
Abstract
Objective
miR-497/195, located at 17p13.1, is a highly conserved miRNA cluster whose abnormal expression is a key regulator of carcinogenesis. We performed a comprehensive analysis of the miR-497/195 cluster to determine its prognostic utility and role in cervical cancer (CC) using publicly available datasets.
Results
In silico analysis and validation revealed that this cluster is downregulated in CC. A total of 60 target genes of miR-497/195 cluster were identified as differentially expressed between normal and CC samples. ShinyGO, STRING, CytoHubba, Timer 2.0, HPA, and HCMBD were used for functional enrichment, PPIN network construction, hub gene identification, immune infiltration correlation, histopathological expression, and determination of the metastatic potential of miR-497/195 cluster and their target genes. PPIN analysis identified CCNE1, CCNE2, ANLN, RACGAP1, KIF23, CHEK1, CDC25A, E2F7, CDK1, and CEP55 as the top 10 hub genes (HGs). Furthermore, the upregulation of RECK, ATD5, and BCL2, downregulation of OSBPL3, RCAN3, and HIST1H3H effected overall survival of CC patients. We identified 6 targets (TFAP2A, CLSPN, RASEF, HIST1H3H, AKT3, and ITPR1) of miR-497/195 cluster to influence metastasis. In addition, 8 druggable genes and 38 potential drugs were also identified. Our study identified miR-497/195 cluster target genes and pathways that could be used for prognostic and therapeutic applications in CC.
Similar content being viewed by others

Introduction
Cervical cancer (CC) remains the leading cancer among female populations in developing countries [1, 2]. In 2022, approximately 661,021 new cases and 348,189 deaths due to CC were reported [3]. Multiple risk factors, such as human papillomavirus (HPV) infection, exposure to tobacco-based products, use of contraceptives, and unhealthy sexual habits, drive CC mortality [1, 4]. Previous genome-wide and mechanistic studies have indicated the role of abnormal genetic and epigenetic changes in CC pathology [5, 6]. The high mortality rate in CC and late detection suggests the need for epigenetic/genetic biomarkers that can be employed for the diagnosis and prognosis of CC [7].
The miR-497/195 cluster located at chromosome 17p13.1 encodes miR-497-5p and miR-195-5p in humans [8]. Clinical and functional studies revealed that this cluster is underexpressed in breast cancer [8], colorectal cancer [9], hepatocellular carcinoma [10], pancreatic cancer [11], ovarian cancer [12], and lymphomas [13]. However, gliomas and chronic lymphocytic leukemia (CLL) exhibit significant overexpression of miR-497, suggesting a possible oncogenic role [14]. In CC, serum miR-497 and miR-195 are proposed as biomarkers to distinguish CC and cervical intraepithelial neoplasia (CIN) patients from healthy individuals [15].
Genome-wide studies are invaluable data for studying molecular events during cancer development and progression [6, 16]. Previous studies have shown that the reanalysis of genome-wide data available in the public domain can identify markers for the clinical management of cancers [17]. The reanalysis of big data via an integrated systems biology approach and validation has been shown to identify molecular markers with high sensitivity and specificity for the diagnosis and prognosis of CC [18, 19]. We and others have demonstrated that the reanalysis of genome-wide studies may identify novel genes and pathways for the clinical management of CC [20, 21]. However, such studies related to miRNA clusters are limited in CC.
The current study aimed to investigate the diagnostic, prognostic, and functional role of the miR-497/195 cluster in CC using an integrated systems biology approach. Most previous studies focused on the functional investigation of individual miRNAs rather than the entire cluster. The advantages of studying the miR-497/195 cluster include a comprehensive understanding of the coordinated regulation and functional interplay between these miRNAs, offering deeper insights into cancer development. Further, this approach could reveal synergistic effects between miRNAs, uncover novel regulatory networks, and identify effective therapeutic targets compared to studying single miRNAs. We initially analysed cluster expression in normal and CC tissue samples from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas-Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma (TCGA-CESC) datasets and subsequently validated the results using a previous study published by us [22]. Next, the target gene interactomes specific to the miR-497/195 cluster were constructed, and their clinical significance was characterized through functional enrichment analyses. Finally, we identified potentially druggable genes targetted by miR-497/195 cluster.
Materials and methods
Data source and identification of the expression of miR-497/195 cluster
The gene expression values of miR-497-5p and miR-195-5p were obtained from the TCGA-CESC and GEO portal (http://www.ncbi.nlm.nih.gov/gds/). The TCGA-CESC dataset consisted of 3 normal and 306 tumor samples. The GEO included the GSE55478 [23] and GSE86100 [24] datasets, which consisted of 10 normal and 10 tumor samples. Differential expression analysis of the clusters in the TCGA-CESC dataset was performed using RNA-seq data (Supplementary Material 2) from UCSC-Xena (https://xenabrowser.net/) [25]. Datasets obtained from GEO were queried using their accession IDs in the GEO portal. The resulting sample data (GSM) obtained were assigned to normal and tumor categories to determine the differential expression of the miR-497/195 cluster using the GEO2R tool. The differentially expressed miRNAs were considered statistically significant if p < 0.05.
Validation of miR-497/195 cluster expression
The findings of the miR-497-5p and miR-195-5p expression were cross-validated using small RNA sequencing of normal (n = 15) and tumor (n = 15) cervical tissue samples published previously by us [22]. To further support our data, we performed a literature search to identify the interaction of miR-497-5p and miR-195-5p with target genes identified in the study.
The materials and methods used in the study are available in Supplementary Material 1.
Results
The miR-497/195 cluster is downregulated in CC
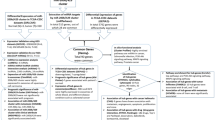
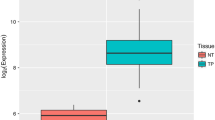
The overall workflow of the study is represented in Fig. 1. The data retrieved from the GEO and TCGA-CESC datasets were evaluated for differential expression of miR-497-5p and miR-195-5p in CC. miR-497-5p and miR-195-5p were significantly downregulated in the GSE55478, GSE84100, and TCGA-CESC datasets (Fig. 2A–C). The cluster downregulation was further confirmed by analysing the expression of the miR-497/195 cluster from our previously published small RNA sequencing data (Fig. 2D) as tabulated in Supplementary Table 1. Supplementary Fig. 1 summarizes the correlation between the cluster and clinicopathological data. The cluster was found to be evolutionarily conserved across vertebrate genomes (Supplementary Fig. 2A). Interestingly, the downregulation of the cluster was significantly correlated with features such as computed tomography (CT) score, pathology, ethnicity, and sample type (Supplementary Fig. 2B and C).

Schematic representation of workflow of in silico analysis

Expression analysis of hsa-miR-497-5p and hsa-miR-195-5p in the datasets. A and B Differentially downregulated miR-497-5p and miR-195-5p (normal vs tumor) in the GSE55478 and GSE86100 datasets, respectively. C Differential expression of the miR-497/miR-195 cluster in the TCGA-CESC dataset. D Validation of the expression of miR-497-5p and miR-195-5p via small RNA sequencing revealed the downregulation of both miRNAs in CC samples
Construction of the miR-497/195 cluster and identification of target genes
CoMeTa tool was used to identify miR-15b, miR-15a, miR-16, miR-195, miR-107, miR-424, and miR-503 as potential coexpressed miRNAs (Fig. 3A). Figure 3B displays the interactome of the miR-497/195 cluster (shown in blue) with its target genes (yellow), lncRNAs (red), sncRNAs (green), and circRNAs (pink), as identified by miRNet (3930 nodes). Analysis of the TCGA-CESC dataset using TACCO database revealed 2020 DEGs (802 upregulated and 1218 downregulated; ± twofold, p < 0.05) (Supplementary Table 2). Among these, 646 genes were identified as targets of the miR-497/195 cluster (Supplementary Table 3). Furthermore, the overlapping analysis identified 60 differentially expressed target genes (DETGs) in CC (Supplementary Table 4). Among the 60 DETGs, 27 were upregulated and 26 were downregulated targets of miR-497-5p, whereas 22 were upregulated and 25 were downregulated targets of miR-195-5p in CC. Furthermore, 40 DETGs were identified as common targets for both miRNAs (18 upregulated and 22 downregulated) (Supplementary Table 4).

The network and interactome of miR-497/195 cluster. A miRNA‒miRNA interactions as predicted by CoMeTa. B Represents the interactome of miR-497/195 cluster (Blue) and its target genes (Yellow), lncRNAs (Red), sncRNAs (Green), and circRNAs (Pink) as identified by miRNet. C Represents the PPIN of 60 DETGs that are targeted by miR-497/195 cluster displaying 57 nodes and 69 edges. D Represents the pathway enrichment analysis of miR-497/195 cluster target genes
PPIN and functional enrichment analysis
The PPIN of the 60 DETGs comprised 57 nodes, 69 edges, and an interaction enrichment with a p value = 5.94e-13 (Fig. 3C). Pathway enrichment analyses of DETGs revealed that the p53 signaling pathway, microRNAs involved in cancer, the cell cycle, EGFR tyrosine kinase inhibitor resistance, gastric cancer, cellular senescence, oocyte meiosis, and Cushing syndrome were the top 10 enriched KEGG pathways (Fig. 3D). Gene Ontology for biological process enrichment included regulation of retinal cell programmed cell death, assembly of the actomyosin apparatus involved in cytokinesis, actomyosin contractile ring assembly, positive regulation of cell migration involved in sprouting angiogenesis, mitotic cytokinesis, embryonic limb morphogenesis, embryonic appendage morphogenesis, sprouting angiogenesis, appendage morphogenesis, and limb morphogenesis (Supplementary Fig. 3A). Cell component enrichment included cyclin E1-CDK2 complex, central spindlin complex, muscle thin filament tropomyosin, cyclin B1-CDK2 complex, cyclin E2-CDK2 complex, bleb, perinuclear endoplasmic reticulum, Flemming body, striated muscle thin filament, and myofilament (Supplementary Fig. 3B).
Similarly, the enriched molecular functions included histone kinase activity, structural constituent of muscle, DNA-binding transcription repressor activity-RNA polymerase II specific, DNA-binding transcription repressor activity, DNA-binding transcription activator activity-RNA polymerase II specific, DNA-binding transcription activator activity, and identical protein binding (Supplementary Fig. 3C).
Prognostic significance of miR-497/195 cluster in CC
The analysis of the 60 DETGs in CC revealed 6 prognostically potential genes (RCAN3, RECK, OSBPL3, ATD5, BCL2, and HIST1H3H) (Fig. 4A). The altered expression of these genes affected the OS of CC patients (Supplementary Fig. 4A). The downregulation of miR-497-5p but not miR-195-5p affected overall survival (Fig. 4B, C). Additionally, 10 DETGs (RECK, HOXA10, PTPRD, KDR, TPM1, MYB, AXIN2, NEGR1, TMEM100, and SLC229A2) were associated with DFS in CC patients. Using the Random Forest model, the OS of DETGs predicted 139 high-risk and 152 low-risk CC with a specificity and sensitivity of 0.92 and 0.94, respectively (Fig. 4D, E). Additionally, the DFS of DETGs identified 150 high-risk and 138 low-risk CC with a specificity of 0.88 and a sensitivity of 0.94 (Supplementary Figs. 5B and C). Furthermore, the model predicted N-stage: N0 (138 entries) and N1, N2, N3 (55 entries) with a specificity of 0.9 and a sensitivity of 0.99 (Supplementary Fig. 5D). Similarly, the model predicted T-stage: T1, T2 (217 entries) and T3, T4 (25 entries) with a specificity of 0.81 and a sensitivity of 1 (Supplementary Fig. 5E).

Identification of survival-significant genes. A Differential expression of 6 genes associated with overall survival. B and C Kaplan‒Meier survival plot of miR-497-5p (significant) and miR-195-5p (nonsignificant). D The survival probability of 60 DETGs for high-risk (red line) and low-risk (blue line) patients. E The sensitivity and specificity of the prognostic model predicted by the random forest method. F OS predicted for Hub Genes by SurvExpress. G Heatmap of Hub gene expression in CC
Identification of DETGs associated with metastasis
Next, we evaluated the associations between the DETGs and metastatic potential. Among the 60 DETGs, genes such as TFAP2A, CLSPN, RASEF, HIST1H3H, AKT3, and ITPR1 were associated with metastasis to secondary sites, including the lungs, and with head and neck carcinoma (Supplementary Table 5). The expression of metastatic genes and their association with OS were verified using GEPIA2 (Supplementary Fig. 5). Interestingly, we found that HIST1H3H has significant prognostic value for OS and metastasis in CC patients.
Identification of Hub genes and functional enrichment analysis
The top 10 highly connected genes identified in our in silico analysis included CCNE1, CCNE2, ANLN, RACGAP1, KIF23, CHEK1, CDC25A, E2F7, CDK1, and CEP55 (Supplementary Fig. 6A). Among that, 9 HGs were experimentally validated targets for miR 497/195 cluster identified using MirTarBase (Supplementary Table 6). Pathway enrichment analysis of HGs using Enrichr (https://maayanlab.cloud/Enrichr/) against the KEGG pathway revealed cell cycle regulation, cellular senescence, the p53 signalling pathway, viral carcinogenesis, regulation of miRNAs in cancer, and other functions as enriched (Supplementary Fig. 6B). The BP, Cell Comp, and MF enrichment of the HG-related genes are provided in Supplementary Fig. 6C–E. Briefly, the enriched BPs included mitotic cytokinesis, protein kinase activity, G1/S-cell cycle transition, mitotic spindle mid-zone assembly, and regulation of cytokinesis. Enriched MFs included cyclin-dependent serine/threonine kinase regulation, histone-threonine kinase activity, microtube binding, RNA polymerase II CTD heptapeptide kinase activity, and tubulin activity. The enriched Cell Comp were cyclin-dependent kinase holoenzyme complex, serine-threonine protein kinase complex, mitotic spindle, nucleus, bleb, microtubules, intracellular membrane, cell-cortex region, and condensed nuclear chromosome.
HG expression and its prognostic application
SurvExpress analysis revealed that the expression of the 10 HGs can affect the OS of patients (p = 0.0005753 and hazard ratio = 3.18) (Fig. 4F, G). The expression of HGs at the protein level in normal and tumor samples was analysed with the HPA tool. Among the 10 HGs, protein expression data were available for only 6 genes (CCNE1, CCNE2, ANLN, RACGAP1, CDK1, and CEP55). The protein expression data were in concordance with the TCGA-CESC hub gene expression data (Fig. 5A). Furthermore, analysis of the role of HGs in immune infiltration using TIMER 2.0 revealed that abnormal expression of HGs may predict CD4 + T cell, CD8 + T cell, and neutrophil infiltration in CC (Supplementary Fig. 7 and Supplementary Table 7).

Identification of protein expression and drug-gene interaction network analysis. A Differential expression of Hub genes at the mRNA level is represented as violin plots, whereas the protein levels are represented as immunohistochemical images. B STITCH analysis depicting the interactions between the 8 target genes and 38 drugs. C STITCH analysis showing the drug-gene interactions of the 10 hub genes. D Gene-score vs. drug-score graph showing the status of drug regimens interacting with 8 target genes. E Classification of drug-target genes interacting by families. F Pie chart showing the drug approval status
Identification of drug–gene interactions
Drug-gene interaction analysis of 60 DETGs in the cluster revealed 38 drugs that interact with 8 target genes, namely, KDR, MYB, CCNE1, CHEK1, RACGAP1, AKT3, BCl2, and FGF2 (Supplementary Table 8). STITCH network analysis of all 8 target genes and interacting drugs revealed a network of 15 nodes and 64 edges (Fig. 5B). The 10 HGs, along with their interacting drug partners, provided 10 nodes and 22 edges (Fig. 5C). We found that few genes were targeted by multiple drugs, such as BCL2 (cisplatin, paclitaxel, etoposide, vincristine, and doxorubicin), CHEK1 (cisplatin, gemcitabine, etoposide, and palbociclib), and MYB (paclitaxel and doxorubicin). Among these drugs, only the gemcitabine-cisplatin combination was found to be approved for CC treatment. Consistent with this observation, PanDrugs analysis identified other potential drugs targeting these genes as either experimental drugs or drugs under clinical trials (Fig. 5D–F).
Discussion
Despite advancements in early detection and availability of the HPV vaccine, CC is a leading gynecological cancer in many underdeveloped and developing countries [2, 4]. The high incidence and mortality of CC may be due to (i) a lack of active implementation of cancer screening programs for early detection, (ii) knowledge, attitudes, and practices related to CC, and (iii) molecular determinants responsible for CC [7]. The availability of genome-wide data in the public domain and advances in the field of bioinformatics have provided an opportunity to reanalyze these data to understand the critical genes and pathways responsible for carcinogenesis and to translate the findings for improved management of cancer patients.
MicroRNAs are a class of noncoding RNAs critical for regulating the expression of protein-coding genes. Several reports have shown that abnormal expression of miRNAs is a critical event during CC [22, 26]. Furthermore, profiling the abnormally expressed miRNAs can be used for diagnostic and prognostic applications in CC. Our previous study demonstrated that integrated bioinformatic analysis can identify miRNAs as diagnostic and prognostic biomarkers and revealed their role in disease-causing mechanisms in conditions such as cancers [19].
Our study showed that miR-497-5p and miR-195-5p were downregulated in CC and may promote cervical carcinogenesis by targeting cellular senescence, the p53 signaling pathway, the cell cycle, and EGFR tyrosine kinase inhibitor resistance. The downregulation of miR-497-5p expression correlated with poor OS. Our in silico analysis using three CC datasets followed by experimental validation from our small RNA-seq data confirmed previous findings that suggested the tumor-suppressor function of the cluster in CC [27,28,29]. However, HCMDB analysis revealed that miR-497/195 cluster expression was not significantly altered between metastatic and nonmetastatic CC. Thus, our in silico analysis identified several known and novel miR-497/195 cluster interactomes with diagnostic and prognostic potential in CC.
A study by Zhang et al. 2015 demonstrated that circulating miR-16–2*, miR-195, miR-2861, and miR-497 can be useful in distinguishing normal from cervical intraepithelial neoplasia (CIN) and CC [15]. This finding suggested that measuring the circulating levels of miRNAs in the cluster might be a reliable diagnostic tool. Although miR-195 and miR-497 are coexpressed, most studies have investigated their functions individually rather than as clusters. For example, a study by Chen and coworkers confirmed that miR-497-5p inhibits CC proliferation by inducing cell cycle arrest by targeting the CBX4 gene [27]. Previous studies using clinical samples and cell lines have reported the tumor growth suppressive functions of the miR-497/195 cluster in CC. In SiHa and HeLa cells, overexpression of miR-497 inhibited the growth, invasion, and migration of cells by activating caspase-mediated apoptosis and inhibiting the insulin-like growth factor 1 receptor [30]. Similarly, miR-497 is known to regulate the expression of FASN [31] and CBX4 [27] in CC. It has also been reported that the HPV-encoded E6 oncoprotein can target the KDM5C/lnc_000231/miR-497-5p/CCNE1 axis to stimulate CC progression [32]. Our in silico analysis of the miR-497/195 cluster interactome also identified CCNE1 as one of the HGs.
miR-195-5p is another downregulated member of the miR-497/195 cluster in CC. miR-195-5p targets MMP14 to suppress CC proliferation and invasion by inhibiting TNF signaling pathways [33]. Furthermore, miR-195-5p prevents malignant progression by inhibiting ATG9A [34] and YAP1-mediated EMT in CC [29]. Furthermore, modulation of the miR-195-5p/MAPK axis contributed to the proliferation and migration of CC cells [32]. Taken together, various functional studies indicate that the downregulation of miR-195-5p and miR-497-5p fuels the growth, migration, proliferation, and invasion of CC cells.
Our study identified 60 differentially expressed target genes and a subclass of hub genes whose upregulation predicted CC prognosis and metastasis. Our risk prediction model using the random forest approach successfully revealed that the 60 DETGs of the miR-497/195 cluster could differentiate 134 high-risk and 136 low-risk individuals with a test specificity of 0.92 and sensitivity of 0.94, suggesting that the DETGs could have a powerful prognostic function in determining high/low-risk CC. Among the 60 DETGs in the cluster, RCAN3, RECK, OSBPL3, ATD5, BCL2, and HIST1H3H affected the OS of CC patients, suggesting the prognostic significance of these genes in CC. Furthermore, analysis of the PPIN identified CCNE1, CCNE2, ANLN, RACGAP1, KIF23, CHEK1, CDC25A, E2F7, CDK1, and CEP55 as the key HGs. All the HGs were significantly upregulated in CC samples in the TCGA-CESC cohort. Notably, the overexpression of a few HGs was associated with OS and DFS as well as the induction of metastatic phenotypes. KM survival analysis revealed that the overexpression of HGs can significantly affect OS among high-risk and low-risk CC patients, with patients with lower HG expression exhibiting better survival than patients with higher HG expression. Thus, testing the expression of the miR-497/195 cluster and its HGs may be useful for predicting the diagnosis, prognosis, and metastasis of CC patients.
To further support our data, we performed a literature search to identify the interaction of miR-497-5p and miR-195-5p with target genes identified in the study. For example, targeting of CCNE1 by miR-497 to suppress cervical cancer cell proliferation is reported [32]. An indirect association between miR-497and CDC25A mediated by LNC00662 is reported by Wei et al. 2020 [35]. Likewise, a direct interaction between miR-195-5p and Clusterin (CLU) is reported in prostate cancer cells through luciferase reporter assay [36]. Similarly targeting of HOXA10 by miR-195-5p and an inverse relationship between them is reported in lung adenocarcinoma [37]. miR-195-5p is shown to regulate AXIN2 in colorectal cancer cells [38]. BCL2 targeting by miR-497 in A549 is reported in lung adenocarcinoma cells [39]. miR-195/ FGF axis and miR-195 as a negative regulator of FGF are reported in prostate cancer cells [40], hepatocellular carcinoma [41], and thyroid cancer [42]. We found a study that reported overexpression of miR-195 inhibits cervical cancer progression by targeting CCND1 and MYB [43]. While few studies support our target predictions with experimental validation, some of the DETGs identified in our study lack validation in cervical cancer. Collectively, our data suggests that miR-497/195 has the potential to target DETGs and requires further validation.
Resistance to therapy is a major concern in cancer treatment, and recurrence affects the quality of life of individuals [44, 45]. Thus, understanding the molecular mechanisms leading to drug resistance is critical for improving the management of CC patients. To identify druggable genes and repurpose drugs, we performed a drug‒gene interaction analysis and identified 38 potential drugs targeting 8 genes. Although several of these drugs, except for the gemcitabine-cisplatin combination, are approved for CC management, the nonconventional drugs identified either as experimental candidates or in clinical trials serve as targets for drug repurposing for better therapeutic outcomes in CC. Our study collectively identified the clinical relevance and significance of the miR-497/195 cluster and its target network in terms of its diagnostic and prognostic applications using an array of statistical tests and bioinformatic predictions, offering novel insights for developing functional studies.
Limitations
-
•
An independent validation of the miR-497/195 cluster was performed with only 15 normal and 15 tumor samples, and the TCGA CESC dataset included only 3 normal samples. Though these samples were able to capture the differences in cluster expressions, a more detailed investigation using a large number of clinical samples is needed before further conclusions are drawn.
-
•
Our study used public datasets and web-based tools to perform the analysis. Although we used only those gene targets that were experimentally validated through different independent studies from target identification databases, many other target genes might be targeted by this miRNA cluster which might need further functional validations.
-
•
While this study provides comprehensive details, limitations about the in-silico nature of the study should be considered by the scientific community to conduct specific studies to validate and confirm these results through further experimental validation.
Data availability
All the data generated or analysed during this study are included in this published article and its supplementary files. The TCGA-CESC miRNA and mRNA data are included in the Additional Data. No datasets were generated or analysed during the current study.
Abbreviations
- NGS:
-
Next-generation sequencing
- PI3/Akt:
-
Phosphatidylinositol 3-kinase/protein kinase B
- EGFR:
-
Epidermal growth factor receptor
- DNA:
-
Deoxyribonucleic acid
- CCNE1 :
-
G1/S-specific cyclin-E1
- CCNE2 :
-
G1/S-specific cyclin-E2
- ANLN :
-
Anillin actin binding protein
- RACGAP1 :
-
Rac GTPase activating protein 1
- KIF23 :
-
Kinesin family member 23
- CHEK1 :
-
Checkpoint kinase1
- CDC25A :
-
Cell division cycle25a
- E2F7 :
-
E2F Transcription factor 7
- CDK1 :
-
Cyclin-dependent kinase1
- CEP55 :
-
Centrosomal Protein 55
- RCAN3 :
-
RCAN family member3 < RCAN-regulator of calcineurin >
- RECK :
-
Reversion-inducing cysteine-rich protein with Kazal motifs
- OSBPL3 :
-
Oxysterol-binding protein-related protein 3
- ATD5 :
-
Asphyxiating thoracic dystrophy 5
- BCL2 :
-
B-cell lymphoma 2
- HIST1H3H:
-
Histone cluster 1 H3 family member H
- HOXA10 :
-
Homeobox A10
References
Arbyn M, Weiderpass E, Bruni L, de Sanjose S, Saraiya M, Ferlay J, Bray F. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health. 2020;8(2):e191–203.
Brisson M, Kim JJ, Canfell K, Drolet M, Gingras G, Burger EA, Martin D, Simms KT, Benard E, Boily MC, et al. Impact of HPV vaccination and cervical screening on cervical cancer elimination: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet. 2020;395(10224):575–90.
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–63.
Singh D, Vignat J, Lorenzoni V, Eslahi M, Ginsburg O, Lauby-Secretan B, Arbyn M, Basu P, Bray F, Vaccarella S. Global estimates of incidence and mortality of cervical cancer in 2020: a baseline analysis of the WHO global cervical cancer elimination initiative. Lancet Glob Health. 2023;11(2):e197–206.
Bowden SJ, Kalliala I, Veroniki AA, Arbyn M, Mitra A, Lathouras K, Mirabello L, Chadeau-Hyam M, Paraskevaidis E, Flanagan JM, et al. The use of human papillomavirus DNA methylation in cervical intraepithelial neoplasia: a systematic review and meta-analysis. EBioMedicine. 2019;50:246–59.
Adebamowo SN, Adeyemo AA, Rotimi CN, Olaniyan O, Offiong R, Adebamowo CA, Group HAAR. Genome-wide association study of prevalent and persistent cervical high-risk human papillomavirus (HPV) infection. BMC Med Genet. 2020;21(1):231.
Canfell K, Kim JJ, Brisson M, Keane A, Simms KT, Caruana M, Burger EA, Martin D, Nguyen DTN, Benard E, et al. Mortality impact of achieving WHO cervical cancer elimination targets: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet. 2020;395(10224):591–603.
Li D, Zhao Y, Liu C, Chen X, Qi Y, Jiang Y, Zou C, Zhang X, Liu S, Wang X, et al. Analysis of MiR-195 and MiR-497 expression, regulation and role in breast cancer. Clin Cancer Res. 2011;17(7):1722–30.
Pidikova P, Reis R, Herichova I. miRNA clusters with down-regulated expression in human colorectal cancer and their regulation. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21134633.
Furuta M, Kozaki K, Tanimoto K, Tanaka S, Arii S, Shimamura T, Niida A, Miyano S, Inazawa J. The tumor-suppressive miR-497-195 cluster targets multiple cell-cycle regulators in hepatocellular carcinoma. PLoS ONE. 2013;8(3):e60155.
Xu J, Wang T, Cao Z, Huang H, Li J, Liu W, Liu S, You L, Zhou L, Zhang T, et al. MiR-497 downregulation contributes to the malignancy of pancreatic cancer and associates with a poor prognosis. Oncotarget. 2014;5(16):6983–93.
Wang W, Ren F, Wu Q, Jiang D, Li H, Shi H. MicroRNA-497 suppresses angiogenesis by targeting vascular endothelial growth factor a through the PI3K/AKT and MAPK/ERK pathways in ovarian cancer. Oncol Rep. 2014;32(5):2127–33.
Hoareau-Aveilla C, Quelen C, Congras A, Caillet N, Labourdette D, Dozier C, Brousset P, Lamant L, Meggetto F. miR-497 suppresses cycle progression through an axis involving CDK6 in ALK-positive cells. Haematologica. 2019;104(2):347–59.
Yang G, Xiong G, Cao Z, Zheng S, You L, Zhang T, Zhao Y. miR-497 expression, function and clinical application in cancer. Oncotarget. 2016;7(34):55900–11.
Zhang Y, Zhang D, Wang F, Xu D, Guo Y, Cui W. Serum miRNAs panel (miR-16-2*, miR-195, miR-2861, miR-497) as novel non-invasive biomarkers for detection of cervical cancer. Sci Rep. 2015;5:17942.
Bowden SJ, Bodinier B, Kalliala I, Zuber V, Vuckovic D, Doulgeraki T, Whitaker MD, Wielscher M, Cartwright R, Tsilidis KK, et al. Genetic variation in cervical preinvasive and invasive disease: a genome-wide association study. Lancet Oncol. 2021;22(4):548–57.
Eswaran S, Padavu M, Kumar D, Kabekkodu SP. Systematic analysis of the therapy resistance genes and their prognostic relevance in cervical cancer. Curr Pharm Des. 2023;29(25):2018–32.
Meneur C, Eswaran S, Adiga D, Sriharikrishnaa S, Nadeem G, Mallya S, Chakrabarty S. Kabekkodu SP analysis of nuclear encoded mitochondrial gene networks in cervical cancer. Asian Pac J Cancer Prev. 2021;22(6):1799–811.
Sriharikrishnaa S, Shukla V, Khan GN, Eswaran S, Adiga D, Kabekkodu SP. Integrated bioinformatic analysis of miR-15a/16–1 cluster network in cervical cancer. Reprod Biol. 2021;21(1):100482.
van Dam PA, van Dam PJ, Rolfo C, Giallombardo M, van Berckelaer C, Trinh XB, Altintas S, Huizing M, Papadimitriou K, Tjalma WA, et al. In silico pathway analysis in cervical carcinoma reveals potential new targets for treatment. Oncotarget. 2016;7(3):2780–95.
Eswaran S, Adiga D, Khan GN. S S, Kabekkodu SP: Comprehensive analysis of the exocytosis pathway genes in cervical cancer. Am J Med Sci. 2022;363(6):526–37.
Shukla V, Varghese VK, Kabekkodu SP, Mallya S, Chakrabarty S, Jayaram P, Pandey D, Banerjee S, Sharan K, Satyamoorthy K. Enumeration of deregulated miRNAs in liquid and tissue biopsies of cervical cancer. Gynecol Oncol. 2019;155(1):135–43.
Park H, Lee MJ, Jeong JY, Choi MC, Jung SG, Joo WD, Lee C, An HJ. Dysregulated microRNA expression in adenocarcinoma of the uterine cervix: clinical impact of miR-363-3p. Gynecol Oncol. 2014;135(3):565–72.
Gao D, Zhang Y, Zhu M, Liu S, Wang X. miRNA expression profiles of HPV-infected patients with cervical cancer in the uyghur population in China. PLoS ONE. 2016;11(10): e0164701.
Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–8.
Kawai S, Fujii T, Kukimoto I, Yamada H, Yamamoto N, Kuroda M, Otani S, Ichikawa R, Nishio E, Torii Y, et al. Identification of miRNAs in cervical mucus as a novel diagnostic marker for cervical neoplasia. Sci Rep. 2018;8(1):7070.
Chen Y, Du J, Wang Y, Shi H, Jiang Q, Wang Y, Zhang H, Wei Y, Xue W, Pu Z, et al. MicroRNA-497-5p induces cell cycle arrest of cervical cancer cells in s phase by targeting CBX4. Onco Targets Ther. 2019;12:10535–45.
Pan SS, Zhou HE, Yu HY, Xu LH. MiR-195-5p inhibits the cell migration and invasion of cervical carcinoma through suppressing ARL2. Eur Rev Med Pharmacol Sci. 2019;23(24):10664–71.
Liu X, Zhou Y, Ning YE, Gu H, Tong Y, Wang N. MiR-195-5p inhibits malignant progression of cervical cancer by targeting YAP1. Onco Targets Ther. 2020;13:931–44.
Luo M, Shen D, Zhou X, Chen X, Wang W. MicroRNA-497 is a potential prognostic marker in human cervical cancer and functions as a tumor suppressor by targeting the insulin-like growth factor 1 receptor. Surgery. 2013;153(6):836–47.
Zhang H, Wang R, Tang X, Li J, Li J, Wang M. FASN targeted by miR-497-5p regulates cell behaviors in cervical cancer. Nutr Cancer. 2022;74(8):3026–34.
Zhang Y, Li X, Zhang J, Mao L. E6 hijacks KDM5C/lnc_000231/miR-497-5p/CCNE1 axis to promote cervical cancer progression. J Cell Mol Med. 2020;24(19):11422–33.
Li M, Ren CX, Zhang JM, Xin XY, Hua T, Wang HB, Wang HB. The effects of miR-195-5p/MMP14 on proliferation and invasion of cervical carcinoma cells through TNF signaling pathway based on bioinformatics analysis of microarray profiling. Cell Physiol Biochem. 2018;50(4):1398–413.
Liu X, Liu Z, Liu Y, Wang N. ATG9A modulated by miR-195-5p can boost the malignant progression of cervical cancer cells. Epigenetics. 2023;18(1):2257538.
Wei J, Wang L, Sun Y, Bao Y. LINC00662 contributes to the progression and the radioresistance of cervical cancer by regulating miR-497-5p and CDC25A. Cell Biochem Funct. 2020;38(8):1139–51.
Ma X, Zou L, Li X, Chen Z, Lin Q, Wu X. MicroRNA-195 regulates docetaxel resistance by targeting clusterin in prostate cancer. Biomed Pharmacother. 2018;99:445–50.
Yuan C, Bai R, Gao Y, Jiang X, Li S, Sun W, Li Y, Huang Z, Gong Y, Xie C. Effects of microRNA-195-5p on biological behaviors and radiosensitivity of lung adenocarcinoma cells via targeting HOXA10. Oxid Med Cell Longev. 2021;2021:4522210.
Gu H, Xu Z, Zhang J, Wei Y, Cheng L, Wang J. circ_0038718 promotes colon cancer cell malignant progression via the miR-195-5p/Axin2 signaling axis and also effect Wnt/beta-catenin signal pathway. BMC Genomics. 2021;22(1):768.
Zhu W, Zhu D, Lu S, Wang T, Wang J, Jiang B, Shu Y, Liu P. miR-497 modulates multidrug resistance of human cancer cell lines by targeting BCL2. Med Oncol. 2012;29(1):384–91.
Liu C, Guan H, Wang Y, Chen M, Xu B, Zhang L, Lu K, Tao T, Zhang X, Huang Y. miR-195 inhibits EMT by targeting FGF2 in prostate cancer cells. PLoS ONE. 2015;10(12): e0144073.
Wang M, Zhang J, Tong L, Ma X, Qiu X. MiR-195 is a key negative regulator of hepatocellular carcinoma metastasis by targeting FGF2 and VEGFA. Int J Clin Exp Pathol. 2015;8(11):14110–20.
Yin Y, Hong S, Yu S, Huang Y, Chen S, Liu Y, Zhang Q, Li Y, Xiao H. MiR-195 inhibits tumor growth and metastasis in papillary thyroid carcinoma cell lines by targeting CCND1 and FGF2. Int J Endocrinol. 2017;2017:6180425.
Du X, Lin LI, Zhang L, Jiang J. microRNA-195 inhibits the proliferation, migration and invasion of cervical cancer cells via the inhibition of CCND2 and MYB expression. Oncol Lett. 2015;10(4):2639–43.
Haider T, Pandey V, Banjare N, Gupta PN, Soni V. Drug resistance in cancer: mechanisms and tackling strategies. Pharmacol Rep. 2020;72(5):1125–51.
Adiga D, Eswaran S, Pandey D, Sharan K, Kabekkodu SP. Molecular landscape of recurrent cervical cancer. Crit Rev Oncol Hematol. 2021;157: 103178.
Chou PH, Liao WC, Tsai KW, Chen KC, Yu JS, Chen TW. TACCO, a database connecting transcriptome alterations, pathway alterations and clinical outcomes in cancers. Sci Rep. 2019;9(1):3877.
Huang HY, Lin YC, Cui S, Huang Y, Tang Y, Xu J, Bao J, Li Y, Wen J, Zuo H, et al. miRTarBase update 2022: an informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022;50(D1):D222–30.
Chang L, Zhou G, Soufan O, Xia J. miRNet 2.0: network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020;48(W1):W244–51.
Gennarino VA, D’Angelo G, Dharmalingam G, Fernandez S, Russolillo G, Sanges R, Mutarelli M, Belcastro V, Ballabio A, Verde P, et al. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res. 2012;22(6):1163–72.
Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36(8):2628–9.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014. https://doi.org/10.1186/1752-0509-8-S4-S11.
Thul PJ, Lindskog C. The human protein atlas: a spatial map of the human proteome. Protein Sci. 2018;27(1):233–44.
Zheng G, Ma Y, Zou Y, Yin A, Li W, Dong D. HCMDB: the human cancer metastasis database. Nucleic Acids Res. 2018;46(D1):D950–5.
Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47(W1):W556–60.
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–14.
Litwin TR, Irvin SR, Chornock RL, Sahasrabuddhe VV, Stanley M, Wentzensen N. Infiltrating T-cell markers in cervical carcinogenesis: a systematic review and meta-analysis. Br J Cancer. 2021;124(4):831–41.
Zou P, Yang E, Li Z. Neutrophil-to-lymphocyte ratio is an independent predictor for survival outcomes in cervical cancer: a systematic review and meta-analysis. Sci Rep. 2020;10(1):21917.
Cotto KC, Wagner AH, Feng YY, Kiwala S, Coffman AC, Spies G, Wollam A, Spies NC, Griffith OL, Griffith M. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018;46(D1):D1068–73.
Pineiro-Yanez E, Reboiro-Jato M, Gomez-Lopez G, Perales-Paton J, Troule K, Rodriguez JM, Tejero H, Shimamura T, Lopez-Casas PP, Carretero J, et al. PanDrugs: a novel method to prioritize anticancer drug treatments according to individual genomic data. Genome Med. 2018;10(1):41.
Szklarczyk D, Santos A, von Mering C, Jensen LJ, Bork P, Kuhn M. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44(D1):D380-384.
Acknowledgements
We thank DST-Ph.D. fellowship, KSTePS, DST, Government of Karnataka (Reference ID- DST/KSTePS/Ph.D. Fellowship/LIF-11: 2019-20), and ICMR, the Government of India, for the Senior Research Fellowship (Reference ID- 2020/8704/CMB/BMS) to Ms. Sangavi Eswaran, Yuvaraja's College, and the Manipal Academy of Higher Education (MAHE) for infrastructure support. All the authors thank Manipal Academy of Higher Education, Manipal, Fund for Improvement of S&T Infrastructure (FIST), and Karnataka Fund for Infrastructure Strengthening in Science and Technology (K-FIST), Government of Karnataka.
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal. This work is supported by the MATRICS, Science and Engineering Research Board (SERB) Department of Science and Technology (DST), Government of India (Grant No: MTR/2021/000182) and SPARC grant (SPARC/2019‐2020/P2297/SL).
Author information
Authors and Affiliations
Contributions
S.P.K.: Conceptualization and Formal analysis. S.H.K., and S.E. Data curation, Investigation, Analysis, Validation and Writing—original draft. S.M.: Methodology, and Analysis. S.C., P.S.S., and S.P.K.: Writing—original draft and Review and Editing. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
13104_2024_6876_MOESM1_ESM.pptx
Additional file 1: Figure 1. miR-497 and miR-195 expression analysis was performed using TCGA-UALCAN. A) Expression analysis of miR-497 in normal tissues and CC tissues at cancer stages 1, 2, and 3. B) Different age groups. C) Ethnicity and race. D) Tumor grade. E) Expression analysis of miR-195 in normal tissues and CC tissues at cancer stages 1, 2, and 3. F) Different age groups. G) Ethnicity and race and H) tumor grade.
13104_2024_6876_MOESM2_ESM.pptx
Additional file 2: Figure 2. Conservation and expression analysis of miR-497/195 cluster. A) miR-497 and miR-195 comprise UTRs (yellow blocks) across species. B) & C) Correlation analysis of miR-497 and miR-195 expression from TCGA-CESC datasets and clinical attributes.
13104_2024_6876_MOESM3_ESM.pptx
Additional file 3: Figure 3. Functional enrichment analysis of miR-497/195 cluster target genes. A) Biological Processes, B) Cellular Components, and C) Molecular Functions.
13104_2024_6876_MOESM4_ESM.pptx
Additional file 4: Figure 4. The survival plots of genes associated with A) Overall Survival B)&C) Disease Free Survival and D)& E) Cancer stages.
13104_2024_6876_MOESM6_ESM.pptx
Additional file 6: Figure 6. Identification and characterization of Hub genes. A) Identification of Hub genes using the STRING database and 10 interacting genes (CCNE1, CCNE2, ANLN, RACGAP1, KIF23, CHEK1, CDC25A, E2F7, CDK1, and CEP55). B) Functional enrichment analysis of the hub genes. B) KEGG pathway analysis. GO enrichment of the component Hub genes (C) Cellular component (D) Molecular functions and (E) Biological processes.
13104_2024_6876_MOESM7_ESM.pptx
Additional file 7: Figure 7. miR-497/195 cluster and its immune infiltrates. Spearman infiltration levels of CD8+ T cells, CD4+ T cells, and neutrophils.
13104_2024_6876_MOESM9_ESM.xlsx
Additional file 9. Supplementary Material 2: miRNA and mRNA expression values in normal and tumor samples of the TCGA-CESC dataset from the UCSC-Xena Browser.
13104_2024_6876_MOESM10_ESM.docx
Additional file 10: Table 1. List of differentially expressed miRNA cluster and its members in CC from small RNA sequencing data.
13104_2024_6876_MOESM14_ESM.docx
Additional file 14: Table 5. The differentially expressed target genes of miR-497/195 cluster and their characteristic metastatic signatures.
13104_2024_6876_MOESM15_ESM.xlsx
Additional file 15: Table 6. The mir497/195 cluster and its experimentally validated gene target interactions from miRTarBase database.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Karunakara, S.H., Eswaran, S., Mallya, S. et al. Analysis of miR-497/195 cluster identifies new therapeutic targets in cervical cancer. BMC Res Notes 17, 217 (2024). https://doi.org/10.1186/s13104-024-06876-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-024-06876-8