accordance with the Helsinki Declaration.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table of APOE genotypes of the study individuals. (XLSX 10 kb)
Additional file 2:
Table of differentially methylated sites associated with Alzheimer’s disease. Differentially methylated sites in Alzheimer’s disease discordant a) monozygotic, b) dizygotic and in c) both twin pair groups and d) in brain of non-twin subjects with Alzheimer’s disease in comparison to reference subjects. (XLSX 415 kb)
Additional file 3:
The effect sizes of the top-ranked differentially methylated sites. The effect sizes of the top-ranked differentially methylated sites from the monozygotic twin pairs (MZ, "Discovery set") were examined for reproducibility in the dizygotic twin pairs ("DZ, Validation set") by comparing the corresponding values in a scatterplot. A) effect size was measured as methylation difference (% estimate by RADMeth) and b) standardised effect size was measured as estimated methylation difference over standard deviation of pairwise methylation difference (n = 172, R = 0.452, p value = 4.754e-10 and R = 0.354, p value = 1.837e-6, respectively, by Pearson correlation test). The top 200 sites in the MZ cohort were initially selected (ranked by q value) of which 172 met the coverage criteria for the DZ cohort. The correlation tests indicate overall consistency in direction of methylation of the top-ranked MZ sites in the DZ cohort. (TIF 4938 kb)
Additional file 4:
Correlation of the DNA methylation in blood and brain. Correlation of two peripheral blood DNA methylation marks associated Alzheimer’s disease was examined in publicly available Illumina 450K data available from blood and different regions of brain by using the online tool: https://epigenetics.essex.ac.uk/bloodbrain/. (PDF 1782 kb)
Additional file 5:
Gene expression level of candidate genes in blood of non-twin subjects with Alzheimer’s disease in comparison to reference subjects. Gene expression levels of the candidate genes from Additional file 2c in GEO NCBI data from blood (GSE63061) of non-twin subjects with Alzheimer’s disease in comparison to reference subjects. (XLSX 12 kb)
Additional file 6:
Proportion of peripheral blood mononuclear cell subtypes in twin pairs discordant for episodic memory as determined by single cell 3' RNA-sequencing. (PDF 4 kb)
Additional file 7:
Expression levels of the candidate genes in the peripheral blood mononuclear cell subtypes in twin pairs discordant for episodic memory as determined by single cell 3' RNA-sequencing. (TIF 37322 kb)
Additional file 8:
Residual plots for linear mixed effects models. CpG methylation of the region chr10:1,405,405-366, in exon three of ADARB2 gene was analyzed with targeted pyrosequencing. Association of disease status with methylation level (outcome) was examined with linear mixed effects model (lme4 R package) including zygosity, age and gender as fixed effects and twin pair information nested with genomic position as random effects. In the Figure is the representative data of the residual plots for the models: a) me ~ dis + zyg + age + sex + dis * zyg + dis * age + dis * sex + (1 | pairid/pos) in the data including both male and female twin pairs, b) me ~ dis + zyg + age + dis * zyg + dis * age + (1 | pairid/pos) in the data including only males. me = CpG methylation level, dis = disease status, zyg = zygosity, pairid = twin pair information, pos = genomic position. (JPG 297 kb)
Additional file 9:
DNA methylation level of the ADARB2 mark is higher in the Alzheimer’s disease cases with a history of smoking. Smoking history was included in the linear mixed effects model as a fixed effect, and interaction term, to determine whether this adjustment attenuates association of the ADARB2 DNA methylation mark with Alzheimer’s disease. The analysis was carried out by including all the 60 twin pairs with smoking data available. According to the results the disease association remained significant also when smoking was included in the model (estimate 64.51%, SE 9.23, Wald t value 6.99, not adjusted for APOE genotype). Smoking did interact with the disease status (estimate 8.10%, SE 1.43, Wald t value 5.68) and the median methylation level was increased in the cases with a smoking history in comparison to non-smoking cases and controls as well as controls with a smoking history. The association remained significant also when APOE genotype was included in the model (estimate 35.41%, SE 9.41 and Wald t value 3.76). A) DNA methylation levels for the ADARB2 mark in non-smokers and past or current smokers are shown separately for females with APOE ε33 genotype who differ from the other groups and show no disease association. B) Residual plot for the full model: me ~ dis + dis * zyg + dis * age + dis * sex + dis * smo + dis * apoe + (1 | pairid/pos). me = CpG methylation level, dis = disease status, zyg = zygosity, smo = smoking history (never, ever), apoe = apoe genotype group, pairid = twin pair information, pos = genomic position. (PDF 715 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Konki, M., Malonzo, M., Karlsson, I.K. et al. Peripheral blood DNA methylation differences in twin pairs discordant for Alzheimer’s disease. Clin Epigenet 11, 130 (2019). https://doi.org/10.1186/s13148-019-0729-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-019-0729-7