
Abstract
Background
Metformin is a widely prescribed antihyperglycemic agent that has been also associated with multiple therapeutic effects in various diseases, including several types of malignancies. There is growing evidence regarding the contribution of the epigenetic mechanisms in reaching metformin’s therapeutic goals; however, the effect of metformin on human cells in vivo is not comprehensively studied. The aim of our study was to examine metformin-induced alterations of DNA methylation profiles in white blood cells of healthy volunteers, employing a longitudinal study design.
Results
Twelve healthy metformin-naïve individuals where enrolled in the study. Genome-wide DNA methylation pattern was estimated at baseline, 10 h and 7 days after the start of metformin administration. The whole-genome DNA methylation analysis in total revealed 125 differentially methylated CpGs, of which 11 CpGs and their associated genes with the most consistent changes in the DNA methylation profile were selected: POFUT2, CAMKK1, EML3, KIAA1614, UPF1, MUC4, LOC727982, SIX3, ADAM8, SNORD12B, VPS8, and several differentially methylated regions as novel potential epigenetic targets of metformin. The main functions of the majority of top-ranked differentially methylated loci and their representative cell signaling pathways were linked to the well-known metformin therapy targets: regulatory processes of energy homeostasis, inflammatory responses, tumorigenesis, and neurodegenerative diseases.
Conclusions
Here we demonstrate for the first time the immediate effect of short-term metformin administration at therapeutic doses on epigenetic regulation in human white blood cells. These findings suggest the DNA methylation process as one of the mechanisms involved in the action of metformin, thereby revealing novel targets and directions of the molecular mechanisms underlying the various beneficial effects of metformin.
Trial registration
EU Clinical Trials Register, 2016-001092-74. Registered 23 March 2017, https://www.clinicaltrialsregister.eu/ctr-search/trial/2016-001092-74/LV.
Similar content being viewed by others

Background
Metformin is the first-line drug for type 2 diabetes (T2D) therapy, used since 1950s [1]. Although there are a great number of various studies on the metformin pharmacogenomics, pharmacokinetics, and lately its interaction with the gut microbiome, the details of the molecular mechanisms of metformin action have not been fully understood.
So far, there are only a few studies within the context of metformin action and changes in one of the most commonly studied epigenetic modifications—DNA methylation. One of the targeted studies has shown that metformin treatment of pregnant rats with gestational diabetes can reduce methylation level of peroxisome proliferator-activated receptor γ coactivator-1A (PPARGC1A), therefore preventing the abnormal glycolipid metabolism in their offspring [2]. In addition, a genome-wide study of metformin effects on lymphoblastoid cell lines has revealed potential biomarkers for metformin’s anticancer response [3]. In the context of possible molecular mechanisms of how metformin induce changes in the methylation profile, a recent study has proved that, in cancer cells, metformin can exert its effects via regulation of the H19/SAHH axis [4]. This has been supported by data showing that metformin promotes global methylation by decreasing S-adenosylhomocysteine (SAH) intracellular levels in various cell types, including non-cancerous [5]. One of the latest studies have specifically shown metformin’s effect on lowering the methylation levels at the metformin transporter genes, resulting in higher expression levels in liver tissue [6]. Studies describing other epigenetic effects of metformin have shown its impact on various histone modifications via multiple mechanisms, mostly AMPK dependent, and effect on expression levels of numerous miRNAs through increase in DICER protein levels as well [7].
Nevertheless, there is a significant lack of information on how metformin affects global epigenetic regulation in non-cancerous cells or in cells obtained from metformin-treated humans. Therefore, our aim was to investigate the short-term effect of metformin on DNA methylation profiles in blood cells from healthy volunteers. Here we compared the changes in DNA methylation in the same subjects before and after the metformin intake.
Results
Characteristics of the study participants
We used Illumina Infinium 450k array to evaluate the effect of metformin on DNA methylation in 12 healthy volunteers. The characteristics of the study group are summarized in Table 1. Samples, for analysis of the methylation levels, from each participant were obtained at three time points, further marked as M0 (before starting a metformin therapy), M10h (10 h after the first metformin intake, before the second tablet), and M7d (time point after 7 days of metformin administration). M10h sample was chosen to evaluate effect of one metformin’s dose; to ensure accuracy of this measurement, all study participants were strictly instructed to take the second metformin tablet only after the M10h blood sampling.
Differentially methylated CpGs
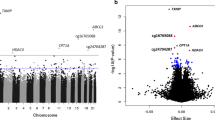
During the data preprocessing stage, 64,512 (13.29%) probes were filtered out, leaving 421,000 probes for downstream analysis. To detect differentially methylated CpG sites/probes (DMPs), we applied limma analysis between contrasts at all three time points, i.e., baseline, after 10 h and 7 days of metformin administration. The model included the methylation values at the contrasted time points, together with the cell-type estimations as covariates. Comparing methylation values at M10h and M0 samples, 72 differentially methylated CpG sites with a false discovery rate (FDR) of < 0.05 were identified after correction for multiple testing using the Benjamini-Hochberg method. In the same way, 52 DMPs were found applying contrast between methylation levels at M7d and M0 and only one (cg07026010—NUDCD3) in case of M7d with M10h comparison (full list of significant CpGs is available in Additional file 1). Of these, 43 (59.72%), 24 (46.15%), and 1 (100%) CpG sites were hypermethylated, and 29 (40.28%), 28 (53.85%), and 0 (0%) CpG sites were hypomethylated when contrast analyses were applied for M10h vs M0, M7d vs M0, and M7d vs M10h respectively (Fig. 1). The median absolute difference in beta values, comparing all contrasts, was 0.013 (interquartile range (IQR), 0.006–0.029) for statistically significant differentially methylated probes. The average estimated genomic inflation factor (λ) for all three contrasts before correction was 1.64 ± 0.28, and after including covariates, it was reduced to 1.30 ± 0.15. Additional evaluation of λ with qq-plots depicted the same improvement ensured by including covariates (data not shown).

Differentially methylated positions in all analyzed contrasts. Volcano plot showing raw − log10 (p value) versus log-fold change of M values and the Manhattan plot showing the position of probes with their corresponding unadjusted p values across the genome in a M10h vs M0, b M7d vs M0, and c M7d vs M10h sample comparisons. The significant CpG sites (after FDR correction) are highlighted in red. M0—before starting a metformin therapy; M10h—10 h after the first metformin intake, before the second tablet; M7d—time point after 7 days of metformin administration
Among the identified DMP, a total of 11 CpGs with the most consistent changes in the DNA methylation profile were emphasized (Fig. 2) based on two additional criteria. First, we included all overlapping DMP at both contrasts M10h vs M0 and M7d vs M0 (n = 5; cg03515060, cg18394557, cg16013966, cg05638165, cg18824330). Second, we selected probes if their median beta values at time points M10h and M7h overlapped IQRs of M7h and M10h, respectively. Also, IQRs of both time points could not overlap with IQR of time point M0 (n = 6; cg12740863, cg16843994, cg12162450, cg19176072, cg01644741, cg02622542). Of these 11 CpGs, 8 (72.73%) CpG sites displayed hypermethylation, while 3 (27.27%) CpG sites showed hypomethylation when comparing methylation levels after the metformin use (at time points M10h and M7d) with methylation levels before the use of metformin (Fig. 2).

Methylation levels of the top 11 most significant CpGs across the investigated three time points, i.e., M0 (before starting a metformin therapy), M10h (10 h after the first metformin intake, before the second tablet), and M7d (time point after 7 days of metformin administration), together with their associated genes. Box plots depict median, maximum, minimum, 25th percentile, and 75th percentile. Dots beyond the bounds of the whiskers represent outliers. * and ** denote significance levels 0.05 and 0.01 respectively
All identified 11 CpG sites corresponded to 11 genes according to the 450k annotation file published by Price [8] (Table 2). One of these CpG sites was located in high-density CpG island, 7 CpG sites—in intermediate-density CpG islands with 1 bordering high-density CpG island, and 3—in non-islands according to HIL CpG classes.
To analyze the possible influence of circadian changes on the methylation profile, firstly, we searched our DMP list for the most common genes associated with regulation of circadian rhythm, such as BMAL1, PER1, PER2, PER3, ARNTL, CRY1, and CRY2. Secondly, we evaluated the main known functional roles of genes associated with the 125 DMPs, and, thirdly, we used the results from pathway enrichment analysis to find any connections with the circadian regulation. In result of these steps, we did not find any significant associations between the DMPs and circadian rhythm.
The correlation between methylation and RNA expression level of the corresponding gene was verified using targeted data form RNA-seq. Out of 11 genes tested, only the expression of UPF1 (p − 0.024), MUC4 (p − 0.029), and KIAA1614 (p − 0.048) showed significant correlation with the methylation of corresponding CpG sites (Table 2).
Differentially methylated regions (DMRs)
During the DMR analysis, we found 13 regions with significant differences in methylation levels (summarized in Table 3). Five of the identified regions overlapped with some of the significant DMPs but not with the 11 sites prioritized by us.
Enrichment analysis
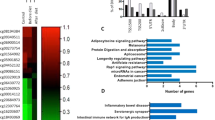
To evaluate the potential biological significance of the impact of differentially methylated CpG sites, we performed a gene set pathway enrichment analysis by using the Ingenuity Pathway Analysis (IPA). All genes associated with significant differentially methylated probes (FDR < 0.05) from different contrasts were selected. Thus, 72 genes were selected from the M10h vs M0 contrast and included in the first pathway analysis, and 52 genes from the M7d vs M0 contrast and included in the second pathway analysis. We did not include the only significant result from the M7d vs M10h contrast. The top enriched canonical pathways are summarized in Table 4.
In addition to the canonical pathways, we identified nine enriched networks in the M10h vs M0 contrast, and four in the M7d vs M0 comparison. The top enriched networks with IPA score > 20 were as follows (score/focus molecules): M10h vs M0—hematological system development and function, cellular movement, cell-to-cell signaling and interaction (28/13); hereditary disorder, neurological disease, organismal injury and abnormalities (23/11). M7d vs M0—cell-to-cell signaling and interaction, cellular assembly and organization, cellular function and maintenance (48/19); cell morphology, cell-to-cell signaling and interaction, cellular assembly and organization (41/17). Two of the most relevant networks are visualized in Fig. 3.

Top enriched networks from IPA. Green nodes—hypermethylated; red nodes—hypomethylated. a Cell-to-cell signaling and interaction, cellular assembly and organization, and cellular function and maintenance (central predicted associated biological functions—tumorigenesis processes) had an IPA score of 48. b Cell morphology, cell-to-cell signaling and interaction, and cellular assembly and organization (central predicted associated biological functions—metabolism processes) had an IPA score of 41
Discussion
The aim of our study was to examine metformin-induced alterations in epigenetic regulation processes by performing genome-wide DNA methylation analysis in human white blood cells followed by estimation of RNA expression levels of identified genes. We conducted our study in order to understand the pathways affected by metformin at real life physiological conditions in humans. This is extremely important taking into account the pleiotropic effects of metformin, and such studies may pinpoint important novel targets not only for treatment of T2D but also for other diseases. Various studies have shown that the evaluated effects in the methylation profile of peripheral blood DNA, that is the only option to access repeated tissue sampling in humans, are highly representative to the changes in other organs [9,10,11]. It is known that the DNA methylation pattern is highly subject specific and is influenced by many factors making it very difficult to identify the metformin-specific effects in a case-control-based type of study. We therefore selected a longitudinal design for this study, using short response time in order to exclude the influence of other factors. We also involved healthy volunteers to avoid a background of any commonly studied diseases so far related with the beneficial effects of metformin. One of our goals was to detect the fastest practically measurable effect of metformin on DNA methylation. Taking into account the known high variability of metformin pharmacokinetics, the time point when to evaluate the immediate and at the same time most profound effect was chosen to be the impact of one dose, and sampling time was selected at 10 h, before the recommended administration time of the second dose. Furthermore, M10h vs M0 sample comparison revealed the highest number or DMPs, representing the significant effect of one metformin dose.
To our best knowledge, this is the first study showing the metformin-mediated change of DNA methylation in healthy individuals already 10 h after administration. From the pool of 125 significantly modified sites, we prioritized 11 differentially methylated CpG with the largest and most consistent changes in beta values at different contrasts.
We assumed that some methylation changes measured at 10 h (M10h) could be caused by the circadian rhythm, which has been well described before and proven to be a driver of dynamic gene expression [12]. To avoid any false conclusions about the epigenetic targets of metformin, we paid specific attention to the presence of genes involved in the circadian rhythm among regions covering DMPs. We also evaluated this possibility by focusing on two contrasts that represent the different methylation profiles of DNA purified from blood samples that were collected in two distinct time points of the day—M7d vs M0 and M7d vs M10h. We did not observe any overlapping DMPs between the particular contrasts, suggesting no significant influence of the circadian rhythm on the DNA methylation in our data. Surprisingly, we observed only one significant DMP comparing M7d and M10h time points, providing a strong support for the fact that observed methylation changes are indeed caused by metformin rather than other factors changing during the trial, such as diet or circadian cycle.
Genes corresponding to the top-ranked DMPs represent the main functional groups associated with previously described targets of metformin therapy: regulatory processes of energy homeostasis, inflammatory responses, tumorigenesis, and neurodegeneration. The criteria based on the comparison of median beta values and IQRs (see the “Results” section) were chosen to avoid bias in prioritization and would allow to include potentially important DMPs in addition to only those being significant at both M10h vs M0 and M7d vs M0 contrasts.
Interestingly, we found DMP within CAMKK1 gene—one of two highly homologous genes coding for Ca2+/calmodulin-dependent protein kinase kinases (CaMKK) [13]—with CaMKK2 being a known regulator of AMP-activated protein kinase (AMPK). Despite the fact that only CaMKK2 has been proven to form a stable complex with AMPK, both isoforms of the CaMKK are capable of phosphorylating the AMPKα subunit at Thr-172 in vitro [14, 15]. From our data, the differentially methylated CpG close to the CAMKK1 TSS together with negatively correlated mRNA expression data as the result of metformin administration suggests a potential contribution of CaMKK1 in the AMPK-mediated mechanism of metformin anti-diabetic action.
Furthermore, it is known that metformin exerts its effects also via AMPK-independent mechanisms [16], as shown by CaMKK1 ability to mediate glucose uptake in muscle cells independently from AMPK and Akt [17], in that way suggesting that methylation level changes in CaMKK1 could be a part from an alternative pathway responsible for the therapeutic effects of metformin.
Additionally, we identified a differentially methylated CpG site near the transcription factor coding gene SIX3 [18]. Downregulation of SIX3 due to the methylation of the SIX3 promoter is observed in lung adenocarcinoma tissues and lung cancer cell lines, where mRNA expression of the gene is also associated with higher survival rate [19]. Some research suggest SIX3 linkage to diabetes from genetic studies [20] and show SIX3 as possible regulator of insulin production in β-cells in an age-dependent manner [21]. Lowered methylation level of CpG near the SIX3 TSS shown in our data suggests the DNA methylation as another potential epigenetic mechanism involved in SIX3 expression regulation. SIX3 is not expressed in human white blood cells [22], explaining the absence of reads corresponding to SIX3 in our RNA-seq data, but gene expression may manifest in other tissues. So far, normalized insulin production itself has not been considered as a therapeutic effect of metformin, although it might be affected along with metformin-induced improvements of insulin sensitivity [23].
Our data also show ADAM8 as a considerable potential contributor in the anti-inflammatory action of metformin, that is, one of the known beneficial effects of this medication [24]. ADAM8 is a cell surface protease, mainly expressed in granulocytes and monocytic cells, where it conducts the regulation of monocyte adhesion and migration [25,26,27]. Its contribution in the inflammatory responses regarding neurodegenerative disorders, allergy, asthma, and acute lung inflammation has been widely described before [28,29,30,31]. Our data justify the anti-inflammatory properties of metformin independently of diabetes status [24] and suggest the potential contribution of ADAM8 in the process. Due to its expression in human white blood cells, ADAM8 might be considered a promising biomarker for the detection of metformin-induced anti-inflammatory responses while reflecting inflammatory processes in adipocytes; however, further experimental evidence is required.
Many of the genes linked to the top-ranked DMPs are functionally associated with various malignancies. The most significant DMP in our study appeared to be situated in the body of POFUT2. O-Fucosyltransferase 2 encoded by POFUT2 is proved to restrict epithelial-mesenchymal transition and affect cell motility in mouse embryos [32], and is considered as a useful prognostic biomarker in patients with glioblastoma and adenocarcinoma [33, 34]. To our knowledge, there are no reports yet describing POFUT2’s association with the beneficial effects of metformin. Our data also show several more DMPs located within or near the TSS of tumor-related genes, including SNORD12B—previously associated with colorectal and breast cancer pathogenesis [35,36,37], MUC4—promising prognostic marker and therapeutic target in the case of pancreatic cancer [38,39,40], KIAA1614 with promoter hypermethylation observed in colon tissues from patients with ulcerative collitis as well as in colon cancer cell lines [41], and UPF1 with indisputably crucial role in the maintenance of genome stability, significantly implicated in various malignancies [42,43,44,45,46,47].
The functions of two genes from the top DMPs’ associated list are poorly defined. We identified increased DNA methylation level close to the TSS of VPS8 gene. VPS8 is an accessory subunit of CORVET complex, necessary for mediating multiple steps in the endocytic pathway and required for fusion of early endosomes [48]. Thus far, there is no conclusive data indicating the possible effects of VPS8 dysregulation on phenotype in humans [49,50,51,52]. Likewise, the function of long intergenic non-protein coding RNA 1249 (LINC01249/LOC727982) is still not clear with only few reports on genetic association of SNPs within the gene with infectious disease and blood pressure [53, 54].
Overall, the DNA methylation has a repressive effect on transcription factor binding; therefore, we used ENCODE data on transcription factor binding sites to identify such possible interactions [55, 56]. We detected transcription factors CTCF, CTCFL, and Egr1 binding to the genomic region overlapping the differentially methylated CpG within EML3 gene; out of these, CTCF is proved to mediate glucagon production [57] and Egr1 is responsible for insulin resistance [58]. Although there are no data available to date, supporting direct metformin impact on EML3 (nuclear microtubule-binding protein) [59] or describing EML3 contribution in metformin therapeutic effects, increased expression of EML3 in cultured human cells as a result of metformin-1816 small molecule perturbation has been reported before [60]. Likewise, the genomic region within UPF1 gene, covering the top-ranked CpG site is associated with CTCF, Egr1, and two more transcription factors: MYC—involved in the pathogenesis of diabetes [61], and PU1—initiating insulin resistance as well as regulating lipolysis [62].
The detected DMRs, as well, could essentially be grouped by connection to the processes currently known to be affected by metformin. For example, the most significant DMR was associated with EPHB1, which together with other Ephrin receptors forms the largest subgroup of the Eph receptor tyrosine kinase (RTK) family [63]. Underexpression of the EphB1 protein is significantly associated with tumor progression in gastric carcinomas and higher invasiveness of colorectal cancer cells, suggesting a tumor-suppressive role of the protein and possible implication in the beneficial effects of metformin [64, 65].
Another noteworthy DMR was associated with APP-cleaving enzyme 2 coding gene (BACE2) encoded protein that cleaves amyloid precursor protein into amyloid beta peptide, and is implicated in the pathogenesis of neurodegenerative diseases including Alzheimer’s disease [66,67,68]. Interestingly, increased β-cell proliferation and glucose-stimulated insulin secretion resulting from reduced Bace2 levels have been previously reported [69]. In a mouse model of T2D, induced by the overexpression of human islet amyloid polypeptide, BACE2 deficiency improved glucose tolerance, suggesting that BACE2 inhibition might serve as a potential therapeutic strategy for T2D treatment [70].
Another DMR is associated with SFRP2, Secreted Frizzled Related Protein 2. Methylation changes in the promoter region of SFRP2 have been proposed as a potential noninvasive biomarker for colorectal cancer [71, 72]. Its mRNA is also expressed in mouse and human adipose tissue, and elevated levels have positive correlation with BMI and with abnormal glucose tolerance [73].
The pathway enrichment analysis revealed metformin’s association with various pathways some of which already has been described in connection with metformin action but not in the context of epigenetic regulation. The top enriched pathway after one dose of metformin—Unfolded Protein Response (UPR)—has been shown to be one of the main mechanisms of inducing apoptosis by metformin in acute lymphoblastic leukemia cells [74], and metformin-induced UPR inhibition in kidney cells can explain metformin’s beneficial effects [75].
One of the products of the top enriched pathway describing changes after week long metformin administration (S-methyl-5-thio-α-d-ribose 1-phosphate Degradation) is l-methionine, an essential amino acid in human organism. Moreover, it is known that l-methionine is used for generation of S-adenosylmethionine (SAM) [76], which has been depicted to be an essential part of metformin-induced increase in global methylation levels as it accumulates in cells during metformin therapy [5]. Taking into account the results from enriched pathways and the fact that we mostly observe metformin-induced hypermethylation than hypomethylation, it is possible that activation of this particular canonical pathway may contribute to the previously described increase in SAM levels.
Although enriched networks (Fig. 3) are not directly related to known metformin effects, the downstream molecules of those associated with differential methylation levels in our study group are known to be involved in various pathways related with T2D (e.g., AKT, ERK1/2, JNK, P13K), insulin regulation processes [77], cancer development mechanisms [78], and other.
The correlation between DNA methylation and gene expression is complex and nonlinear [79]. The generally accepted consequence of DNA methylation is transcriptional repression; however, methylation in the transcribed region might also demonstrate positive correlation with mRNA expression [80]. In our study, we did not detect a convincing correlation between DNA methylation of top-ranked loci and transcription level of corresponding genes; however, the influence of methylation as well as gene expression itself are tissue-specific and might be missed by focusing on single type of cells only. Nevertheless, the significant correlation observed between the expression levels of UPF1, MUC4, KIAA1614, and the methylation level of the corresponding CpG sites provide evidence for a crucial contribution of epigenetic regulation in the mechanism of action of metformin, which results in specific alterations of gene expression profiles.
Currently, it is not fully known whether metformin has only an indirect effect on the epigenetic regulation processes in the human organism via the previously described H19/SAHH axis or through linking cellular metabolism to the mechanisms needed for DNA methylation [4, 5]. However, the methylation profile and concentration of metformin used in cell type specific in vitro experiments may significantly differ from the physiological levels and observations in the affected cells in human body. The large variation of SAH and SAM levels in various cell types has been described [5]. In addition, the previous studies evaluating the metformin-induced methylation profile changes mostly have been targeted; thus, it is not surprising that we did not observe the DMPs at the same genes or pathways.
Major limitation of this study is the low sample size even though there are number of reports using the same number of individuals in their studies [81,82,83,84]. On the other hand, we believe that this weakness is compensated by the number of strengths in our design. First, we used a longitudinal study design and it has been recognized that, in similar time series studies, individuals can be treated as their own controls before and during treatment and sufficiently increase the power of the study [85] compared to case-control design especially accounting for the inter-individual variability among study participants. Secondly, the longitudinal design combined with observation of methylation changes in the shortest possible time allows us to minimize the effects of other factors that can induce changes in methylation unrelated to the metformin treatment. Thirdly, inclusion of healthy subjects should have minimized false associations and conclusions arising from unaccounted treatment status by metformin or other medications in T2D patients, including the unknown true duration of T2D before diagnosis. Finally, the use of genome-wide methylation analysis allows us to observe unbiased effects and find new metformin targets.
Another limitation in our study is the lack of clinical and biochemical measures at all time points. In the same time, it has been previously shown that metformin has small or no effect of such measures as plasma glucose level in healthy individuals [86, 87], and we decided not to include those in study protocol.
Unfortunately, due to the lack of similar studies, we were not able to support our findings from literature and replication in other cohorts is needed.
Conclusions
This is the first study showing the immediate effect of metformin on white blood cell DNA methylation in humans at therapeutic doses. The gained knowledge about the metformin-induced methylation profile changes in healthy individuals can be used as basis for further in vitro and in vivo studies, which are important due to the growing number of various metformin therapeutic application possibilities in non-diabetic patients.
Methods
Study design
Study group involved 12 healthy metformin-naïve voluntary individuals. The involvement and sample collection was organized in collaboration with the Genome Database of Latvian Population (LGDB) [88]. Exclusion/inclusion criteria (Additional file 2) were defined according to the requirements of concurrently ongoing clinical trial (registration number: 2016-001092-74 (www.clinicaltrialsregister.eu)), which also involves gut microbiome analysis. Participants were included if they matched the following criteria: have not used antibiotics, immunosuppressive medicaments, corticosteroids, or pharmaceutical-grade probiotics during the time period of the past 2 months; have not been diagnosed with oncological, autoimmune, chronical gastrointestinal tract diseases, or T2D; have not had diarrhea in the past week; and are not taking any other medications incompatible with metformin. The research subjects received an 850-mg metformin tablet (Berlin-Chemie AG) twice a day for a week. Samples for hematological, biochemical tests were collected in certified clinical laboratory at fasting state 1–3 days before starting the metformin administration. Whole blood samples for methylation analysis were collected at three time points: (1) before starting metformin therapy (morning, fasting state)—M0, (2) 10 h after first metformin intake, before the second tablet (evening)—M10h, and (3) after 7 days of metformin administration (morning, fasting state)—M7d. Throughout the article, we have defined the measurement of 10-h sample as the immediate effect of metformin.
Sample analysis
DNA isolation from whole blood samples using the phenol-chloroform extraction method was performed by Genome Database of Latvian Population (briefly described before [89]). DNA samples were quantified with Qubit® 2.0 Fluorometer using Qubit dsDNA HS Assay Kit (TherfmoFisher Scientific, USA). For the bisulfite conversion, the EZ DNA Methylation-Gold TM kit (Zymo research, USA) was used according to the manufacturer’s instructions. DNA methylation was determined by the Illumina Infinium HumanMethylation450 BeadChip Array (Illumina, USA), using 500 ng of each bisulfite-treated DNA sample.
Total RNA was isolated from whole blood samples using PerfectPure RNA Blood Kit (5Prime GmbH, Hamburg, Germany). Ribosomal RNS depletion was done with Low Input RiboMinus™ Eukaryote System v2 (Thermo Fisher Scientific, USA). For cDNA library preparation, we used Ion Total RNA-Seq Kit v2 (Thermo Fisher Scientific, USA), and sequencing was performed on the Ion Proton™ System and Ion PI™ Chip (Thermo Fisher Scientific, USA).
Data preprocessing and statistical analysis
IDAT files were imported using R package minfi [90]. Cell counts were estimated from methylation data using Houseman algorithm [91] implemented in minfi.
Data preprocessing and normalization was done using Enmix [92]. Briefly, probes with detection p value > 0.05 and probes with a multimodal distribution were filtered out. Background correction was performed with the function preprocessENmix using unused color channels as a background parameter estimate. Probe intensities were normalized using a quantile normalization method, and probe type bias was adjusted using the Regression on Correlated Probes (RCP) method [93]. Probes having a SNP or single base extension annotation in CpG site were excluded. Due to interrupted use of metformin by one of the study subjects, the sample taken after 1 week of metformin administration for that particular subject was discarded.
Batch effect was removed from data using slide and subsequently subjects as covariates as they showed the strongest influence on the probe methylation variability. Batch effect was removed using ComBat [94] wrapped in the Enmix package. Differentially methylated probes between time points were identified using limma [95] on ComBat preprocessed data, adjusting for the following cell types estimated by minfi: CD8T, CD4T, NK, and Gran. Inflation factor of p-value distribution was estimated using R package GenABEL [96]. All analyses were performed using R (3.3.3).
Statistically significant DMRs were identified with DMRcate software [97], FDR < 0.05. Threshold for minimum number of probes within the region was set to three. DMRs were estimated from methylation M values using the individual CpG site significance threshold at FDR < 0.05. The interval between individual significant CpG sites had to be less than 1000 bp in the regions. The bandwidth scaling factor was set as suggested in the manual (C = 2). Regulatory information from Ensembl 91 regulation resources was added to identified DMPs and DMRs using Ensembl Regulation API [98].
Pathway enrichment analysis was performed with the IPA tool [99]. Information about enriched canonical pathways and networks was obtained performing the core analysis on all significant DMPs with FDR < 0.05.
RNA-seq data analysis
Reads were mapped against human reference genome GRCh38, and read quantification was performed using STAR (2.5.3a) [100]. Obtained per-gene read counts were normalized using trimmed mean normalization (TMM), and counts per million (CPM) values were calculated with edgeR [101]. ComBat [94] implemented in R package sva [102] was used to adjust CPM values for subject-specific effects, and the Spearman correlation was estimated for the adjusted CPM values and the beta values for 11 selected CpG sites with SciPy [103].
Abbreviations
- CPM:
-
Counts per million
- DMP:
-
Differentially methylated CpG site/probe
- DMR:
-
Differentially methylated region
- IPA:
-
Ingenuity Pathway Analysis
- IQR:
-
Interquartile range
- RCP:
-
Regression on Correlated Probes
- T2D:
-
Type 2 diabetes
- TMM:
-
Trimmed mean normalization
- TSS:
-
Transcription starting site
References
Marshall SM. 60 years of metformin use: a glance at the past and a look to the future. Diabetologia. 2017;60:1561–5.
Song AQ, Sun LR, Zhao YX, Gao YH, Chen L. Effect of insulin and metformin on methylation and glycolipid metabolism of peroxisome proliferator-activated receptor gamma coactivator-1A of rat offspring with gestational diabetes mellitus. Asian Pac J Trop Med. 2016;9:91–5.
Niu N, Liu T, Cairns J, Ly RC, Tan X, Deng M, Fridley BL, Kalari KR, Abo RP, Jenkins G, et al. Metformin pharmacogenomics: a genome-wide association study to identify genetic and epigenetic biomarkers involved in metformin anticancer response using human lymphoblastoid cell lines. Hum Mol Genet. 2016;25:4819–34.
Zhong T, Men Y, Lu L, Geng T, Zhou J, Mitsuhashi A, Shozu M, Maihle NJ, Carmichael GG, Taylor HS, Huang Y. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene. 2017;36:2345–54.
Cuyas E, Fernandez-Arroyo S, Verdura S, Garcia RA, Stursa J, Werner L, Blanco-Gonzalez E, Montes-Bayon M, Joven J, Viollet B, et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene. 2017;37:963.
Garcia-Calzon S, Perfilyev A, Mannisto V, de Mello VD, Nilsson E, Pihlajamaki J, Ling C. Diabetes medication associates with DNA methylation of metformin transporter genes in the human liver. Clin Epigenetics. 2017;9:102.
Bridgeman SC, Ellison GC, Melton PE, Newsholme P, Mamotte CDS. Epigenetic effects of metformin: from molecular mechanisms to clinical implications. Diabetes Obes Metab. 2018;20:1553–62.
Price ME, Cotton AM, Lam LL, Farre P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6:4.
Crujeiras AB, Diaz-Lagares A, Sandoval J, Milagro FI, Navas-Carretero S, Carreira MC, Gomez A, Hervas D, Monteiro MP, Casanueva FF, et al. DNA methylation map in circulating leukocytes mirrors subcutaneous adipose tissue methylation pattern: a genome-wide analysis from non-obese and obese patients. Sci Rep. 2017;7:41903.
Farre P, Jones MJ, Meaney MJ, Emberly E, Turecki G, Kobor MS. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin. 2015;8:19.
Barault L, Ellsworth RE, Harris HR, Valente AL, Shriver CD, Michels KB. Leukocyte DNA as surrogate for the evaluation of imprinted Loci methylation in mammary tissue DNA. PLoS One. 2013;8:e55896.
Lim AS, Srivastava GP, Yu L, Chibnik LB, Xu J, Buchman AS, Schneider JA, Myers AJ, Bennett DA, De Jager PL. 24-hour rhythms of DNA methylation and their relation with rhythms of RNA expression in the human dorsolateral prefrontal cortex. PLoS Genet. 2014;10:e1004792.
Okuno S, Kitani T, Fujisawa H. Studies on the substrate specificity of Ca2+/calmodulin-dependent protein kinase kinase alpha. J Biochem. 1997;122:337–43.
Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33.
Green MF, Anderson KA, Means AR. Characterization of the CaMKKbeta-AMPK signaling complex. Cell Signal. 2011;23:2005–12.
Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–86.
Witczak CA, Fujii N, Hirshman MF, Goodyear LJ. Ca2+/calmodulin-dependent protein kinase kinase-alpha regulates skeletal muscle glucose uptake independent of AMP-activated protein kinase and Akt activation. Diabetes. 2007;56:1403–9.
Granadino B, Gallardo ME, Lopez-Rios J, Sanz R, Ramos C, Ayuso C, Bovolenta P, Rodriguez de Cordoba S. Genomic cloning, structure, expression pattern, and chromosomal location of the human SIX3 gene. Genomics. 1999;55:100–5.
Mo ML, Okamoto J, Chen Z, Hirata T, Mikami I, Bosco-Clement G, Li H, Zhou HM, Jablons DM, He B. Down-regulation of SIX3 is associated with clinical outcome in lung adenocarcinoma. PLoS One. 2013;8:e71816.
Hwang JY, Sim X, Wu Y, Liang J, Tabara Y, Hu C, Hara K, Tam CH, Cai Q, Zhao Q, et al. Genome-wide association meta-analysis identifies novel variants associated with fasting plasma glucose in East Asians. Diabetes. 2015;64:291–8.
Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, Spitale RC, Dai C, Gu X, Qu K, et al. Age-dependent pancreatic gene regulation reveals mechanisms governing human beta cell function. Cell Metab. 2016;23:909–20.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 2012;22:1760–74.
Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 2003;29:6S28–35.
Cameron AR, Morrison VL, Levin D, Mohan M, Forteath C, Beall C, McNeilly AD, Balfour DJ, Savinko T, Wong AK, et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ Res. 2016;119:652–65.
Yoshiyama K, Higuchi Y, Kataoka M, Matsuura K, Yamamoto S. CD156 (human ADAM8): expression, primary amino acid sequence, and gene location. Genomics. 1997;41:56–62.
Yoshida S, Setoguchi M, Higuchi Y, Akizuki S, Yamamoto S. Molecular cloning of cDNA encoding MS2 antigen, a novel cell surface antigen strongly expressed in murine monocytic lineage. Int Immunol. 1990;2:585–91.
Hodgkinson CP, Ye S. Microarray analysis of peroxisome proliferator-activated receptor-gamma induced changes in gene expression in macrophages. Biochem Biophys Res Commun. 2003;308:505–10.
Dreymueller D, Pruessmeyer J, Schumacher J, Fellendorf S, Hess FM, Seifert A, Babendreyer A, Bartsch JW, Ludwig A. The metalloproteinase ADAM8 promotes leukocyte recruitment in vitro and in acute lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2017;313:L602–14.
Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor alpha induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron-glia interactions during neurodegeneration. J Neurosci. 2000;20:7964–71.
Fourie AM, Coles F, Moreno V, Karlsson L. Catalytic activity of ADAM8, ADAM15, and MDC-L (ADAM28) on synthetic peptide substrates and in ectodomain cleavage of CD23. J Biol Chem. 2003;278:30469–77.
Chen J, Jiang X, Duan Y, Long J, Bartsch JW, Deng L. ADAM8 in asthma. Friend or foe to airway inflammation? Am J Respir Cell Mol Biol. 2013;49:875–84.
Du J, Takeuchi H, Leonhard-Melief C, Shroyer KR, Dlugosz M, Haltiwanger RS, Holdener BC. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev Biol. 2010;346:25–38.
Dong S, Nutt CL, Betensky RA, Stemmer-Rachamimov AO, Denko NC, Ligon KL, Rowitch DH, Louis DN. Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J Neuropathol Exp Neurol. 2005;64:948–55.
Aramburu A, Zudaire I, Pajares MJ, Agorreta J, Orta A, Lozano MD, Gurpide A, Gomez-Roman J, Martinez-Climent JA, Jassem J, et al. Combined clinical and genomic signatures for the prognosis of early stage non-small cell lung cancer based on gene copy number alterations. BMC Genomics. 2015;16:752.
Gaedcke J, Grade M, Camps J, Sokilde R, Kaczkowski B, Schetter AJ, Difilippantonio MJ, Harris CC, Ghadimi BM, Moller S, et al. The rectal cancer microRNAome--microRNA expression in rectal cancer and matched normal mucosa. Clin Cancer Res. 2012;18:4919–30.
Xu L, Ziegelbauer J, Wang R, Wu WW, Shen RF, Juhl H, Zhang Y, Rosenberg A. Distinct profiles for mitochondrial t-RNAs and small nucleolar RNAs in locally invasive and metastatic colorectal cancer. Clin Cancer Res. 2016;22:773–84.
Askarian-Amiri ME, Crawford J, French JD, Smart CE, Smith MA, Clark MB, Ru K, Mercer TR, Thompson ER, Lakhani SR, et al. SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA. 2011;17:878–91.
Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol. 2013;10:607–20.
Andrianifahanana M, Moniaux N, Schmied BM, Ringel J, Friess H, Hollingsworth MA, Buchler MW, Aubert JP, Batra SK. Mucin (MUC) gene expression in human pancreatic adenocarcinoma and chronic pancreatitis: a potential role of MUC4 as a tumor marker of diagnostic significance. Clin Cancer Res. 2001;7:4033–40.
Gautam SK, Kumar S, Cannon A, Hall B, Bhatia R, Nasser MW, Mahapatra S, Batra SK, Jain M. MUC4 mucin- a therapeutic target for pancreatic ductal adenocarcinoma. Expert Opin Ther Targets. 2017;21:657–69.
Kang K, Bae JH, Han K, Kim ES, TO K, Yi JM. A genome-wide methylation approach identifies a new hypermethylated gene panel in ulcerative colitis. Int J Mol Sci. 2016;17:1291.
Azzalin CM, Lingner J. The human RNA surveillance factor UPF1 is required for S phase progression and genome stability. Curr Biol. 2006;16:433–9.
Chawla R, Redon S, Raftopoulou C, Wischnewski H, Gagos S, Azzalin CM. Human UPF1 interacts with TPP1 and telomerase and sustains telomere leading-strand replication. EMBO J. 2011;30:4047–58.
Chang L, Li C, Guo T, Wang H, Ma W, Yuan Y, Liu Q, Ye Q, Liu Z. The human RNA surveillance factor UPF1 regulates tumorigenesis by targeting Smad7 in hepatocellular carcinoma. J Exp Clin Cancer Res. 2016;35:8.
Li L, Geng Y, Feng R, Zhu Q, Miao B, Cao J, Fei S. The human RNA surveillance factor UPF1 modulates gastric cancer progression by targeting long non-coding RNA MALAT1. Cell Physiol Biochem. 2017;42:2194–206.
Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, Xu G, Lu L, Wang C, Song M, et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med. 2014;20:596–8.
Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, Harding H, Ron D, Gardner LB. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol. 2011;31:3670–80.
Perini ED, Schaefer R, Stoter M, Kalaidzidis Y, Zerial M. Mammalian CORVET is required for fusion and conversion of distinct early endosome subpopulations. Traffic. 2014;15:1366–89.
Lunetta KL, D'Agostino RB Sr, Karasik D, Benjamin EJ, Guo CY, Govindaraju R, Kiel DP, Kelly-Hayes M, Massaro JM, Pencina MJ, et al. Genetic correlates of longevity and selected age-related phenotypes: a genome-wide association study in the Framingham Study. BMC Med Genet. 2007;8(Suppl 1):S13.
Pankratz N, Dumitriu A, Hetrick KN, Sun M, Latourelle JC, Wilk JB, Halter C, Doheny KF, Gusella JF, Nichols WC, et al. Copy number variation in familial Parkinson disease. PLoS One. 2011;6:e20988.
Antoni G, Oudot-Mellakh T, Dimitromanolakis A, Germain M, Cohen W, Wells P, Lathrop M, Gagnon F, Morange PE, Tregouet DA. Combined analysis of three genome-wide association studies on vWF and FVIII plasma levels. BMC Med Genet. 2011;12:102.
Cai DC, Fonteijn H, Guadalupe T, Zwiers M, Wittfeld K, Teumer A, Hoogman M, Arias-Vasquez A, Yang Y, Buitelaar J, et al. A genome-wide search for quantitative trait loci affecting the cortical surface area and thickness of Heschl’s gyrus. Genes Brain Behav. 2014;13:675–85.
Manjurano A, Sepulveda N, Nadjm B, Mtove G, Wangai H, Maxwell C, Olomi R, Reyburn H, Drakeley CJ, Riley EM, et al. USP38, FREM3, SDC1, DDC, and LOC727982 gene polymorphisms and differential susceptibility to severe malaria in Tanzania. J Infect Dis. 2015;212:1129–39.
Simino J, Shi G, Bis JC, Chasman DI, Ehret GB, Gu X, Guo X, Hwang SJ, Sijbrands E, Smith AV, et al. Gene-age interactions in blood pressure regulation: a large-scale investigation with the CHARGE, global BPgen, and ICBP Consortia. Am J Hum Genet. 2014;95:24–38.
Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, Stamatoyannopoulos JA. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 2015;12:1184–95.
Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. ENCODE data in the UCSC genome browser: year 5 update. Nucleic Acids Res. 2013;41:D56–63.
Saxena R, Saleheen D, Been LF, Garavito ML, Braun T, Bjonnes A, Young R, Ho WK, Rasheed A, Frossard P, et al. Genome-wide association study identifies a novel locus contributing to type 2 diabetes susceptibility in Sikhs of Punjabi origin from India. Diabetes. 2013;62:1746–55.
Shen N, Yu X, Pan FY, Gao X, Xue B, Li CJ. An early response transcription factor, Egr-1, enhances insulin resistance in type 2 diabetes with chronic hyperinsulinism. J Biol Chem. 2011;286:14508–15.
Tegha-Dunghu J, Neumann B, Reber S, Krause R, Erfle H, Walter T, Held M, Rogers P, Hupfeld K, Ruppert T, et al. EML3 is a nuclear microtubule-binding protein required for the correct alignment of chromosomes in metaphase. J Cell Sci. 2008;121:1718–26.
Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–35.
Cheung L, Zervou S, Mattsson G, Abouna S, Zhou L, Ifandi V, Pelengaris S, Khan M. c-Myc directly induces both impaired insulin secretion and loss of beta-cell mass, independently of hyperglycemia in vivo. Islets. 2010;2:37–45.
Lin L, Pang W, Chen K, Wang F, Gengler J, Sun Y, Tong Q. Adipocyte expression of PU.1 transcription factor causes insulin resistance through upregulation of inflammatory cytokine gene expression and ROS production. Am J Physiol Endocrinol Metab. 2012;302:E1550–9.
Himanen JP, Nikolov DB. Eph receptors and ephrins. Int J Biochem Cell Biol. 2003;35:130–4.
Wang JD, Dong YC, Sheng Z, Ma HH, Li GL, Wang XL, Lu GM, Sugimura H, Jin J, Zhou XJ. Loss of expression of EphB1 protein in gastric carcinoma associated with invasion and metastasis. Oncology. 2007;73:238–45.
Sheng Z, Wang J, Dong Y, Ma H, Zhou H, Sugimura H, Lu G, Zhou X. EphB1 is underexpressed in poorly differentiated colorectal cancers. Pathobiology. 2008;75:274–80.
Hussain I, Powell DJ, Howlett DR, Chapman GA, Gilmour L, Murdock PR, Tew DG, Meek TD, Chapman C, Schneider K, et al. ASP1 (BACE2) cleaves the amyloid precursor protein at the beta-secretase site. Mol Cell Neurosci. 2000;16:609–19.
Basi G, Frigon N, Barbour R, Doan T, Gordon G, McConlogue L, Sinha S, Zeller M. Antagonistic effects of beta-site amyloid precursor protein-cleaving enzymes 1 and 2 on beta-amyloid peptide production in cells. J Biol Chem. 2003;278:31512–20.
Murphy MP, LeVine H 3rd. Alzheimer's disease and the amyloid-beta peptide. J Alzheimers Dis. 2010;19:311–23.
Esterhazy D, Stutzer I, Wang H, Rechsteiner MP, Beauchamp J, Dobeli H, Hilpert H, Matile H, Prummer M, Schmidt A, et al. Bace2 is a beta cell-enriched protease that regulates pancreatic beta cell function and mass. Cell Metab. 2011;14:365–77.
Alcarraz-Vizan G, Castano C, Visa M, Montane J, Servitja JM, Novials A. BACE2 suppression promotes beta-cell survival and function in a model of type 2 diabetes induced by human islet amyloid polypeptide overexpression. Cell Mol Life Sci. 2017;74:2827–38.
Wang DR, Tang D. Hypermethylated SFRP2 gene in fecal DNA is a high potential biomarker for colorectal cancer noninvasive screening. World J Gastroenterol. 2008;14:524–31.
Pehlivan S, Artac M, Sever T, Bozcuk H, Kilincarslan C, Pehlivan M. Gene methylation of SFRP2, P16, DAPK1, HIC1, and MGMT and KRAS mutations in sporadic colorectal cancer. Cancer Genet Cytogenet. 2010;201:128–32.
Crowley RK, O'Reilly MW, Bujalska IJ, Hassan-Smith ZK, Hazlehurst JM, Foucault DR, Stewart PM, Tomlinson JW. SFRP2 is associated with increased adiposity and VEGF expression. PLoS One. 2016;11:e0163777.
Leclerc GM, Leclerc GJ, Kuznetsov JN, DeSalvo J, Barredo JC. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS One. 2013;8:e74420.
Theriault JR, Palmer HJ, Pittman DD. Inhibition of the unfolded protein response by metformin in renal proximal tubular epithelial cells. Biochem Biophys Res Commun. 2011;409:500–5.
Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–12.
Frojdo S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochim Biophys Acta. 2009;1792:83–92.
Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. 2014;2:57.
Lim YC, Li J, Ni Y, Liang Q, Zhang J, Yeo GSH, Lyu J, Jin S, Ding C. A complex association between DNA methylation and gene expression in human placenta at first and third trimesters. PLoS One. 2017;12:e0181155.
Jones PA. The DNA methylation paradox. Trends Genet. 1999;15:34–7.
Benton MC, Johnstone A, Eccles D, Harmon B, Hayes MT, Lea RA, Griffiths L, Hoffman EP, Stubbs RS, Macartney-Coxson D. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 2015;16:8.
Alkhaled Y, Laqqan M, Tierling S, Lo Porto C, Amor H, Hammadeh ME. Impact of cigarette-smoking on sperm DNA methylation and its effect on sperm parameters. Andrologia. 2018;50:e12950.
Cheng Q, Zhao B, Huang Z, Su Y, Chen B, Yang S, Peng X, Ma Q, Yu X, Zhao B, Ke X. Epigenome-wide study for the offspring exposed to maternal HBV infection during pregnancy, a pilot study. Gene. 2018;658:76–85.
Urdinguio RG, Torro MI, Bayon GF, Alvarez-Pitti J, Fernandez AF, Redon P, Fraga MF, Lurbe E. Longitudinal study of DNA methylation during the first 5 years of life. J Transl Med. 2016;14:160.
Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, Knight R, Ley RE. Conducting a microbiome study. Cell. 2014;158:250–62.
Sambol NC, Chiang J, O'Conner M, Liu CY, Lin ET, Goodman AM, Benet LZ, Karam JH. Pharmacokinetics and pharmacodynamics of metformin in healthy subjects and patients with noninsulin-dependent diabetes mellitus. J Clin Pharmacol. 1996;36:1012–21.
Tokubuchi I, Tajiri Y, Iwata S, Hara K, Wada N, Hashinaga T, Nakayama H, Mifune H, Yamada K. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS One. 2017;12:e0171293.
Rovite V, Wolff-Sagi Y, Zaharenko L, Nikitina-Zake L, Grens E, Klovins J. Genome Database of the Latvian Population (LGDB): design, goals, and primary results. J Epidemiol. 2018;28:353–60.
Ignatovica V, Latkovskis G, Peculis R, Megnis K, Schioth HB, Vaivade I, Fridmanis D, Pirags V, Erglis A, Klovins J. Single nucleotide polymorphisms of the purinergic 1 receptor are not associated with myocardial infarction in a Latvian population. Mol Biol Rep. 2012;39:1917–25.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Xu Z, Niu L, Li L, Taylor JA. ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016;44:e20.
Niu L, Xu Z, Taylor JA. RCP: a novel probe design bias correction method for Illumina Methylation BeadChip. Bioinformatics. 2016;32:2659–63.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–6.
Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, R VL, Clark SJ, Molloy PL. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6.
Zerbino DR, Johnson N, Juetteman T, Sheppard D, Wilder SP, Lavidas I, Nuhn M, Perry E, Raffaillac-Desfosses Q, Sobral D, et al. Ensembl regulation resources. Database (Oxford). 2016;2016:bav119.
Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–30.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–3.
Olivier BG, Rohwer JM, Hofmeyr JH. Modelling cellular processes with Python and Scipy. Mol Biol Rep. 2002;29:249–54.
Acknowledgements
We acknowledge the Genome Database of Latvian Population (LGDB) and Latvian Biomedical Research and Study Centre for organization of participants’ recruitment and bio-sample processing and data storage. The authors thank all the volunteers who participated in the clinical trial.
Funding
The work was supported by the European Regional Development Fund under the project “Investigation of interplay between multiple determinants influencing response to metformin: search for reliable predictors for efficacy of type 2 diabetes therapy” (Project Nr.: 1.1.1.1/16/A/091).
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
IKalnina, LZ, DF, HBS, and JK designed the research; IKonrade and VP oversaw patient recruitment; IE, IKalnina, DG, IRS, conducted the research; IE, IS, MU, and RP performed the analyses; IE, IS, MU wrote the manuscript; IK, DMC, CZ, HBS, and JK oversaw the research and reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Study has been carried out in accordance with the Declaration of Helsinki and approved by Central Medical Ethics Committee (1/16-05-12) and The State Agency of Medicines of the Republic of Latvia (17-1723), clinical trial registration number: 2016-001092-74 (www.clinicaltrialsregister.eu).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Full results representing all CpGs within the analyzed contrasts with significantly changed methylation levels, identified after correction for multiple testing using the Benjamini-Hochberg method. (XLSX 21 kb)
Additional file 2:
List of inclusion/exclusion criteria. (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Elbere, I., Silamikelis, I., Ustinova, M. et al. Significantly altered peripheral blood cell DNA methylation profile as a result of immediate effect of metformin use in healthy individuals. Clin Epigenet 10, 156 (2018). https://doi.org/10.1186/s13148-018-0593-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0593-x