
Abstract
Background
Related to genetic alteration, frequent promoter hypermethylation is also a contributing factor in the development of human cancers. Recently, we discovered numerous novel genes that were aberrantly methylated in hepatocellular carcinoma (HCC) by using Infinium HumanMethylation27 BeadChip array. We utilized a quantitative methylation-specific PCR (Q-MSP) system for the evaluation of PAX6 methylation in 29 normal controls and 160 paired HCC tissues and their adjacent non-tumor tissues. We verified the correlation between the methylation status of PAX6 and clinical characteristics with different viral status.
Results
Paired-box 6 promoter methylation was observed in 39.4 %, 15.6 %, and 3.4 % in primary HCCs, adjacent non-tumors, and normal control tissues, respectively. Methylation of the PAX6 promoter region in HCCs significantly increased compared with control tissues. PAX6 was frequently methylated in HCV-positive HCC tissues (61.3 %) and rarely methylated in HBV-positive (22.1 %) and double-negative HCC tissues (33.3 %).
Conclusions
Our data suggests that promoter hypermethylation of PAX6 is a common event in HCCs and the association of PAX6 methylation in clinicopathological features is divergent with different viral status.
Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC) is one of the most malignant tumors worldwide [1, 2]. It is a fatal disease due to difficulties in early detection which leads to poor prognosis and high mortality rates. Currently, therapeutic options are not effective, as indicated by data results for reaching a global survival rate of 20–65 % for 1 year, 10–30 % for 3 years, and 10–20 % for 5 years after diagnosis [3, 4]. The most significant risk factors associated with HCC include chronic hepatitis B and C virus (HBV and HCV) infections, cryptogenic liver cirrhosis, aflatoxin-B1-contaminated food, excessive alcohol consumption, and obesity [1]. The recent increase in the incidence of HCC in Western countries is predominantly due to the HCV endemic and the lack of effective vaccination. Despite significant effort made over the past decade to understand the development of HCC, the genetic alterations that lead to the initiation and progression of HCC still remain largely unknown. In addition to genetic alterations, epigenetic alterations have been progressively recognized as a key impetus for carcinogenesis. Methylation of CpG islands in promoter regions is frequently associated with transcriptional silencing and is implicated in tumor suppressor gene (TSG) inactivation in cancer cells [5]. Some studies have investigated the methylation profiles associated with different viral infections but without the yield of any definitive conclusions.
Recently, technical advances in array systems, such as the Infinium assay [6] can detect altered methylation patterns and other epigenetic changes in cancer. It has also been successfully applied in the study of HCC [7–10]. An advantage of adapting array-based platforms is that researchers can analyze DNA methylation of regions with no prior knowledge of the sequence. Furthermore, by validating the results from the high-throughput screening approach, researchers can effectively discover more novel genes that may have potential applications in clinical practice.
In our recent analysis [11], we recognized several aberrantly methylated genes in HCC by using the Infinium HumanMethylation27 BeadChip and then verified 34 genes by methylation-specific PCR (MSP). Of these genes, we further proved that frequent methylation of homeobox A9 (HOXA9) in HCC tissues and plasma samples from patients could be a useful biomarker to assist in HCC detection. IRAK3 methylation was associated with tumor stage and poor prognosis of patients [12]. However, several novel genes in our array data were not further validated by quantitative MSP (Q-MSP), such as PAX6. Paired-box 6 (PAX6), located on chromosome 11p13, a gene coding for a DNA binding/transcription control protein, participates in normal eye, nose, pancreas, and brain development [13]. PAX6 exerts a tumor suppressor effect and is frequently silenced by promoter methylation in human cancers, including bladder, breast, gastric, and non-small cell lung cancer [14–17], suggesting that it may play a role in carcinogenesis. Until now, there are no data regarding the PAX6 methylation in HCC. Moreover, there are no quantitative data about the methylation levels of PAX6 in HCC.
In the present study, the hypermethylation of CpG islands (CGI) of PAX6 was quantitatively investigated in HCC by Q-MSP, with particular attention made to the changes in methylation intensities in primary HCC tissues and their corresponding non-tumor liver tissues. The correlation between the methylation status of PAX6 and clinical disease was also addressed. Finally, we analyzed the promoter methylation level of PAX6 to be significantly higher in HCV-positive HCC.
Results
Distinct DNA methylation patterns and the association with diseases in HCC with different viral infections
In our recent study, we analyzed the methylation profiles of HCC with three different viral etiologies (HBV-positive, HCV-positive, and double-negative) by using the Infinium HumanMethylation27 BeadChip and reported 2924 genes which were aberrantly methylated in non-tumor or tumor tissues of HCC as compared with the normal controls [11]. To further investigate the DNA methylation profiles of unique etiologic-driven HCC, a Venn diagram was utilized to compare the genes identified in each of the etiological groups. Among the 2924 genes, 430, 590, and 331 genes are shown to be aberrantly methylated in HBV-positive, HCV-positive, and double-negative non-tumor or tumor tissues of HCC, respectively (Fig. 1a; Additional file 1: Table S1). To better understand the association between the etiologic-driven changes in DNA methylation patterns and diseases, DAVID disease analysis was used for genes in each etiology. Genes changing in HBV-positive HCC were primarily found to be involved in alcoholic diseases and cirrhosis. Genes identified in HCV-positive HCC were relative to immune deficiency diseases, including type 1 diabetes, atopy, systemic sclerosis, and cardiovascular diseases. Genes in the HCC without hepatitis B and hepatitis C showed the association with age-associated diseases and metabolic diseases (Fig. 1b; Additional file 2: Table S2). Then, to investigate the interest in genes that the changes in DNA methylation status are driven by specific risk factors, the PAX6 gene was selected as our hypermethylated candidate gene for validation based on the results of the methylation array (data not shown).

Genes and the association of genes with diseases in HCC with different etiologic factors. The Venn diagram drawing the unique and overlapping genes in HBV-positive (HBV), HCV-positive (HCV), and double-negative (NBC) HCC (a). DAVID ontology (disease) for genes abnormally methylated in HBV, HCV, and NBC-HCC (b)
PAX6 methylation is associated with altered mRNA expression in HCC cell lines
We first investigated the PAX6 expression in nine HCC cell lines. The reverse transcription polymerase chain reaction (RT-PCR) data showed that there was down-regulation of PAX6 messenger RNA (mRNA) in HCC cell lines with PAX6 hypermethylation (Fig. 2a). To confirm that the lack of expression of PAX6 mRNA in the HCC lines was due to promoter hypermethylation, we treated cells with 5-aza-2′-deoxycytidine (5DAC), an inhibitor of DNA methylation. After treatment with 5 μM of 5DAC, the unmethylated promoter DNA was detected by MSP and bisulfite sequencing (Fig. 2b, c); PAX6 mRNA was restored or increased in HCC cell lines (Fig. 2a). These data indicate that hypermethylation of PAX6 may be responsible for the absence or down-regulation of mRNA transcription.

Expression analysis and methylation analysis of PAX6 in cell lines. PAX6 and GAPDH expression were analyzed by the RT-PCR in the normal control, the normal liver cell line THLE-3, and the HCC cell lines treated with or without DNMT inhibitors (5-aza-2′-deoxycytidine; 5DAC) (a). Promoter methylation of PAX6 and COL2A was analyzed by MS-PCR with methylation-specific primers in the normal control, THLE-3, and HCC cell lines treated with or without 5DAC. CpG methylated human genomic DNA and DNA extracted from normal peripheral blood lymphocytes (PBL) which were modified by sodium bisulfite to generate a positive control, 1/5 diluted positive control, 1/20 diluted positive control, and a negative control, respectively (b). PAX6 methylation was also analyzed by bisulfite sequencing in Mahlavu cells treated with or without 5DAC. Each clone is represented by a row, and 30 CpG sites are represented as circles. Black and white circles represent methylated cytosine and unmethylated cytosine, respectively. Gray regions indicate the CpG sites that the MS-PCR/Q-MSP primer set covered (c)
Frequent methylation of the PAX6 gene in HCC tissues
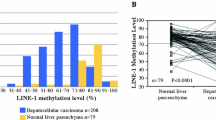
The methylation status of the PAX6 gene was analyzed by Q-MSP in 349 liver tissues, including 29 normal controls and 160 pairs of HCC tissues and their adjacent non-tumor tissues. The results showed a significantly increased methylation level in HCC tissues as compared with non-tumor tissues (p < 0.0001) (Fig. 3a). By using receiver operating characteristic (ROC) curve analysis to discriminate 160 HCC tissues and 29 normal controls, the best cutoff value of PAX6 methylation was MI > 1.13. (Fig. 3b). Under the best cutoff value, 3.4 % of 29 normal controls, 15.6 % of 160 non-tumor tissues, and 39.4 % of 160 HCC tissues were reported as positive for PAX6 methylation (p < 0.0001) (Table 2). An association between gene methylation and clinicopathological features was also analyzed among the 160 HCC patients. Interestingly, there was a statistically positive association between PAX6 methylation and HCV infection (OR = 4.46; 95 % CI = 2.26–8.80). There was a statistically negative association between PAX6 methylation and HBV infection (OR = 0.29; 95 % CI = 0.15–0.58), as well as no significance in the association with other clinical parameters (Table 3).

Methylation levels and ROC curve analysis of PAX6 in liver tissues. Gene methylation was determined in 29 normal controls (NC) and 160 paired hepatocellular carcinoma (HCC) tissues and their adjacent non-tumor tissues (NT) by Q-MSP. The results are represented as the difference in the methylation index. The black lines indicate the mean of the methylation index. The p value for the methylation levels among the groups was computed from Wilcoxon rank-sum test (NC vs HCC and NC vs NT) and Wilcoxon signed-rank test (NT vs HCC) (a). The best cutoff value for the methylation index (MI) and the area under the receiver operating characteristic curve (AUC) were calculated to discriminate 29 normal controls and 160 HCC (b). Gene methylation was respectively determined in normal controls (NC) and paired hepatocellular carcinoma (HCC) tissues and their adjacent non-tumor tissues (NT) by etiologic factors. The p value for methylation levels among the groups was computed from Wilcoxon rank-sum test (NC vs HCC and NC vs NT) and Wilcoxon signed-rank test (NT vs HCC) (c)
The association of PAX6 methylation with clinicopathological features in HCC with different viral status
The methylation status and methylation frequency of the PAX6 gene was further examined in HCC with different viral etiologies. To test whether the PAX6 methylation in HCC tissues was associated with different virus infections, we analyzed the methylation level in 29 normal controls (NC) and 160 paired HCC tissues and their adjacent non-tumor tissues. Among the 68 pairs of HCC tissues with HBV infection, 62 pairs exhibited HCV infection and 30 pairs showed neither HBV nor HCV infections. The methylation levels of the PAX6 gene were significantly increased in HCV-positive HCC tissues (p < 0.0001) as compared with their adjacent non-tumor tissues (Fig. 3c). Moreover, the PAX6 methylation level was significantly higher in HCV-positive HCC as compared with HBV-positive (p < 0.0001) and double-negative HCC tissues (p = 0.003). Under the best cutoff value (MI > 1.13), PAX6 was frequently methylated in HCV-positive HCC tissues (61.3 %) and rarely methylated in HBV-positive (22.1 %) and double-negative HCC tissues (33.3 %) (Table 2). In addition, logistic regression analysis showed PAX6 methylation was not associated with any clinical characteristics in HCV-positive HCC, but it was positively associated with cirrhosis in double-negative HCC tissues (OR = 12.00; 95 % CI = 1.11–129.42) (Table 3).
Discussion
Epigenetic modifications include DNA methylation, and covalent modification of histones, including methylation, acetylation, and phosphorylation. Alterations in DNA methylation affect the structure of DNA and do not materially affect the genetic code. Methylation of CpG islands in gene promoter regions are known to be an essential mechanism for guiding normal cellular development and play a critical role in the regulation of tumor suppressor gene expression.
Recently, high-resolution methods for genome-wide methylation analysis have been used in the study of HCC, such as in Illumina’s array-based Infinium Methylation Assay [7–10]. These results provide evidence that HCC tumors with specific DNA methylation patterns associated with risk factors or progression of HCC have important clinical applications. In our recent study, we also used a genome-wide methylation array, the Infinium HumanMethylation27 BeadChip to analyze 1968 genes that were hypermethylated in non-tumor tissue and/or tumor tissue from patients with HCC of different viral etiologies.
Molecular signatures causing HCC from chronic infection of HBV or HCV are not clearly known. Some studies suggest that different pathways are preferentially inactivated epigenetically in HCCs caused by different viral infections [18]. To better understand the association between the etiologic-driven changes in DNA methylation patterns and diseases, we use DAVID ontology disease analysis for genes in each etiology. Immune deficiency diseases, including type 1 diabetes, atopy, systemic sclerosis, and cardiovascular diseases were predominantly enriched in HCV-positive HCC, whereas alcoholic diseases and cirrhosis were mainly enriched in HBV-positive HCC. Genes in HCC without hepatitis B and hepatitis C show the association with age-associated diseases and metabolic diseases (Fig. 1b).
The paired-box (PAX) genes are a family of transcription factors composed of nine members with crucial roles in tissue development, cellular differentiation, migration, and proliferation [19]. PAX6, a gene coding for a DNA binding/transcription factor, participates in normal eye, nose, pancreas, and brain development [13]. PAX6 functions as either oncogene or tumor suppressors which seem to be tissue specific and is discussed controversially [20, 21]. However, methylation promoters of PAX6 in HCCs have never been illustrated.
In this study, we observed that the PAX6 expression was frequently down-regulated in HCC cell lines, and the reduced expression is associated with promoter methylation, as confirmed by promoter methylation analyses and pharmacological demethylation treatment. This implicates that DNA methylation is the principle regulatory mechanism of PAX6 inactivation in HCC, suggesting the PAX6 would be a candidate tumor suppressor in the pathogenesis of HCC. Our findings suggest that hypermethylation of PAX6 is a common event in HCC but its potential clinical application value needs to be further clarified.
This is the first study to analyze the relationship between PAX6 methylation status and different viral etiologies in HCC. In accordance with our data, PAX6 methylation was significantly increased in HBV-positive, HCV-positive, and double-negative HCC tissues as compared with their adjacent non-tumor tissues irrespective of the hepatitis virus status. However, there was no relationship with other clinical characteristics, including age, gender, tumor size, TNM stage, and invasion in HCV-positive HCC (Table 1). Moreover, it was positively associated with invasion in HBV-positive HCC and positively associated with cirrhosis in double-negative HCC tissues. Interestingly, we found that the highest number of methylated genes in HCV-positive HCC tissue (Fig. 1b) and PAX6 was frequently methylated in HCV-positive HCC tissues (Tables 2 and 3, Fig. 3c). This is consistent with previous studies showing that methylation levels were higher in the HCV-positive HCCs than in the HBV-positive or double-negative HCC [22, 23].
The HCV core contributes to HCV-mediated carcinogenesis through alteration of various signaling pathways, transcriptional activation, immune modulation, apoptosis, and lipid metabolism [24, 25]. HCV was demonstrated to affect CpG island methylation patterns especially for those genes responsible for DNA mismatch repair (MMR) [26] and cell cycle regulation [27]. Furthermore, core down-regulates the levels of p16 in HCC cells, and via core up-regulates the levels of DNMT1 and DNMT3b to induce promoter hypermethylation [28]. PAX5, a member of the PAX family, is frequently inactivated by promoter methylation in HCC and slightly decreases its proliferation rates in HCC cell lines through interaction with a p53 signaling pathway [29]. We therefore reasonably presumed that the HCV core-induced PAX6 methylation consequently contributed to down-regulate PAX6 expression.
In summary, by using genome-wide methylation analysis, we identified the frequently methylated gene PAX6 in HCC. The quantitative methylation level of PAX6 was higher in HCC tissues than that in corresponding non-tumor liver tissues irrespective of the virus infection background. In addition, PAX6 was frequently methylated in HCV-positive HCC tissues. We suggest that the HCV core plays the role of inducing PAX6 methylation. Further investigation is needed to better understand the PAX6 functional pathway in hepatocarcinogenesis.
Conclusions
Our data suggests that promoter hypermethylation of PAX6 is a common event in HCCs and the association of PAX6 methylation in clinicopathological features is divergent with different viral status.
Methods
Clinical samples
Twenty-nine specimens of normal livers from hemangiomas, obtained from the Taiwan Liver Cancer Network (TLCN), were used as normal controls in this study. For validation, a total of 160 paired samples of HCC and adjacent non-tumor tissues were used as subjects, including 40 paired samples which were obtained from Tri-Service General Hospital, and 120 paired samples of HCC and adjacent non-tumor tissues were obtained from TLCN. The TLCN is funded by the National Science Council to provide researchers in Taiwan with primary liver cancer tissue specimens and their associated clinical information. The use of clinical samples in this study was approved by our Institutional Review Board and the TLCN User Committee. These specimens were obtained during surgery and were frozen immediately in liquid nitrogen and/or at −80 °C until DNA extraction. The diagnosis of HCC was confirmed by histology. The clinicopathological characteristics of the patients are summarized in Table 1.
DNA extraction and bisulfite conversion
Genomic DNA from tissues was extracted by using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Genomic DNA extracted from tissues was treated with sodium bisulfite by using an EZ DNA methylation kit (Zymo Research, Orange, CA, USA). For quantitative methylation analysis, CpG methylated human genomic DNA (Thermo Fisher Scientific Inc.) and DNA extracted from normal peripheral blood lymphocytes (PBL) which were modified by sodium bisulfite to generate a positive control and a negative control, respectively.
Q-MSP
Q-MSP was performed in the TaqMan probe system using the LightCycler 480 system (Roche Applied Science, Mannheim, Germany) and prepared as previously described [12]. Each sample was analyzed in duplicate. The testing results of each sample were assessed as quantification cycle values (Cp value). The COL2A gene was used as an internal reference gene by amplifying non-CpG sequences. The results with a Cp value of COL2A > 38 were defined as detection failure. The results with a Cp value of PAX6 > 45 were defined as undetermined after PCR analysis. The DNA methylation level was assessed as a methylation index (MI), using the formula 100 × 2−[(Cp of PAX6) − (Cp of COL2A)] [30]. The primer and probe sequences are as follows: COL2A forward: gggaagatgggatagaagggaatat, reverse: tctaacaattataaactccaaccaccaa, probe: ttcattctaacccaatacct; PAX6 forward: agggagtatttaatcggttggc, reverse: ctcctacgcctaaaccaaaacg, probe: aaataaaaccgaaccacgatt.
Statistical analysis
The Wilcoxon rank-sum test and Wilcoxon signed-rank test were used to determine the differences between gene methylation level and disease status. The χ 2 test for trend and logistic regression was used to evaluate the association between gene methylation, disease status, and clinical characteristics. ROC curves were generated to determine the optimal cutoff point of gene methylation for discriminating HCC tissues and normal controls.
Abbreviations
- 5DAC:
-
5-aza-2′-deoxycytidine
- CGI:
-
CpG islands
- HBV:
-
hepatitis B virus
- HCC:
-
hepatocellular carcinoma
- HCV:
-
hepatitis C virus
- PAX6:
-
paired-box 6
- Q-MSP:
-
quantitative methylation-specific PCR
- TSG:
-
tumor suppressor gene
References
Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis. 2005;9(2):191–211. v.
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–917.
Arrieta O, Cacho B, Morales-Espinosa D, Ruelas-Villavicencio A, Flores-Estrada D, Hernandez-Pedro N. The progressive elevation of alpha fetoprotein for the diagnosis of hepatocellular carcinoma in patients with liver cirrhosis. BMC Cancer. 2007;7:28.
Nguyen VT, Law MG, Dore GJ. Hepatitis B-related hepatocellular carcinoma: epidemiological characteristics and disease burden. J Viral Hepat. 2009;16(7):453–63.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. Epub 2007/02/27.
Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics. 2009;1(1):177–200. Epub 2009/10/01.
Liu BB, Zheng D, Liu YK, Kang XN, Sun L, Guo K, et al. Array-based profiling of the differential methylation status of CpG islands in hepatocellular carcinoma cell lines. Oncol Lett. 2010;1(5):815–20.
Shin SH, Kim BH, Jang JJ, Suh KS, Kang GH. Identification of novel methylation markers in hepatocellular carcinoma using a methylation array. J Korean Med Sci. 2010;25(8):1152–9.
Shen J, Wang S, Zhang YJ, Kappil M, Wu HC, Kibriya MG, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012;55(6):1799–808. Epub 2012/01/12.
Shen J, Wang S, Zhang YJ, Wu HC, Kibriya MG, Jasmine F, et al. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium HumanMethylation 450 BeadChips. Epigenetics. 2013;8(1):34–43.
Kuo CC, Lin CY, Shih YL, Hsieh CB, Lin PY, Guan SB, et al. Frequent methylation of HOXA9 gene in tumor tissues and plasma samples from human hepatocellular carcinomas. Clin Chem Lab Med. 2014;52(8):1235–45.
Kuo CC, Shih YL, Su HY, Yan MD, Hsieh CB, Liu CY, et al. Methylation of IRAK3 is a novel prognostic marker in hepatocellular carcinoma. World J Gastroenterol. 2015;21(13):3960–9.
Mansouri A, Hallonet M, Gruss P. Pax genes and their roles in cell differentiation and development. Curr Opin Cell Biol. 1996;8(6):851–7.
Hellwinkel OJ, Kedia M, Isbarn H, Budaus L, Friedrich MG. Methylation of the TPEF- and PAX6-promoters is increased in early bladder cancer and in normal mucosa adjacent to pTa tumours. BJU Int. 2008;101(6):753–7.
Moelans CB, Verschuur-Maes AH, van Diest PJ. Frequent promoter hypermethylation of BRCA2, CDH13, MSH6, PAX5, PAX6 and WT1 in ductal carcinoma in situ and invasive breast cancer. J Pathol. 2011;225(2):222–31.
Yang Q, Shao Y, Shi J, Qu Y, Wu K, Dang S, et al. Concomitant PIK3CA amplification and RASSF1A or PAX6 hypermethylation predict worse survival in gastric cancer. Clin Biochem. 2014;47(1-2):111–6.
Zhang X, Yang X, Wang J, Liang T, Gu Y, Yang D. Down-regulation of PAX6 by promoter methylation is associated with poor prognosis in non small cell lung cancer. Int J Clin Exp Pathol. 2015;8(9):11452–7.
Feng Q, Stern JE, Hawes SE, Lu H, Jiang M, Kiviat NB. DNA methylation changes in normal liver tissues and hepatocellular carcinoma with different viral infection. Exp Mol Pathol. 2010;88(2):287–92. Epub 2010/01/19.
Schafer BW. Emerging roles for PAX transcription factors in cancer biology. Gen Physiol Biophys. 1998;17(3):211–24.
Shyr CR, Tsai MY, Yeh S, Kang HY, Chang YC, Wong PL, et al. Tumor suppressor PAX6 functions as androgen receptor co-repressor to inhibit prostate cancer growth. Prostate. 2010;70(2):190–9.
Zhao X, Yue W, Zhang L, Ma L, Jia W, Qian Z, et al. Downregulation of PAX6 by shRNA inhibits proliferation and cell cycle progression of human non-small cell lung cancer cell lines. PLoS One. 2014;9(1), e85738.
Deng YB, Nagae G, Midorikawa Y, Yagi K, Tsutsumi S, Yamamoto S, et al. Identification of genes preferentially methylated in hepatitis C virus-related hepatocellular carcinoma. Cancer Sci. 2010;101(6):1501–10.
Shitani M, Sasaki S, Akutsu N, Takagi H, Suzuki H, Nojima M, et al. Genome-wide analysis of DNA methylation identifies novel cancer-related genes in hepatocellular carcinoma. Tumour Biol. 2012;33(5):1307–17.
Liang TJ, Heller T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl 1):S62–71.
Koike K. Hepatitis C, virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J Gastroenterol Hepatol. 2007;22 Suppl 1:S108–11.
Matsukura S, Soejima H, Nakagawachi T, Yakushiji H, Ogawa A, Fukuhara M, et al. CpG methylation of MGMT and hMLH1 promoter in hepatocellular carcinoma associated with hepatitis viral infection. Br J Cancer. 2003;88(4):521–9.
Lim JS, Park SH, Jang KL. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012;321(2):154–61.
Park SH, Lim JS, Lim SY, Tiwari I, Jang KL. Hepatitis C virus Core protein stimulates cell growth by down-regulating p16 expression via DNA methylation. Cancer Lett. 2011;310(1):61–8.
Liu W, Li X, Chu ES, Go MY, Xu L, Zhao G, et al. Paired box gene 5 is a novel tumor suppressor in hepatocellular carcinoma through interaction with p53 signaling pathway. Hepatology. 2011;53(3):843–53.
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28(8), E32. Epub 2000/03/29.
Acknowledgements
The authors would like to thank the Taiwan Liver Cancer Network (TLCN) for providing the hepatocellular carcinoma tissue samples and related clinical data (all are anonymous) for their research work. This work was supported in part by the following Grants: MOST 104-2314-B-016-047, MOST 104-2321-B-016-003, and MOST 103-2314-B-016-008 from the Ministry of Science and Technology, Taiwan; MAB-104-004; MAB-104-005; MAB-104-006; MAB-105-001; MAB-105-002; MAB-105-003 from the Ministry of National Defense, Taiwan. This work was also supported in part by the Liver Disease Prevention and Treatment Research Foundation, Taiwan, Republic of China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SYL and KCC performed the majority of the experiments. YMD and LYW provided the vital reagents and analytical tools and also helped to edit the manuscript. HCB and HTY coordinated and provided the collection of the human resources and also provided financial support for this work. SYL and KCC designed the study and wrote the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1: Table S1.
Top 50 abnormally methylated genes in HCC with different etiologic factors. (PDF 27.4 kb)
Additional file 2: Table S2.
Genes in DAVID ontology that abnormally methylated in HBV, HCV, and NBC-HCC. (PDF 6.77 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shih, YL., Kuo, CC., Yan, MD. et al. Quantitative methylation analysis reveals distinct association between PAX6 methylation and clinical characteristics with different viral infections in hepatocellular carcinoma. Clin Epigenet 8, 41 (2016). https://doi.org/10.1186/s13148-016-0208-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-016-0208-3