
Abstract
Background
Smoking has been reported to be associated with peripheral blood DNA methylation, but the causal aspects of the association have rarely been investigated. We aimed to investigate the association and underlying causation between smoking and blood methylation.
Methods
The methylation profile of DNA from the peripheral blood, collected as dried blood spots stored on Guthrie cards, was measured for 479 Australian women including 66 monozygotic twin pairs, 66 dizygotic twin pairs, and 215 sisters of twins from 130 twin families using the Infinium HumanMethylation450K BeadChip array. Linear regression was used to estimate associations between methylation at ~ 410,000 cytosine-guanine dinucleotides (CpGs) and smoking status. A regression-based methodology for twins, Inference about Causation through Examination of Familial Confounding (ICE FALCON), was used to assess putative causation.
Results
At a 5% false discovery rate, 39 CpGs located at 27 loci, including previously reported AHRR, F2RL3, 2q37.1 and 6p21.33, were found to be differentially methylated across never, former and current smokers. For all 39 CpG sites, current smokers had the lowest methylation level. Our study provides the first replication for two previously reported CpG sites, cg06226150 (SLC2A4RG) and cg21733098 (12q24.32). From the ICE FALCON analysis with smoking status as the predictor and methylation score as the outcome, a woman’s methylation score was associated with her co-twin’s smoking status, and the association attenuated towards the null conditioning on her own smoking status, consistent with smoking status causing changes in methylation. To the contrary, using methylation score as the predictor and smoking status as the outcome, a woman’s smoking status was not associated with her co-twin’s methylation score, consistent with changes in methylation not causing smoking status.
Conclusions
For middle-aged women, peripheral blood DNA methylation at several genomic locations is associated with smoking. Our study suggests that smoking has a causal effect on peripheral blood DNA methylation, but not vice versa.
Similar content being viewed by others

Background
Epigenetics is a mechanism modifying gene expression without changing underlying DNA sequence. DNA methylation, a phenomenon that typically a methyl group (-CH3) is added to a cytosine-guanine dinucleotide (CpG) at which the cytosine is converted to a 5-methylcytosine, has been proposed to play a role in the aetiology of complex traits and diseases [1, 2].
At least 21 epigenome-wide association studies (EWASs) have reported that methylation in the blood of adults at a great many CpGs is associated with smoking status [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23]. A recent, and the largest meta-analysis so far, reported 18,760 CpGs annotated to 7201 genes, which account for approximately one third of the known human genes, were differentially methylated between 2433 current smokers and 6956 never smokers [11]. Associations for several loci, such as AHRR, F2RL3, GPR15, GFI1, 2q37.1 and 6p21.33, have been consistently reported, and a systematic review published in 2015 found that associations for 62 CpGs had been reported at least three times [24]. Apart from smoking status, other smoking exposures such as cumulative smoking [3, 4, 8,9,10,11,12, 16,17,18, 20, 22] and years since quitting [4, 9,10,11,12, 15, 16, 19, 20, 22] have also been found to be associated with blood DNA methylation.
Most of the reported associations are from cross-sectional designs; thus, the causal nature of the association, i.e. whether DNA methylation has a causal effect on smoking or vice versa, is unknown. There is also a possibility that cross-sectional epigenetic associations are due to familial confounding [25]. Studies have suggested that smoking-related blood DNA methylation mediates the effects of smoking on lung cancer [26, 27], death [28], leukocyte telomere length [29], and subclinical atherosclerosis [30]. These studies assume that smoking has a causal effect on methylation without evidence of causality. To the best of our knowledge, the only causal evidence comes from a study using a two-step Mendelian randomisation (MR) approach to investigate the mediating role of methylation between smoking and inflammation [31]. This study found that smoking had a causal effect on methylation at CpGs located at F2RL3 and GPR15 genes.
In this study, we aimed to investigate association between smoking and blood DNA methylation, to replicate associations previously reported and to investigate putative causal nature of the association using regression methods for related individuals.
Methods
Study sample
The sample comprised women from the Australian Mammographic Density Twins and Sisters Study [32]. A total of 479 women including 66 monozygotic twin pairs, 66 dizygotic twin pairs and 215 sisters from 130 families were selected [33].
Smoking data collection
A telephone-administered questionnaire was used to collect participants’ self-reported information on smoking. Participants were asked the question ‘Have you ever smoked at least one cigarette per day for 3 months or longer?’ Participants who answered ‘No’ were classified as never smokers, and the rest ever smokers. Ever smokers were further questioned for age at starting smoking, the average number of cigarettes smoked per day, and age at stopping smoking, if any. Ever smokers who had stopped smoking before the interview were classified as former smokers, and the rest current smokers.
DNA methylation data
DNA was extracted from dried blood spots stored on Guthrie cards using a method previously described [34]. Methylation was measured using the Infinium HumanMethylation450K BeadChip array. Raw intensity data were processed by Bioconductor minfi package [35], which included normalisation of data using Illumina’s reference factor-based normalisation methods (preprocessIllumina) and subset-quantile within array normalisation (preprocessSWAN) [36] for type I and II probe bias correction. An empirical Bayes batch-effects removal method ComBat [37] was applied to minimise technical variation across batches. Probes with missing values (detection P value> 0.01) in one or more samples, with documented SNPs at the target CpG, with beadcount < 3 in more than 5% samples, binding to multiple locations [38] or binding to X chromosome, and the 65 control probes were excluded, leaving 411,219 probes included in the analysis; see Li et al. [33] for more details.
Epigenome-wide association analysis
We investigated the association using a linear mixed-effects model in which the methylation M value, a logit transformation of the percentage of methylation, as the outcome and smoking status (never, former and current smokers) as the predictor. The model was adjusted for age and estimated cell-type proportions [39] as fixed effects and for family and zygosity as random effects, fitted using the lmer() function from the R package lme4 [40]. The likelihood ratio test was used to make inference, that is, a nested model without smoking status was fitted and a P value was calculated based on that, twice the difference in the log likelihoods between the full and nested models approximately follows the chi-squared distribution with two degrees of freedom. To account for multiple testing, associations with a false discovery rate (FDR) [41] < 0.05 were considered statistically significant and the corresponding CpGs were referred to as ‘identified CpGs’.
For identified CpGs, we investigated their associations with cumulative smoke exposure indicated by pack-years for ever smokers and with years since quitting for former smokers. Pack-years were calculated as the average number of cigarettes smoked per day divided by 20 and multiplied by the number of years smoked, and were log-transformed to be approximately normal distributed. Years since quitting were calculated as age at interview minus age at stopping smoking. The covariates adjusted and statistical inference were the same as those for smoking status, except that the model for pack-years was additionally adjusted for smoking status (former and current smokers) to investigate associations independent of smoking status.
Replication of previously reported associations
After quality control, 18,671 CpGs reported from the largest meta-analysis performed by Joehanes et al. [11] were included in our study. For these CpGs, we investigated their associations with smoking status in our study. Given the sample size of our study and not to miss any potential replication, associations with a nominal P < 0.05 and the same direction as that reported by Joehanes et al. were considered to be replicated, and the corresponding CpGs were referred to as ‘replicated CpGs’.
Familial confounding analysis
For the identified CpGs and replicated CpGs, we performed between- and within-sibship analyses [25, 42] to investigate if familial factors confound the associations. Given that never and former smokers had similar methylation levels for most of the CpGs, we combined them into one group. The new smoking status was thus analysed with current smokers as ‘1’ and the rest as ‘0’.
In the analysis, the methylation M values, smoking exposures and covariates were orthogonally transformed within sibships to obtain sibship means and within-sibship differences for these variables; see Stone et al. [42] for more details about the transformation. The between-sibship analyses investigated associations between sibship means for methylation levels and those for smoking exposures, and the within-sibship analyses investigated associations between within-sibship differences for methylation levels and those for smoking exposures. Associations estimated from the within-sibship analyses are independent of familial confounding, as the confounding effects of familial factors shared by siblings, both known and unknown, were cancelled out when using within-sibship differences. Evidence for familial confounding can be obtained by comparing between-sibship coefficient (βB) and within-sibship coefficient (βW). When βB ≠ βW and βW ≈ 0, i.e. the association disappears when familial factors are adjusted, the observation is consistent with the association being due to familial confounding. When βB ≈ βW ≠ 0, i.e. the association is similar regardless of whether familial factors are adjusted, the observation is consistent with absence of evidence for familial confounding; see Carlin et al. [43] for more details about the implications from comparing βB and βW.
Causal inference analysis
We performed causal inference between smoking status and methylation using Inference about Causation through Examination of FAmiliaL CONfounding (ICE FALCON), a regression-based methodology for analysing twin data [44,45,46,47,48]. By causal is meant, that if it were possible to vary a predictor measure experimentally, the expected value of the outcome measure would change.
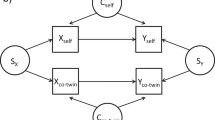
As shown in Fig. 1, suppose there are two variables, X and Y, measured for pairs of twins, and for example, let X refer to smoking status and Y refer to methylation. Assume that X and Y are positively associated within an individual. Let S denote the unmeasured familial factors that affect both twins, SX represents those factors that influence X values only, SY those that influence Y values only, and SXY those that influence both X and Y values. For the purpose of explanation, let ‘self’ refer to an individual and ‘co-twin’ refer to the individual’s twin, but recognise that these labels can be exchanged and both twins within a pair are used in the analysis.

Some possible directed acyclic graphs for the cross-twin cross-trait correlation. a The cross-twin cross-trait correlation is due to familial confounding. b The cross-twin cross-trait correlation is due to the causal effect of X on Y. c The cross-twin cross-trait correlation is due to the causal effect of Y on X
If there is a correlation between Yself and Xco-twin, it might be due to a familial confounder, SXY (Fig. 1a). It could also be due to X having a causal effect on Y within an individual, provided Xself and Xco-twin are correlated (Fig. 1b), or to Y having a casual effect on X, provided Yself and Yco-twin are correlated (Fig. 1c). Note that the confounders specific to an individual, Cself and Cco-twin, do not of themselves result in a correlation between Yself and Xco-twin.
Using the Generalised Estimating Equations (GEE), fitted using the geeglm() function from R package geepack [49], to take into account any correlation in Y between twins within the same pair, three models are fitted:
Model 1: E(Yself) = α + βselfXself
Model 2: E(Yself) = α + βco-twinXco-twin
Model 3: E(Yself) = α + β′selfXself + β′co-twinXco-twin
If the correlation between Yself and Xco-twin is solely due to familial confounders (Fig. 1a), the marginal association between Yself and Xself (βself in model 1) and the marginal association between Yself and Xco-twin (βco-twin in model 2) must both be non-zero. Adjusting for Xself, however, the conditional association between Yself and Xco-twin (β′co-twin in model 3) is expected to attenuate from βco-twin in model 2 towards the null. Similarly, adjusting for Xco-twin (model 3), the conditional association between Yself and Xself (β′self in model 3) is expected to attenuate from βself in model 1 towards the null.
If the correlation between Yself and Xco-twin is solely due to a causal effect from X to Y (Fig. 1b), Yself and Xco-twin in model 2 will be associated through two pathways: the confounder SX, and conditioning on the collider Yco-twin (GEE analysis in effect conditions on Yco-twin). Conditioning on Yco-twin induces a negative correlation between Xco-twin and Yself (note that we assume X and Y are positively associated within an individual), so that βco-twin in model 2 depends on the within-pair correlations in X (ρX) and in Y (ρY): if ρX > ρY, βco-twin is expected to be positive; otherwise βco-twin to be negative. Conditioning on Xself (model 3), both pathways are blocked and the conditional association (β′co-twin in model 3) is expected to attenuate towards the null.
If the correlation between Yself and Xco-twin is solely due to a causal effect from Y to X (Fig. 1c), in model 2 the pathway through SX is blocked due to Xself as a collider, and the pathway through SY is blocked due to that GEE analysis in effect conditions on Yco-twin, so there is no marginal association between Yself and Xco-twin, and βco-twin of model 2 is expected to be zero.
We studied methylation at the identified CpGs and replicated CpGs, respectively. For each group of CpGs, methylation was analysed as a weighted methylation score, calculated as the sum of the products of methylation level and weight of each CpG. For a locus containing multiple CpGs, only the CpG with the smallest P value was included in the methylation score. For the identified CpGs, the methylation level was the standardised M value and the weight was the log odds ratio for smoking status. For the replicated CpGs, the methylation level was the Beta value, the scale used in the meta-analysis, and the weight was the Z statistic reported by Joehanes et al. [11]. Smoking status was analysed as a binary variable with current smokers as ‘1’ and the rest as ‘0’. We first used smoking status to be X and methylation score to be Y and regressed methylation score on smoking status. We then exchanged X and Y to regress smoking status on methylation score and undertook the same analyses. The data for 132 twin pairs were used. We made statistical inference about the change in regression coefficient using one-sided t test with a standard error computed using nonparametric bootstrap method. That is, twin pairs were randomly sampled with replacement to generate 1000 new datasets with the same sample size as the original dataset. ICE FALCON was then applied to each dataset to calculate the change in regression coefficient for that dataset and standard error was then estimated by computing the standard deviation.
Results
Characteristics of the sample
The mean (standard deviation [SD]) age for the 479 women was 56.4 (7.9) years. The women included 291 (60.8%) never smokers, 147 (30.7%) former smokers and 41 (8.5%) current smokers. Ever smokers had a median (interquartile range) of 7.0 (13.8) pack-years. Former smokers had an average (SD) of 21.5 (11.4) years since quitting.
Epigenome-wide analysis results
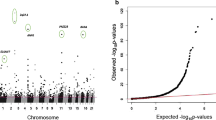
Methylation at 39 CpGs located at 27 loci was found to be associated with smoking status (Table 1; Q-Q plot and Manhattan plot in Fig. 2). Associations for 37 of the 39 CpGs have been reported by at least two studies and associations for two CpGs, cg06226150 (SLC2A4RG) and cg21733098 (12q24.32), have only been reported from the meta-analysis performed by Joehanes et al. [11]. For all 39 CpGs, current smokers had the lowest methylation level (Table 1). The 27 loci included several consistently reported loci, such as AHRR (9 CpGs), 2q37.1 (3 CpGs), 6p21.33 (3 CpGs), and F2RL3 (1 CpG).

Q-Q plot (a) and Manhattan plot (b) for the results from the epigenome-wide association analysis between DNA methylation and smoking status
Of the 39 CpGs and at a 5% FDR, methylation at 18 CpGs was negatively associated with pack-years and at 20 CpGs was positively associated with years since quitting. Methylation at 15 CpGs was associated with pack-years and years since quitting both (Table 2).
Replication for previously reported associations
For the associations for 18,671 CpGs reported by Joehanes et al. [11], 1882 were replicated with a nominal P < 0.05 and in the same direction, and the 133 most significant associations also had a FDR < 0.05. Of the 1882 replications, 1154 were for the novel CpGs reported by Joehanes et al. (Additional file 1: Table S1).
Between- and within-sibship analyses results
For the 39 identified CpGs, no evidence for a difference between βB and βW was found for any CpG (Table 3; all P values > 0.05 from the βB and βW comparison). The same results were found from the analyses of pack-years and years since quitting (Table 3).
For the 1882 replicated CpGs, no evidence for a difference between βB and βW was found for any CpG (Additional file 2: Table S2; the smallest P value = 1.3 × 10− 3 and the smallest FDR = 0.99 from the β B and βW comparison).
ICE FALCON analysis results
Within twin pairs, the correlation in smoking status was 0.11 (95% confidence interval (CI) − 0.06, 0.27), smaller than the correlations in methylation scores for the replicated CpGs and for the identified CpGs, which were 0.37 (95% CI 0.23, 0.50) and 0.22 (95% CI 0.05, 0.37), respectively.
The ICE FALCON results for methylation at the replicated CpGs are shown in Table 4. From the analysis in which smoking status was the predictor and methylation score the outcome, a women’s methylation score was associated with her own smoking status (model 1; βself = 74.6, 95% CI 55.3, 93.9), and negatively associated with her co-twin’s smoking status (model 2; βco-twin = − 30.8, 95% CI − 57.7, − 4.0). Conditioning on her co-twin’s smoking status (model 3), β′self remained unchanged (P = 0.41) compared with βself in model 1, while conditioning on her own smoking status (model 3), βco-twin in model 2 attenuated by 123.3% (95% CI 49.6%, 185.2%; P = 0.002) to be β′co-twin of 2.5 (95% CI − 16.3, 21.3). From the analysis in which methylation score was the predictor and smoking status the outcome, a woman’s smoking status was associated with her own methylation score (model 1; βself = 4.1, 95% CI 2.7, 5.4), but not with her co-twin’s methylation score (model 2; βco-twin = 0.4, 95% CI − 1.0, 1.8). In model 3, β′self and β′co-twin remained unchanged (both P > 0.1) compared with βself in model 1 and βco-twin in model 2, respectively. These results were consistent with that smoking has a causal effect on the overall methylation level at these CpGs, but not in the opposite direction. Similar results were found and a similar causality was inferred for smoking status and the overall methylation level at the identified CpGs (Table 4).
Discussion
We performed an EWAS of smoking for a sample of middle-aged women and found 39 CpGs at which methylation was associated with smoking status. Our study confirmed the associations for several previously consistently reported loci including AHRR, F2RL3, 2q37.1, and 6p21.33, and for two novel CpGs, cg06226150 (SLC2A4RG) and cg21733098 (12q24.32), reported by the largest meta-analysis [11] so far. In addition, we replicated the associations for 1882 CpGs reported by the meta-analysis. The investigation of causation suggests that smoking has a causal effect on DNA methylation, not vice versa or being due to familial confounding.
To the best of our knowledge, our study is the first study to confirm the associations for cg06226150 and cg21733098. cg06226150 is located at the promoter of, and potentially regulates the expression of, SLC2A4RG (solute carrier family 2 member 4 regulator gene). SLC2A4RG is involved in the Gene Ontology pathway for regulation of transcription (GO:0006355). Protein encoded by SLC2A4RG regulates the activation of SLC2A4 (solute carrier family 2 member 4). SLC2A4 is involved in the glucose transportation across cell membranes stimulated by insulin. Genetic variants at SLC2A4RG have been found to be associated with inflammatory bowel disease [50] and prostate cancer [51]. cg21733098 is located at an intergenic region on 12q24.32. The region contains several long non-coding RNA genes. Little is known about the regulatory function of cg21733098. The biological relevance of smoking to blood methylation at these two CpGs is largely unknown, and more research are warranted.
We found evidence that 18 and 20 of the identified CpGs were also associated with pack-years and years since quitting, respectively. Given that smokers have lower methylation levels at the identified CpGs, the negative associations with pack-years imply that there appear to be dose-relationships between smoking and methylation at the 18 CpGs, and the positive associations with years quitting smoking imply that methylation changes at the 20 CpGs tend to reverse after cessation. The dose-relationship and reversion have also been reported by several studies [4, 9,10,11,12, 15, 16, 19, 20, 22].
Our study, as one of the first studies, provides insights into the causality underlying the cross-sectional association between smoking and blood DNA methylation. Our results are inconsistent with the proposition that the cross-sectional association is due to familial confounding, e.g. shared genes and/or environment. The roles of shared genes and/or environment are also in part unsupported by that certain smoking-related loci, such as AHRR and F2RL3, are observed across Europeans [3, 5, 8,9,10,11, 16, 19, 20, 22], South Asians [8], Arabian Asians [21], East Asians [12, 23], and African Americans [7, 11, 13, 18], who have different germline genetic backgrounds and environments. Our results support that smoking has a causal effect on the overall methylation at the identified CpGs and at the replicated CpGs, but not vice versa. Results from the two-step MR analysis performed by Jhun et al. [31] also suggest that differential methylation at cg03636183 (F2RL3) and cg19859270 (GPR15) between current and never smokers are consequential to smoking under the assumptions of MR.
That smoking causes changes in methylation is also supported to some extent by other evidence. The ‘reversion’ phenomenon is in line with the ‘experimental evidence’ criterion proposed by Bradford Hill, i.e. ‘reducing or eliminating a putatively harmful exposure and seeing if the frequency of disease subsequently declines’ [52]. The associations between cord blood methylation for newborns at some active-smoking-related loci, such as AHRR and GFI1, and maternal smoking in pregnancy [53] also imply that smoking is likely to cause methylation changes at these loci. Additionally, some smoking-related loci are involved in the metabolism of smoking-released chemicals. AHRR gene encodes a repressor of the aryl hydrocarbon receptor (AHR) gene, the protein encoded by which is involved in the regulation of biological response to planar aromatic hydrocarbons. Polycyclic aromatic hydrocarbons, one main smoking-related toxic and carcinogenic substance, trigger AHR signalling cascade [16, 22]. Protein coded by the AHR gene activates the expression of the AHRR gene, which in turn represses the function of AHR through a negative feedback mechanism [54]. That hypomethylation at AHRR gene caused by smoking is biologically plausible.
That smoking causes changes in blood methylation has great clinical and etiological implications: methylation might mediate the effects of smoking on smoking-related health outcomes. As introduced above, there have been a few studies [26,27,28,29] investigating the mediating role of methylation. A better understanding of the mechanisms of smoking affecting health is expected with more investigations on methylation.
Our study shows the value of ICE FALCON in causality assessment for observational associations. Associations from observational studies can be due to confounding and, although analyses of measured potential confounders can eliminate some confounding, there is always the possibility of unmeasured confounding, even with prospective studies. With recent discoveries of genetic markers that predict variation in risk factors, the MR concept has been explored by epidemiologists. MR uses measured genetic variants as the instrumental variable and the results of MR might be biased due to several factors such as strengthen of instrumental variable, directional pleiotropy, and unmeasured confounding [55]. ICE FALCON is a novel approach to making inference about causation. It in effect uses the familial causes of exposure and of outcome as instrumental variables. The familial causes are not measured but surrogated by co-twin’s measured exposure and outcome. Thus, ICE FALCON resembles a bidirectional MR approach [56]. The instrumental variables consider all familial causes in exposure and in outcome, thus potentially less biased by their strengths than a finite number of genetic markers. More importantly, even should directional pleiotropy exist, the attenuation in the coefficient for co-twin’s exposure after adjusting for an individual’s own exposure also supports a causal effect.
Conclusions
We found evidence that in the peripheral blood from middle-aged women, DNA methylation at several loci is associated with smoking. By investigating causation underlying the association, our study found evidence consistent with smoking having a causal effect on methylation, but not vice versa.
Abbreviations
- AHRR:
-
Aryl hydrocarbon receptor repressor gene
- CI:
-
Confidence interval
- CpG:
-
Cytosine-guanine dinucleotide
- EWAS:
-
Epigenome-wide association study
- F2RL3:
-
F2R-like thrombin or trypsin receptor 3 gene
- FDR:
-
False discovery rate
- GEE:
-
Generalised estimating equations
- GFI1:
-
Growth factor independent 1 transcriptional repressor gene
- GPR15:
-
G protein-coupled receptor 15 gene
- ICE FALCON:
-
Inference about causation through examination of familial confounding
- MR:
-
Mendelian randomisation
- SD:
-
standard deviation
- SLC2A4RG:
-
Solute carrier family 2 member 4 regulator gene
References
Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–7.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59.
Allione A, Marcon F, Fiorito G, Guarrera S, Siniscalchi E, Zijno A, Crebelli R, Matullo G. Novel epigenetic changes unveiled by monozygotic twins discordant for smoking habits. PLoS One. 2015;10:e0128265.
Ambatipudi S, Cuenin C, Hernandez-Vargas H, Ghantous A, Le Calvez-Kelm F, Kaaks R, Barrdahl M, Boeing H, Aleksandrova K, Trichopoulou A, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8:599–618.
Besingi W, Johansson A. Smoke-related DNA methylation changes in the etiology of human disease. Hum Mol Genet. 2014;23:2290–7.
Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88:450–7.
Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, Monick M, Brody GH, Tan K, Beach SR, Philibert RA. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151.
Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, Davey Smith G, Hughes AD, Chaturvedi N, Relton CL. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics. 2014;6:4.
Guida F, Sandanger TM, Castagne R, Campanella G, Polidoro S, Palli D, Krogh V, Tumino R, Sacerdote C, Panico S, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet. 2015;24:2349–59.
Harlid S, Xu Z, Panduri V, Sandler DP, Taylor JA. CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the Sister Study. Environ Health Perspect. 2014;122:673–8.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–47.
Lee MK, Hong Y, Kim SY, London SJ, Kim WJ. DNA methylation and smoking in Korean adults: epigenome-wide association study. Clin Epigenetics. 2016;8:103.
Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics. 2012;7:1331–8.
Philibert RA, Beach SR, Lei MK, Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics. 2013;5:19.
Sayols-Baixeras S, Lluis-Ganella C, Subirana I, Salas LA, Vilahur N, Corella D, Munoz D, Segura A, Jimenez-Conde J, Moran S, et al. Identification of a new locus and validation of previously reported loci showing differential methylation associated with smoking. The REGICOR study. Epigenetics. 2015;10:1156–65.
Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013;22:843–51.
Su D, Wang X, Campbell MR, Porter DK, Pittman GS, Bennett BD, Wan M, Englert NA, Crowl CL, Gimple RN, et al. Distinct epigenetic effects of tobacco smoking in whole blood and among leukocyte subtypes. PLoS One. 2016;11:e0166486.
Sun YV, Smith AK, Conneely KN, Chang Q, Li W, Lazarus A, Smith JA, Almli LM, Binder EB, Klengel T, et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet. 2013;132:1027–37.
Tsaprouni LG, Yang TP, Bell J, Dick KJ, Kanoni S, Nisbet J, Vinuela A, Grundberg E, Nelson CP, Meduri E, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics. 2014;9:1382–96.
Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, Agusti A, Anderson W, Lomas DA, Demeo DL. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012;21:3073–82.
Zaghlool SB, Al-Shafai M, Al Muftah WA, Kumar P, Falchi M, Suhre K. Association of DNA methylation with age, gender, and smoking in an Arab population. Clin Epigenetics. 2015;7:6.
Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8:e63812.
Zhu X, Li J, Deng S, Yu K, Liu X, Deng Q, Sun H, Zhang X, He M, Guo H, et al. Genome-wide analysis of DNA methylation and cigarette smoking in a Chinese population. Environ Health Perspect. 2016;124:966–73.
Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics. 2015;7:113.
Li S, Wong EM, Southey MC, Hopper JL. Association between DNA methylation at SOCS3 gene and body mass index might be due to familial confounding. Int J Obes. 2017;41:995–6.
Fasanelli F, Baglietto L, Ponzi E, Guida F, Campanella G, Johansson M, Grankvist K, Johansson M, Assumma MB, Naccarati A, et al. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat Commun. 2015;6:10192.
Zhang Y, Elgizouli M, Schottker B, Holleczek B, Nieters A, Brenner H. Smoking-associated DNA methylation markers predict lung cancer incidence. Clin Epigenetics. 2016;8:127.
Zhang Y, Schottker B, Florath I, Stock C, Butterbach K, Holleczek B, Mons U, Brenner H. Smoking-associated DNA methylation biomarkers and their predictive value for all-cause and cardiovascular mortality. Environ Health Perspect. 2016;124:67–74.
Gao X, Mons U, Zhang Y, Breitling LP, Brenner H. DNA methylation changes in response to active smoking exposure are associated with leukocyte telomere length among older adults. Eur J Epidemiol. 2016;31:1231–41.
Reynolds LM, Wan M, Ding J, Taylor JR, Lohman K, Su D, Bennett BD, Porter DK, Gimple R, Pittman GS, et al. DNA methylation of the aryl hydrocarbon receptor repressor associations with cigarette smoking and subclinical atherosclerosis. Circ Cardiovasc Genet. 2015;8:707–16.
Jhun MA, Smith JA, Ware EB, Kardia SL, Mosley TH, Turner ST, Peyser PA, Kyun Park S. Modeling the causal role of DNA methylation in the association between cigarette smoking and inflammation in African Americans: a two-step epigenetic Mendelian randomization study. Am J Epidemiol. 2017;
Odefrey F, Stone J, Gurrin LC, Byrnes GB, Apicella C, Dite GS, Cawson JN, Giles GG, Treloar SA, English DR, et al. Common genetic variants associated with breast cancer and mammographic density measures that predict disease. Cancer Res. 2010;70:1449–58.
Li S, Wong EM, Joo JE, Jung CH, Chung J, Apicella C, Stone J, Dite GS, Giles GG, Southey MC, Hopper JL. Genetic and environmental causes of variation in the difference between biological age based on DNA methylation and chronological age for middle-aged women. Twin Res Hum Genet. 2015;18:720–6.
Joo JE, Wong EM, Baglietto L, Jung CH, Tsimiklis H, Park DJ, Wong NC, English DR, Hopper JL, Severi G, et al. The use of DNA from archival dried blood spots with the Infinium HumanMethylation450 array. BMC Biotechnol. 2013;13:23.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Maksimovic J, Gordon L, Oshlack A. SWAN: subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13:R44.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Price ME, Cotton AM, Lam LL, Farre P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6:4.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:48.
Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300.
Stone J, Gurrin LC, Hayes VM, Southey MC, Hopper JL, Byrnes GB. Sibship analysis of associations between SNP haplotypes and a continuous trait with application to mammographic density. Genet Epidemiol. 2010;34:309–18.
Carlin JB, Gurrin LC, Sterne JA, Morley R, Dwyer T. Regression models for twin studies: a critical review. Int J Epidemiol. 2005;34:1089–99.
Hopper JL, Bui QM, Erbas B, Matheson MC, Gurrin LC, Burgess JA, Lowe AJ, Jenkins MA, Abramson MJ, Walters EH, et al. Does eczema in infancy cause hay fever, asthma, or both in childhood? Insights from a novel regression model of sibling data. J Allergy Clin Immunol. 2012;130:1117–22. e1111
Stone J, Dite GS, Giles GG, Cawson J, English DR, Hopper JL. Inference about causation from examination of familial confounding: application to longitudinal twin data on mammographic density measures that predict breast cancer risk. Cancer Epidemiol Biomark Prev. 2012;21:1149–55.
Bui M, Bjornerem A, Ghasem-Zadeh A, Dite GS, Hopper JL, Seeman E. Architecture of cortical bone determines in part its remodelling and structural decay. Bone. 2013;55:353–8.
Dite GS, Gurrin LC, Byrnes GB, Stone J, Gunasekara A, McCredie MR, English DR, Giles GG, Cawson J, Hegele RA, et al. Predictors of mammographic density: insights gained from a novel regression analysis of a twin study. Cancer Epidemiol Biomark Prev. 2008;17:3474–81.
Davey CG, Lopez-Sola C, Bui M, Hopper JL, Pantelis C, Fontenelle LF, Harrison BJ. The effects of stress-tension on depression and anxiety symptoms: evidence from a novel twin modelling analysis. Psychol Med. 2016;46:3213–8.
Højsgaard S, Halekoh U, Yan J. The R package geepack for generalized estimating equations. J Stat Softw. 2005;15:11.
de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez-Achury J, Ji SG, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49:256–61.
Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Ghoussaini M, Luccarini C, Dennis J, Jugurnauth-Little S, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet. 2013;45:385–91. 391e381-382
Rothman KJ, Greenland S, Lash TL. Modern epidemiology. 3rd. Philadephia: Lippincott Williams & Wilkins; 2008.
Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, Reese SE, Markunas CA, Richmond RC, Xu CJ, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98:680–96.
Mimura J, Ema M, Sogawa K, Fujii-Kuriyama Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999;13:20–5.
VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological challenges in Mendelian randomization. Epidemiology. 2014;25:427–35.
Smith GD, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–98.
Acknowledgements
We would like to thank all women participating in this study. The data analysis was facilitated by Spartan, the High Performance Computer and Cloud hybrid system of the University of Melbourne.
Funding
The Australian Mammographic Density Twins and Sisters Study was facilitated through the Australian Twin Registry, a national research resource in part supported by a Centre for Research Excellence Grant from the National Health and Medical Research Council (NHMRC) APP 1079102. The AMDTSS was supported by NHMRC (grant numbers 1050561 and 1079102) and Cancer Australia and National Breast Cancer Foundation (grant number 509307).
SL is supported by the Australian Government Research Training Program Scholarship and the Richard Lovell Travelling Scholarship from the University of Melbourne. TLN is supported by a NHMRC Post-Graduate Scholarship and the Richard Lovell Travelling Scholarship from the University of Melbourne. MCS is a NHMRC Senior Research Fellow. JLH is a NHMRC Senior Principal Research Fellow.
Availability of data and materials
The dataset analysed during the current study is available on Gene Expression Omnibus (GEO) under the accession number GSE100227.
Author information
Authors and Affiliations
Contributions
SL and JLH conceived and designed the study. SL performed the statistical analyses. SL and JLH wrote the first draft of the manuscript. EMW, TLN, JEJ, JS, GSD, GGG, MCS, and JLH contributed to the data collection. MB contributed to the ICE FALCON analyses. RS contributed to the data interpretation. All authors participated in the manuscript revision and have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Human Research Ethics Committee of the University of Melbourne. All participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. This file includes Table S1: Associations for the 1882 replicated CpGs. (XLSX 156 kb)
Additional file 2:
Table S2. This file includes Table S2: Associations of methylation at the 1882 replicated CpGs with smoking status from the between- and within-sibship analyses. (XLSX 100 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, S., Wong, E.M., Bui, M. et al. Causal effect of smoking on DNA methylation in peripheral blood: a twin and family study. Clin Epigenet 10, 18 (2018). https://doi.org/10.1186/s13148-018-0452-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0452-9