
Abstract
Background
Fanconi-Bickel syndrome (FBS, OMIM 227810) is a rare autosomal recessive disease caused by a deficiency of glucose transporter 2 (GLUT2), a member of the facilitative glucose transporter family (Santer et al. J Inherit Metab Dis 21:191–194, 1998). The typical clinical picture is characterized by hepatorenal glycogen accumulation resulting in hepato- and nephromegaly, impaired utilization of glucose and galactose, proximal renal tubular dysfunction, rickets and severe short stature.
Case presentation
We report 2 Palestinian patients from 2 families who were homozygous for the mutation p.R301X (C>T) in exon 7of GLUT2 gene. Patient 1 showed clinical and laboratory improvement with age characterized by normal growth and resolution of rickets. Patient 2 had severe phenotype characterized by progressive weight loss, persistent metabolic acidosis, marked polyuria and clinical and laboratory findings of rickets progressing to death at age 10 months.
Conclusion
This report further expands the clinical spectrum of FBS even with identical mutations. Other yet unknown genetic, environmental or stochastic factors may be responsible for phenotypic variability
Similar content being viewed by others


Background
A functional loss of GLUT2 is compatible with the clinical symptoms observed in FBS patients [1]. GLUT2 is expressed in hepatocytes, enterocytes and pancreatic β-cell membranes and is involved in the transcellular monosaccharide transport of renal tubular cells and enterocytes [1, 2]. The main features of this disorder are hepatorenal glycogen accumulation, proximal renal tubular dysfunction, impaired utilization of glucose and galactose, rickets and severe short stature [3]. Biochemically, the disease is characterized by a general renal proximal tubular defect (glucosuria, bicarbonate wasting, aminoaciduria, renal tubular acidosis, hyperphosphaturia) and carbohydrate abnormalities that include postprandial hyperglycemia, fasting hypoglycemia and hypergalactosemia [3, 4]. Mutations of GLUT2 (SLC2A2) are the basic defect in FBS patients with characteristic clinical features and also in patients with atypical clinical signs such as intestinal malabsorption, failure to thrive, the absence of hepatomegaly or renal hyperfiltration [5]. These mutations are scattered over the whole coding sequence of the SLC2A2 gene and none of these mutations is particularly frequent which makes molecular genetic diagnosis laborious [5, 6]. FBS is generally considered a well-defined clinical condition and the overall prognosis seems to be favorable [2, 7]. Phenotypic variability in FBS has been reported and includes atypical phenotype, unusually mild clinical course, severe phenotype and also variable clinical severity in patients with identical mutations [2–8].
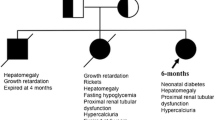
We report the first 2 Palestinian patients with FBS from 2 different families who are unrelated and from different geographic location. They are homozygous for the mutation p.R301X in exon 7 of the GLUT2 gene. Both presented with the typical phenotype of FBS but showed marked variation in the clinical progression of the disease.
Case presentation
Case 1
The 6 year old female was born at term to consanguineous parents after uneventful pregnancy. Birth weight was 2800 g (−1.2 SD), length and occipitofrontal circumference at birth were not recorded. Other 3 male siblings are healthy with no symptoms suggestive of FBS and no other family member with diabetes mellitus. First hospitalization was at age 17 month for evaluation of inadequate weight gain and abdominal distension. Her weight was 6600 g (−5.3 SD), length 69 cm (−3.3 SD) and occipitofrontal circumference 43 cm (−2.6 SD). Physical examination showed hepatomegaly and doll-like face. Laboratory findings included high serum triglycerides, mildly elevated serum aspartate aminotransferase, alanine aminotransferase, low phosphorous and elevated alkaline phosphatase. She had polyuria >6 ml/kg/h, massive generalized aminoaciduria, phosphaturia and glucosuria. Serum ammonia, lactic acid, calcium, parathyroid hormone and 25-hydroxyvitamin D were normal (Table 1). Abdominal ultrasound revealed hepatomegaly and minimal calcification of both renal calyces. Radiological findings included generalized osteopenia and rachitic changes. Genetic analysis showed that the patient is homozygous for the mutation R301X (C>T) in exon 7 of the GLUT2 gene. Treatment included anhydrous phosphate (Joulie’s) solution 5 ml given every 4 h, 5 times daily (0.75 g/24 h), alpha D3 drops (alphacalcidol) 1 µg/day, galactose-restricted diet and uncooked cornstarch 1.6 g/kg given every 6 h during night time to prevent hypoglycemia. Metabolic acidosis was mild and did not require treatment.
The clinical phenotype was characterized by improvement of growth parameters with age. Rickets resolved with normalization of serum phosphate, alkaline phosphatase and triglyceride. She still shows laboratory findings of renal tubular dysfunction including polyuria, massive aminoaciduria, glucosuria and phosphaturia.
At age 6 years, her weight was 18 kg (−0.8 SD) and height 107 cm (−1.6 SD) (Fig. 1). Physical exam showed mild hepatomegaly and renal ultrasound showed resolution of calyceal calcifications.

Showing normal growth of case 1 at age 6 years
Case 2
The patient was the first born child to consanguineous parents at term after uneventful pregnancy at 36 weeks gestation. His birth weight was 2200 g (−2 SD). Length and occipitofrontal circumference at birth were not recorded. No other family members with symptoms suggestive of FBS or other family member with diabetes mellitus. At age 2 months, noted to have inadequate weight gain and enlarged anterior fontanel. Different milk formulas were tried but did not result in weight gain. Hospitalized at age 5 months for evaluation of severe failure to thrive, Hyperlipidemia and metabolic acidosis. His weight was 4500 g (−4.3 SD), length 50 cm (−8.8 SD). Physical examination showed abdominal distension, hepatomegaly and doll-like face. He also had rachitic rosaries and wide wrist joints. Laboratory findings included very low serum phosphate, high serum alkaline phosphatase and triglycerides, mildly elevated serum cholesterol, alanine aminotransferase, aspartate aminotransferase and metabolic acidosis. He had massive generalized aminoaciduria, phosphaturia, proteinuria and glucosuria. Serum calcium, parathyroid hormone and 25-hydroxyvitamin D3 were normal as well as urinary excretion of calcium. Other normal laboratory findings included coagulation studies, serum ammonia and lactic acid (Table 1).
Radiological findings showed generalized osteopenia and rachitic changes in the wrist and knee joints. Genetic analysis also showed that the patient is homozygous for the mutation R301X (C>T) in exon 7 of the GLUT2 gene.
The clinical course was characterized by severe polyuria, inability to gain weight, metabolic acidosis despite treatment with uncooked cornstarch 1.6 g/kg given every 5 h during night time, lactose-free infant formula, oral phosphate supplement at initial dose 0.5 g/24 h given every 4 h, 5 times daily, 1,25-hydroxyvitamin D3 0.5 µg/day and oral sodium bicarbonate supplement 1 mEq/kg given 4 times daily. The medications were given appropriately in terms of both dosage and frequency. Rehospitalized at age 8 months for weight loss, persistent metabolic acidosis and polyuria. Weight was 3.5 kg (−8.9 SD). Indomethacin was added to reduce polyuria but urine output remained persistently elevated >8 ml/kg/h.
The patient died at age 10 months.
Discussion
The clinical picture of FBS has been thought to be rather homogenous and characterized as a combination of hepatomegaly secondary to glycogen accumulation, galactose intolerance, impaired glucose homeostasis with fasting hypoglycemia and postprandial hyperglycemia, proximal tubular nephropathy and, very typically, severely stunted growth [2, 4, 7].
Our patients presented with the typical phenotype described in the introduction. However, a distinctive feature is the wide variability of clinical course showing normal stature and normal physical exam at age 6 years in patient 1 and a rapidly progressive course characterized by weight loss, massive polyuria, metabolic acidosis and persistence of radiological and biochemical signs of rickets progressing to death in patient 2.
Phenotypic variability has been reported in eight patients from a single Bedouin sibship, all were homozygous for the p.R301X mutation. All had failure to thrive and/or hepatomegaly, fluctuations in blood glucose levels and proximal tubular dysfunction evidenced by massive glucosuria, generalized aminoaciduria, hypercalciuria and hyperphosphaturia. Skeletal involvement ranged from none to radiological and/or clinical signs of rickets and osteopenia. A spectrum of diseases severity was evident during follow up regarding the growth parameters, hospitalizations for disease exacerbations, mean amount of electrolyte replacement therapy, and skeletal and renal complications [4]. 3 patients had mild phenotype; two needed small amounts of replacement therapy and one patient did not require replacement therapy since he was under the authors’ follow up at age 16 years. On the other hand, two patients had severe phenotype characterized by hypocalcemic titanic events in one patient and recurrent symptomatic nocturnal hypoglycemia necessitating gastrostomy for continuous night feedings until age 3 years in the other patient.
Our patients had similar phenotype initially and both had clinical and radiological signs of rickets and osteopenia. The main distinction was the rapidly progressive deterioration in patient 2 in our report despite adequate replacement therapy.
The R301X mutation has also been reported in four unrelated families among 33 different SLC2A2 mutations in 49 patients with a clinical diagnosis of FBS [5]. No specific case descriptions was provided and the diagnosis was based on the combination of typical clinical and laboratory signs. The authors postulated that it is premature to discuss a genotype–phenotype correlation in FBS and there is no indication that FBS having missense mutations show a milder clinical course when compared with those having truncated mutations.
Unusually mild phenotype has been described in a 9-year old boy characterized by absence of hepato- and nephromegaly, normal growth, normal glucose tolerance even after glucose loading and very mild glucosuria, aminoaciduria and proteinuria [2]. Another mild phenotype was described in a patient with FBS harboring a novel mutation in the GLUT2 gene detected by neonatal screening for galactosemia. He was found to be homozygous for a 3 bp deletion (425-7/delta) within exon 3. Treatment with phosphate and bicarbonate at age 16 months resulted in marked growth improvement and on a Mediterraniean free diet with fractionated meals, he has never shown symptomatic hypoglycemia [8]. Patient 1 in our report has a much similar phenotype showing improvement of growth parameters and absence of carbohydrate abnormalities.
A significant combined defect of the muscle respiratory chain complexes I, III and IV has been described in an 8 year old boy with FBS who had a novel homozygous base exchange at position IVS5+5 of the GLUT2 gene [7]. The authors hypothesized that FBS patients are prone to a severe secondary respiratory chain defect that could in part explain the heterogeneity of the clinical symptoms. Patient 2 in our report had persistent metabolic acidosis which may be explained by this hypothesis but muscle biopsy for respiratory chain complexes was not performed.
Diabetic ketoacidosis has been described in a female infant with FBS at age 33 days [9]. Although FBS is characterized by carbohydrate abnormalities including posprandial hyperglycemia and fasting hypoglycemia, our patients did not show significant hyperglycemia at presentation or during follow up.
Conclusion
There is an increasingly recognized evidence that FBS is clinically heterogenous. This report further expands the clinical spectrum of FBS even with identical mutations. Other yet unknown genetic, environmental or stochastic factors may be responsible for phenotypic variability.
Abbreviations
- FBS:
-
Fanconi-Bickel syndrome
- GLUT2:
-
glucose transporter 2
- FTT:
-
failure to thrive
References
Santer R, Schneppenheim R, Dombrowski A, Gotze H, Steinmann B, Schaub J. Fanconi-Bickel syndrome—a congenital defect of the liver-type facilitative glucose transporter. J Inherit Metab Dis. 1998;21:191–4.
Grünert SC, Schwab KO, Pohl M, Sass JO, Santer R. Fanconi-Bickel syndrome: GLUT2 mutations associated with mild phenotype. Mol Genet Metab. 2012;105:433–7.
Abbasi F, Azizi F, Javaheri M, Mosallanejad A, Ebrahim-Habibi A, Ghafouri-Fard S. Segregation of a novel homozygous 6 nucleotide deletion in GLUT2 gene in a Fanconi-Bickel syndrome family. Gene. 2015;557:103–5.
Fridman E, Zaharia A, Markus-Eidlitz T, Cohen YH. Phenotypic variability in patients with Fanconi-Bickel syndrome with identical mutations. JIMD Rep. 2014;15:95–104.
Santer R, Groth S, Kinner M, Dombrowski A, Berry GT, Brodehl J, Leonard JV, Moses S, Norgren S, Skovby F, Schneppenheim R, Steinmann B, Schaub J. The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi-Bickel syndrome. Hum Genet. 2002;110:21–9.
Al-Haggar M, Sakamoto O, Shaltout A, El-Hawary A, Wahba Y, Abdel-Hadi D. Fanconi BAickel syndrome: Novel mutations in GLUT2 gene causing a distinguished form of renal tubular acidosis in two unrelated Egyptian families. Case Rep Nephrol. 2011:754369.
Odievre MH, Lombes A, Dessemme P, Santer R, Brivet M, Chevallier B, Lagardere B, Odievre M. A secondary respiratory chain defect in a patient with Fanconi-Bickel syndrome. J Inherit Metab Dis. 2002;25:379–84.
Peduto A, Spada M, Alluto A, La Dolcetta M, Ponzone A, Santer R. A novel mutation in the GLUT2 gene in a patient with Fanconi-Bickel syndrome detected by neonatal screening for galactosemia. J Inherit Metab Dis. 2004;27:279–80.
Seetodeh A, Rabbani A. Transient neonatal diabetes as a presentation of Fanconi-Bickel syndrome. Acta Med Iran. 2012;50:836–8.
Authors’ contributions
ID carried out manuscript preparation, literature search, data acquisition and data analysis. IA provided data about the history of the two patients and prepared the table and the figure. SB contributed to interpretation of biochemical data and manuscript review. MS contributed to treatment of the two patients and manuscript review. All authors read and approved the final manuscript.
Acknowledgements
We like to thank the metabolic and nephrology staff in our hospital for taking care of the patients. A special thanks to the laboratory staff in our hospital for their efforts in the reporting of biochemical data.
Competing interests
The authors declare that they have no competing interests and have received no funding for this article.
Availability of data and materials
The data supporting the conclusions of this paper are included in the paper.
Consent
Written informed consent was obtained from the parents of the two patients for publication of this case report. Parental consent for publication of non-covered images was also obtained.
Ethical clearance
Permission for publication of this article was obtained by An-Najah National University Institutional Review Board.
Funding
There was no research grant for all authors of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dweikat, I.M., Alawneh, I.S., Bahar, S.F. et al. Fanconi-Bickel syndrome in two Palestinian children: marked phenotypic variability with identical mutation. BMC Res Notes 9, 387 (2016). https://doi.org/10.1186/s13104-016-2184-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-016-2184-2