
Abstract
Background
The high prevalence of obesity among US adults has resulted in significant increases in associated metabolic disorders such as diabetes, dyslipidemia, and high blood pressure. Together, these disorders constitute metabolic syndrome, a clinically defined condition highly prevalent among African-Americans. Identifying epigenetic alterations associated with metabolic syndrome may provide additional information regarding etiology beyond current evidence from genome-wide association studies.
Methods
Data on metabolic syndrome and DNA methylation was assessed on 614 African-Americans from the Hypertension Genetic Epidemiology Network (HyperGEN) study. Metabolic syndrome was defined using the joint harmonized criteria, and DNA methylation was assessed using the Illumina HumanMethylation450K Bead Chip assay on DNA extracted from buffy coat. Linear mixed effects regression models were used to examine the association between CpG methylation at > 450,000 CpG sites and metabolic syndrome adjusted for study covariates. Replication using DNA from a separate sample of 69 African-Americans, as well as meta-analysis combining both cohorts, was conducted.
Results
Two differentially methylated CpG sites in the IGF2BP1 gene on chromosome 17 (cg06638433; p value = 3.10 × 10− 7) and the ABCG1 gene on chromosome 21 (cg06500161; p value = 2.60 × 10− 8) were identified. Results for the ABCG1 gene remained statistically significant in the replication dataset and meta-analysis.
Conclusion
Metabolic syndrome was consistently associated with increased methylation in the ABCG1 gene in the discovery and replication datasets, a gene that encodes a protein in the ATP-binding cassette transporter family and is involved in intra- and extra-cellular signaling and lipid transport.
Similar content being viewed by others
Background
The prevalence of obesity among US adults has increased steadily over the past few decades, and currently over two thirds of US adults are either obese or overweight [1]. Excess adipose tissue is linked with chronic inflammation, immune system activation, and oxidative stress, as well as hypertension, dyslipidemia, and insulin resistance. This cluster of inter-related conditions is defined clinically as metabolic syndrome (MetS), and the prevalence of this condition has increased concurrently with obesity rates in the USA [2]. The link between MetS and increased risk and mortality due to many chronic diseases such as cardiovascular disease (CVD), arthritis, chronic kidney disease, schizophrenia, and cancer [3,4,5,6,7,8,9,10,11,12] has been reported in published studies. More recently, there has been renewed interest in better understanding the biological mechanisms underlying the pathogenesis of MetS.
Epigenetic studies provide a unique lens for clarifying molecular pathways through which lifestyle and environmental factors such as diet and physical activity coupled with individual genetic background interact to influence health outcomes. Alterations in epigenetic patterns, such as DNA methylation, which are modifiable, provide a potential mechanism through which MetS and other environmental factors may influence gene expression and ultimately increasing disease risk or mortality. Like other chronic diseases, marked racial differences exist in the prevalence of MetS and its components [2], and the biological mechanisms underlying such disparities remain unclear. Studies of DNA methylation alterations provide an opportunity to systematically interrogate the epigenome to identify population-specific variation associated with disease risk and health outcomes.
The goal of the present study is to examine DNA methylation signatures measured across the genome associated with MetS using DNA from African-American adults included in the Hypertension Genetic Epidemiology Network (HyperGEN) study, with replication in an independent cohort of participants in the REasons for Geographic and Racial Differences in Stroke (REGARDS) study. Results will enhance understanding of the pathophysiology of this condition among African-Americans and potentially lead to the development of novel biomarkers or molecular targets for clinical studies.
Methods
Study population and baseline variables
The Hypertension Genetic Epidemiology Network (HyperGEN) study is a cross-sectional, population-based study of 1224 hypertensive African-American sibships initially recruited between 1996 and 1999. The study was later extended to other siblings and offspring of the original sib-pair. Using stored samples, an ancillary epigenetic study was conducted among cohort participants belonging to the highest and lowest quartiles (total N = 636) of left ventricular mass (LV mass). The family structure of this HyperGEN epigenetic study population, including the number of individuals per family, parent-child, and sibling pairs, is presented in Additional file 1: Table S1. Detailed lifestyle, biometric, and laboratory measures were obtained in HyperGEN, including blood pressure, medications, fasting blood glucose, triglycerides, HDL cholesterol, height, weight, and waist circumference. BMI was calculated using standard measures.
Metabolic syndrome
This was defined using the recently published joint harmonized criteria [13]. The criteria defined MetS as having three or more of the following components: (1) elevated waist circumference (≥ 88 cm for women and ≥ 102 cm for men), (2) elevated triglycerides (≥ 150 mg/dL) or on treatment for dyslipidemia (statin and/or fibric acid derivative), (3) reduced high-density lipoprotein (HDL) cholesterol (< 40 mg/dL in men and < 50 mg/dL in women) or on treatment for dyslipidemia (statin and/or fibric acid derivative), (4) elevated blood pressure (systolic ≥ 130 and/or diastolic ≥ 85 mmHg) or antihypertensive drug treatment in a patient with a history of hypertension), and (5) elevated fasting glucose (≥ 100 mg/dL) or drug treatment for elevated glucose. Of the 636 adults evaluated from the HyperGen cohort, 614 had methylation and MetS data for the basic model; an additional 8 had missing covariate data, leaving 606 for the fully adjusted model.
Epigenomic analysis
The Illumina HumanMethylation450 array was used to analyze DNA extracted from buffy coat. These arrays can measure DNA methylation at > 480,000 cytosine-phosphate-guanine (CpG) sites. Briefly, 500 ng of buffy coat DNA was hybridized to the Methyl450 array after bisulfite conversion with EZ DNA kits (Zymo Research, Irvine, CA) prior to standard Illumina amplification, hybridization, and imaging steps. The resulting intensity files were analyzed with Illumina’s GenomeStudio, which generated beta (β) scores (i.e., the proportion of total signal from the methylation-specific probe or color channel) and “detection p values” (probability that the total intensity for a given probe falls within the background signal intensity). Quality control (QC) measures were conducted by removing samples having more than 1% of CpG sites with a detection p value > 0.05, removing CpG sites having more than 5% of samples with a detection p value > 0.01, and CpG sites with detection p value > 0.01 set as missing. After these QC filters, 484,366 CpG sites were eligible for analysis. We normalized the β scores using the Subset-quantile Within Array Normalization (SWAN) method in minifi package to correct for differences between batches and the type I and type II assay designs within a single 450K array [14]. Cell count proportions (CD8 T lymphocytes, CD4 T lymphocytes, natural killer cells, B cells, and monocytes) were created using the algorithm developed by Houseman, which predicts underlying cellular composition of each sample from DNA methylation patterns [15].
Available SNP data
GWAS data from HyperGEN has been previously described [16]. African-Americans were genotyped with the Affymetrix Genome Wide Human SNP Array 6.0 Array for 872,242 SNPs (median call rate = 99.97%). This SNP set was used for imputation with ~ 3.01 million HapMap SNPs (release 22, phasing data, “revised union” of CEU and YRI populations) using MACH [17, 18]. The GWAS data described was used to investigate cis-methylation quantitative trait loci (cis-meQTL) in the region of the statistically significant CpG association findings. HyperGEN-imputed GWAS data was also used to create principal components for ancestry included in epigenetic association models to adjust for subpopulation structure [19].
Statistical analysis
We determined the proportion of study participants meeting the criteria for MetS and used chi-square tests and t tests to examine differences in baseline socio-demographic variables. We fit linear mixed effects regression models with the CpG β score (approximating % DNA methylation at a CpG site) as the outcome, MetS as the predictor, and adjusted for age, sex, center, first four principal components (PCs) for ancestry (ancestry PCs), estimated cell proportions, batch effects (array, row, and column to account for residual batch effects after normalization), and family structure as a random effect (R kinship package (lmekin function)) [20]. In the fully adjusted model, we additionally adjusted for smoking and alcohol use. A sensitivity model for our statistically significant findings was further adjusted for high versus low left ventricular mass. We corrected for multiple comparisons using a Bonferroni correction for 0.05/460,000 tests setting alpha to 1.1 × 10− 7. Given that Bonferroni correction tends to be conservative, we also used FDR correction for multiple testing. We constructed Manhattan plots to present results of our epigenome-wide association analysis of MetS and used mixed models to compare methylation of the top CpG sites by MetS and component status adjusting for family structure (R kinship package (lmekin function)) [20]. To evaluate the DNA sequence contributions to methylation at the top CpG loci, we conducted cis-meQTL analyses by interrogating associations between all SNPs within 20 kb of the CpG site via linear mixed models with the CpG β score as the outcome, adjusted for age, sex, study site, ancestry PCs, blood cell counts, and familial relationship. For the cis-meQTL analysis, we used the Bonferroni correction for multiple testing as 0.05/the number of SNPs within a 20-kb window. Subsequently, significant SNPs from cis-meQTL analysis were tested for association with MetS via mixed linear models with MetS as the outcome, adjusted for age, sex, study site, ancestry PCs, and familial relationship. We also conducted lookups of the significant MetS SNPs in the catalog of genome-wide association studies (www.gwascentral.org) to evaluate evidence for association with cardiometabolic traits in other populations.
Replication analysis
To assess whether similar findings are replicated in a separate study population, we analyzed DNA methylation data obtained from genomic DNA samples of 100 African-American participants in the REasons for Geographic and Racial Differences in Stroke (REGARDS) study also assayed on the Illumina HumanMethylation450 array. In the REGARDS study, blood pressure, fasting blood glucose, triglycerides, HDL cholesterol, and waist circumference were obtained by trained health professionals during in-home visits at study baseline. Details of the REGARDS study have been published previously [21]. In brief, REGARDS is a nationally representative sample of 32,239 adults ages 45 years and older (45% males and 41% African-American) recruited between 2003 and 2007 and followed for incident stroke and other related cardiovascular outcomes. MetS was defined in REGARDS following the joint harmonized criteria, and epigenomic analysis was conducted using data from 69 participants with complete information for both methylation and MetS. Linear models were used to test the association between CpG site methylation and MetS, adjusting for age, sex, four principal components for ancestry, and cell count proportions in the basic model and adding alcohol and smoking in a fully adjusted model. Statistically significant CpGs from the HyperGEN discovery analyses were tested for replication in REGARDS, and a Bonferroni correction was applied (of 0.05/2 significant CpGs = 2.5 × 10− 2).
Meta-analysis
Top results from the HyperGEN and REGARDS studies were meta-analyzed using random effects models implemented in the METASOFT program based on the method of Han and Eskin [22] to ensure validity in the presence of effect heterogeneity.
Results
Among 614 African-American adults in the HyperGen study (67% female), about half (51%) met the criteria for MetS (Table 1). MetS+ participants were older on average compared with MetS− participants, 52 ± 10 years and 46 ± 11 years, respectively. Body weight (98.5 vs. 84.3 kg), waist circumference (112.4 vs. 96.6 cm), and BMI (35.4 vs. 29.8) were significantly higher in MetS+ compared with MetS− participants. In addition, MetS+ participants had higher levels of triglycerides (125.5 vs. 77 mg/dL) and LDL cholesterol (128 vs. 118.5 mg/dL), lower levels of HDL cholesterol (46.9 vs. 58.8 mg/dL), and higher prevalence of hypertension (87 vs. 49%) compared with MetS− participants. Among MetS+ participants, 51% had three of the five components, 28% had four, and 20% had five, and among MetS− participants, 17% had none of the MetS components, 34% had one, and 49% had two.
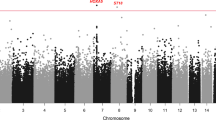
Two differentially methylated CpG sites—one in the IGF2BP1 gene on chromosome 17 (cg06638433) and the other in the ABCG1 gene on chromosome 21 (cg06500161)—were identified in the HyperGEN study in the basic (Fig. 1) and fully adjusted (Additional file 1: Figure S1) models. In the basic model adjusting for age, sex, cell count proportions, ancestry, and family relationship, MetS was associated with increased methylation at cg06500161 (β = 0.017, p value = 1.08 × 10− 8) and cg06638433 (β = 0.01, p value = 2.05 × 10− 8) (Table 2). After additionally adjusting for alcohol use and smoking, the associations remained consistent and statistically significant for cg06500161 (β = 0.02, p value = 2.60 × 10− 8) and closely approached statistical significance for cg06638433 (β = 0.01, p value = 3.10 × 10− 7). The results were consistent, but no longer statistically significant, in a sensitivity model additionally adjusted for left ventricular mass (cg06500161: β = 0.02, p value = 2.22 × 10− 7; cg06638433: β = 0.01, p value = 2.19 × 107). The LVM adjusted results were similar after the FDR correction (adjusted p value = 0.054 for both cg06500161 and cg06638433). Mean DNA methylation level at each of the two CpG sites was significantly higher among HyperGEN participants meeting the criteria for an individual MetS component as well as overall for MetS status (Additional file 1: Figure S2).

Epigenome-wide association for metabolic syndrome among African-Americans in HyperGEN study (N = 614). Basic model includes age, sex, center, first four principal components, five estimated cell proportions, batch effect, and family structure
Both cg06500161 and cg06638433 were examined for association with MetS in the REGARDS cohort. REGARDS participants with relevant data were overall 59.5 ± 4.5 years of age and 47.8% female (see Additional file 1: Table S2 for a description of the replication cohort by MetS status). Similar to HyperGEN, we observed increased DNA methylation levels at cg06500161 in both the basic (β = 0.029, p value = 0.001) and fully adjusted (β = 0.032, p value 4.7 × 10− 4) models in REGARDS. In meta-analysis of the combined HyperGEN and REGARDS cohorts, the association for cg06500161 remained consistent (meta-analysis for fully adjusted model: p value = 2.15 × 10− 10; Table 2). IGF2BP1 cg06638433 was not associated with MetS in REGARDS, and the effect was in the opposite direction (Table 2).
In the cis-meQTL analysis, 16 SNPs were significantly associated with methylation at the cg06500161 locus upon Bonferroni correction for multiple testing (3.2 × 10− 4 = 0.05/154 SNPs within 20 kb of cg06500161), reflecting potential contributions to the methylation signal from underlying common DNA sequence variants (Fig. 2). Among those 16 SNPs, 7 were in turn associated with MetS (p < 0.05) with the top SNP association signals being for rs17114493 and rs4547619 (Table 3). None of the 7 SNPs had previously been identified in GWAS of MetS or other metabolic traits. Those SNPs were associated with decreased methylation at cg06500161 yet increased MetS risk. In our primary analysis, MetS was associated with increased methylation at cg06500161. These results suggest these SNPs are not associated with MetS through this methylation site. Data from ENCODE shows that cg06500161 is a downstream of a predicted promoter region (Fig. 3).

Associations between 154 ABCG1 genetic variants within 20 kb (upstream and downstream) of the cg06500161 chromosome 21 locus and its methylation status (cis-meQTL). SNPs above the dashed line have p value for association > 0.05/154

ENCODE plot with MetS signal of cg06500161. Genome segmentations from ENCODE color coding: bright red, predicted promoter region including transcription start site (TSS); light red, predicted promoter flanking region; orange, predicted enhancer; yellow, predicted weak enhancer or open chromatin cis-regulatory element; blue, CTCF (CCCTC-binding factor)-enriched element; dark green, predicted transcribed region; and gray, predicted low activity region
Discussion
In a sample of African-American adults with a 51% prevalence of MetS, we observed that MetS was associated with increased methylation on the cg06500161 locus in the ABCG1 gene. This finding was successfully replicated in an independent sample of African-American adults in the REGARDS cohort, and meta-analysis of data from the two studies strengthened the association of the CpG with MetS. Methylation at cg06500161 (within a CpG island shore) [23] was associated with each MetS component suggesting this locus has pleiotropic effects. Evaluation of the region in ENCODE revealed the CpG is in a strong regulatory region which suggests that differential methylation at this locus could impact gene expression. An analysis of common SNPs in the region did not confirm SNPs influence on the relationship between the CpG and MetS.
This study is one of the firsts to report a potential epigenetic mechanism associated with a highly prevalent condition among African-American adults. Other studies have identified loci associated with MetS traits [24, 25], including central obesity, insulin responsiveness, and type 2 diabetes [26, 27], as well as lipid profiles in diverse populations [28, 29]. Das et al. identified decreased methylation at two loci of the CPT1A gene, involved in fatty acid oxidation, associated with MetS status in a Caucasian population, which corroborates findings by a prior study observing differential methylation at the same loci associated with triglycerides and very low-density lipoprotein cholesterol levels [25]. Methylation of the cg18181703 locus in the SOCS3 gene, involved in leptin and insulin signaling, was also identified in a separate study, inversely correlated with BMI, triglycerides, and MetS but positively correlated with HDL-c levels [24]. Relevant to our findings, Yoon et al. independently identified ABCG1 (cg06500161) as being associated with MetS (in a Caucasian Veteran population) using an Iterative Sure Independence Screening analysis approach [30]. ABCG1 cg06500161 has also been reported to be associated with fasting insulin (in Caucasian non-diabetic adults) [26], blood lipids, and adiposity traits [29, 31]. Additionally, our results are consistent with a recent study of epigenetic determinants of type 2 diabetes in Mexican-Americans, showing differential DNA methylation levels at ABCG1 [27]. Taken together, these studies suggest that epigenetic variations are strongly associated with MetS and its components and that ABCG1 has been a consistent finding across diverse ethnic populations, strengthening the credibility of this gene’s relationship with metabolic traits.
ABCG1 encodes for a protein included in the ATP-binding cassette (ABC) transporters superfamily, involved in the transport of molecules across extra- and intra-cellular membranes. ABCG1 in particular plays an important role in macrophage cholesterol and phospholipid transport, and the regulation of cellular lipid homeostasis. Downregulation of ABCG1 and ABCA1 has been associated with reduced cholesterol efflux and has been shown to increase the risk of atherosclerosis [32]. Other studies have reported an association between ABCG1 methylation and coronary artery disease [33, 34], and several studies report an association between liver X receptor, an inducer of ABC transporter gene expression, and cholesterol efflux which results in the inhibition of cell proliferation and stimulation of apoptosis in cancer [35, 36]. This evidence, taken together with the observation that alterations in the ABCG1 gene were observed in 40% of invasive breast cancer tumors in the Cancer Genome Atlas [37, 38], provides a potential epigenetic link between MetS and chronic diseases including cancer that deserves further study. The higher methylation levels of ABCG1 observed with MetS provide evidence for potential therapeutic targets that may be useful in drug development, and/or prevention of chronic diseases including CVD and cancer.
There are several limitations inherent in this study. First, HyperGen was a cross-sectional study of African-American adults, and this study design limits our ability to infer causality between MetS and DNA methylation. Prospective studies will be needed to definitively determine whether increased methylation of ABCG1 is a consequence or cause of MetS. At least one Mendelian randomization study in Caucasian populations reported that lipids change methylation at cg06500161 suggesting the potential for a feedback loop mechanism that warrants further investigation [39]. In fact, if MetS affects methylation at ABCG1, this could help explain why our results did not suggest cis-meQTL SNPs influence the CpG-MetS relationship. Although we were able to identify significantly altered methylation on an ABCG1 locus in relation to MetS among African-Americans that is consistent with epigenetic studies of metabolic traits in other ethnicities, improved understanding of racial differences in the biological mechanisms leading to significantly higher prevalence of MetS and components such as type 2 diabetes in African-Americans relative to other racial groups will require a larger study with adequate representation of multiple racial groups. DNA methylation was measured on stored and frozen buffy coat samples, which may have been altered during the freezing and thawing process; however, buffy coat samples are widely used for methylation analysis due to the high concentration of DNA. A major strength of the study is the use of epigenome-wide methylation analysis using a wide panel of CpG markers on understudied African-Americans and replication in a separate independent sample of African-American participants.
Conclusion
In conclusion, MetS in African-American adults was associated with increased methylation at the cg06500161 locus in the ABCG1 gene, located in a highly conserved regulatory region of the genome. ABCG1, involved in cholesterol and phospholipid transport, has been associated with MetS and component traits in other studies. The high prevalence of MetS among African-Americans and the consistent associations between MetS and many chronic diseases such as cancer highlight the need for future prospective confirmatory studies that may inform clinical strategies and interventions.
References
Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD, Ogden CL. Trends in obesity among adults in the United States, 2005 to 2014. JAMA. 2016;315(21):2284–91.
Moore JX, Chaudhary N, Akinyemiju T. Metabolic syndrome prevalence by race/ethnicity and sex in the United States, National Health and Nutrition Examination Survey, 1988-2012. Prev Chronic Dis. 2017;14:E24.
Bjorge T, Lukanova A, Jonsson H, Tretli S, Ulmer H, Manjer J, Stocks T, Selmer R, Nagel G, Almquist M, et al. Metabolic syndrome and breast cancer in the me-can (metabolic syndrome and cancer) project. Cancer Epidemiol Biomarkers Prev. 2010;19(7):1737–45.
Borena W, Strohmaier S, Lukanova A, Bjorge T, Lindkvist B, Hallmans G, Edlinger M, Stocks T, Nagel G, Manjer J, et al. Metabolic risk factors and primary liver cancer in a prospective study of 578,700 adults. Int J Cancer. 2012;131(1):193–200.
Borena W, Edlinger M, Bjorge T, Haggstrom C, Lindkvist B, Nagel G, Engeland A, Stocks T, Strohmaier S, Manjer J, et al. A prospective study on metabolic risk factors and gallbladder cancer in the metabolic syndrome and cancer (Me-Can) collaborative study. PLoS One. 2014;9(2):e89368.
Stocks T, Bjorge T, Ulmer H, Manjer J, Haggstrom C, Nagel G, Engeland A, Johansen D, Hallmans G, Selmer R, et al. Metabolic risk score and cancer risk: pooled analysis of seven cohorts. Int J Epidemiol. 2015;44(4):1353–63.
Ulmer H, Bjorge T, Concin H, Lukanova A, Manjer J, Hallmans G, Borena W, Haggstrom C, Engeland A, Almquist M, et al. Metabolic risk factors and cervical cancer in the metabolic syndrome and cancer project (Me-Can). Gynecol Oncol. 2012;125(2):330–5.
Almquist M, Johansen D, Bjorge T, Ulmer H, Lindkvist B, Stocks T, Hallmans G, Engeland A, Rapp K, Jonsson H, et al. Metabolic factors and risk of thyroid cancer in the metabolic syndrome and cancer project (Me-Can). Cancer Causes Control. 2011;22(5):743–51.
Johansen D, Stocks T, Jonsson H, Lindkvist B, Bjorge T, Concin H, Almquist M, Haggstrom C, Engeland A, Ulmer H, et al. Metabolic factors and the risk of pancreatic cancer: a prospective analysis of almost 580,000 men and women in the metabolic syndrome and cancer project. Cancer Epidemiol Biomarkers Prev. 2010;19(9):2307–17.
Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, Rinfret S, Schiffrin EL, Eisenberg MJ. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010;56(14):1113–32.
Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28(7):1769–78.
Wu SH, Liu Z, Ho SC. Metabolic syndrome and all-cause mortality: a meta-analysis of prospective cohort studies. Eur J Epidemiol. 2010;25(6):375–84.
Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC Jr, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–5.
Maksimovic J, Gordon L, Oshlack A. SWAN: subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13(6):R44.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Arnett DK, Meyers KJ, Devereux RB, Tiwari HK, Gu CC, Vaughan LK, Perry RT, Patki A, Claas SA, Sun YV, et al. Genetic variation in NCAM1 contributes to left ventricular wall thickness in hypertensive families. Circ Res. 2011;108(3):279–83.
de Las Fuentes L, Sung YJ, Schwander KL, Kalathiveetil S, Hunt SC, Arnett DK, Rao DC. The role of SNP-loop diuretic interactions in hypertension across ethnic groups in HyperGEN. Front Genet. 2013;4:304.
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–9.
Price AL, Weale ME, Patterson N, Myers SR, Need AC, Shianna KV, Ge D, Rotter JI, Torres E, Taylor KD, et al. Long-range LD can confound genome scans in admixed populations. Am J Hum Genet. 2008;83(1):132–5. author reply 135-139
Lourenco VM, Pires AM, Kirst M. Robust linear regression methods in association studies. Bioinformatics. 2011;27(6):815–21.
Howard VJ, Cushman M, Pulley L, Gomez CR, Go RC, Prineas RJ, Graham A, Moy CS, Howard G. The reasons for geographic and racial differences in stroke study: objectives and design. Neuroepidemiology. 2005;25(3):135–43.
Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011;88(5):586–98.
Aronica L, Levine AJ, Brennan K, Mi J, Gardner C, Haile RW, Hitchins MP. A systematic review of studies of DNA methylation in the context of a weight loss intervention. Epigenomics. 2017;9(5):769–87.
Ali O, Cerjak D, Kent JW Jr, James R, Blangero J, Carless MA, Zhang Y. Methylation of SOCS3 is inversely associated with metabolic syndrome in an epigenome-wide association study of obesity. Epigenetics. 2016;11(9):699–707.
Das M, Sha J, Hidalgo B, Aslibekyan S, Do AN, Zhi D, Sun D, Zhang T, Li S, Chen W, et al. Association of DNA methylation at CPT1A locus with metabolic syndrome in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study. PLoS One. 2016;11(1):e0145789.
Hidalgo B, Irvin MR, Sha J, Zhi D, Aslibekyan S, Absher D, Tiwari HK, Kabagambe EK, Ordovas JM, Arnett DK. Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network study. Diabetes. 2014;63(2):801–7.
Kulkarni H, Kos MZ, Neary J, Dyer TD, Kent JW Jr, Goring HH, Cole SA, Comuzzie AG, Almasy L, Mahaney MC, et al. Novel epigenetic determinants of type 2 diabetes in Mexican-American families. Hum Mol Genet. 2015;24(18):5330–44.
Frazier-Wood AC, Aslibekyan S, Absher DM, Hopkins PN, Sha J, Tsai MY, Tiwari HK, Waite LL, Zhi D, Arnett DK. Methylation at CPT1A locus is associated with lipoprotein subfraction profiles. J Lipid Res. 2014;55(7):1324–30.
Hedman AK, Mendelson MM, Marioni RE, Gustafsson S, Joehanes R, Irvin MR, Zhi D, Sandling JK, Yao C, Liu C, et al. Epigenetic patterns in blood associated with lipid traits predict incident coronary heart disease events and are enriched for results from genome-wide association studies. Circ Cardiovasc Genet. 2017;10(1). https://doi.org/10.1161/CIRCGENETICS.116.001487.
Yoon G, Zheng Y, Zhang Z, Zhang H, Gao T, Joyce B, Zhang W, Guan W, Baccarelli AA, Jiang W, et al. Ultra-high dimensional variable selection with application to normative aging study: DNA methylation and metabolic syndrome. BMC Bioinformatics. 2017;18(1):156.
Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou YH, Hedman AK, Sandling JK, Li LA, Irvin MR, et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet. 2015;24(15):4464–79.
van Eck M, Bos IS, Kaminski WE, Orso E, Rothe G, Twisk J, Bottcher A, Van Amersfoort ES, Christiansen-Weber TA, Fung-Leung WP, et al. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc Natl Acad Sci U S A. 2002;99(9):6298–303.
Sayols-Baixeras S, Subirana I, Lluis-Ganella C, Civeira F, Roquer J, Do AN, Absher D, Cenarro A, Munoz D, Soriano-Tarraga C, et al. Identification and validation of seven new loci showing differential DNA methylation related to serum lipid profile: an epigenome-wide approach. The REGICOR study. Hum Mol Genet. 2016;25:4556.
Guay SP, Brisson D, Munger J, Lamarche B, Gaudet D, Bouchard L. ABCA1 gene promoter DNA methylation is associated with HDL particle profile and coronary artery disease in familial hypercholesterolemia. Epigenetics. 2012;7(5):464–72.
El Roz A, Bard JM, Huvelin JM, Nazih H. LXR agonists and ABCG1-dependent cholesterol efflux in MCF-7 breast cancer cells: relation to proliferation and apoptosis. Anticancer Res. 2012;32(7):3007–13.
Trasino SE, Kim YS, Wang TT. Ligand, receptor, and cell type-dependent regulation of ABCA1 and ABCG1 mRNA in prostate cancer epithelial cells. Mol Cancer Ther. 2009;8(7):1934–45.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Dekkers KF, van Iterson M, Slieker RC, Moed MH, Bonder MJ, van Galen M, Mei H, Zhernakova DV, van den Berg LH, Deelen J, et al. Blood lipids influence DNA methylation in circulating cells. Genome Biol. 2016;17(1):138.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: TA, SA, MRI. Performed the experiments: TA, AD, SA, MRI. Analyzed the data: TA, AD, SA, MRI, HKT, DZ, AP, DA, XG, DA. Wrote the paper: TA, SA, MRI. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. HyperGEN family structure. Table S2. Baseline characteristics of REGARDS study participants. Figure S1. Epigenome-wide association for metabolic syndrome among African-Americans in HyperGEN study (N = 606). Figure S2. Differences in mean DNA methylation (%) of (A) cg06500161 in ABCG1 and of (B) cg06638433 in IGF2BP1 gene by metabolic syndrome and components. (PDF 1194 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Akinyemiju, T., Do, A.N., Patki, A. et al. Epigenome-wide association study of metabolic syndrome in African-American adults. Clin Epigenet 10, 49 (2018). https://doi.org/10.1186/s13148-018-0483-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0483-2