
Abstract
Background
New sequencing technologies have made it possible to explore genetic diversity at higher resolution in microbial populations. However, our understanding evolutionary relationships, and comparison of closely and distantly related bacterial genomes from these massive datasets remains a formidable challenge. Numerous clustering algorithms that group genomic data based on homology have been developed, but new tools are still required to analyse the resultant orthologous maps to understand functional genetic similarities and their phyletic patterns (patterns of presence of absence of genes).
Findings
Bacterial Makeup eXplorer (BMX) implements an algorithm that swiftly and efficiently facilitates the determination of the number of orthologs in prokaryotic genomes employing a reference free approach, which may be further exploited to transfer of gene annotations. BMX is able to integrate orthologous maps of highly diverse prokaryotic genomes therefore making it possible to perform robust and scalable, multi-platform, high quality annotation transfer and gene-by-gene composition assessment method. In addition results are presented in the form of publication quality figures.
Conclusions
BMX allows extensive data analysis of orthologous map databases to understand underlying biological relationships. Furthermore, BMX is portable across different platforms and can be installed easily. In summary, BMX allows higher resolution analysis of genomes from diverse bacterial populations
Similar content being viewed by others


Findings
Background
The concept of orthology provides an important basis for studying mechanisms of bacterial genome evolution, functional genetics, and biological networks. Orthologs are genes descended from a common ancestor as a result of speciation, which are likely to retain the same function in the course of evolution [1,2]. They contrast with paralogs, which may retain the same function or evolve new functions that are related to or different from the original function due to lack of the original selective pressure on one or more copies of the duplicated genes [1-3]. However, it is important to note that function retention among orthologs has some notable exceptions [4]. Determination of orthology between bacterial genomes provides a detailed understanding of bacterial population genetics and evolutionary biology [5,6]. This has greatly been enhanced by recent rapid and cost-effective advances in sequencing technologies, which have provided highly accurate and reproducible large-scale datasets required to unravel the broad spectrum of genetic diversity from different microbes [7]. These datasets have allowed higher resolution analysis of large bacterial populations, and significantly improved our understanding of diversity between and across species.
Numerous computational methods that accurately and efficiently infer orthology from sequenced genomes have been developed [8,9]. However, effective algorithms are still required to gain functional insight from the extensive generated homologous maps [10]. Recently, more emphasis has been redirected towards identification of orthologs, which are crucial elements of comparative genomics applications [6,11,12]. Ortholog annotation allows easy comparison of genomes and can highlight key conserved antigens and virulence complements found in the genomes of clinically important bacterial strains under study, which could be exploited as novel potential targets for therapeutic approaches [13-15].
We describe Bacterial Makeup eXplorer (BMX), a highly efficient, stand alone tool that can be used to easily and effectively compute the size and composition of the genes common to all strain genomes, within each gene cluster under study (referred to henceforth as the core genome) from OrthoMCL [16] generated orthologous gene maps. Most genome alignment approaches require high quality reference sequences and are problematic when comparing distantly related species or species with high rates of recombination [6]. BMX is a robust reference-free highly scalable approach capable of integrating datasets containing highly divergent genomes. Moreover, BMX can also be exploited as an annotation transfer tool by including a known well-annotated reference genome in the dataset, thereby allowing inference of a gene cluster function. Its features include: data reorganization, genome clusters quality assessment, iterative random genome sampling, and core and accessory genome distinction, and size and composition computation; generating accurate molecular barcodes from whole genome sequence data.
Implementation
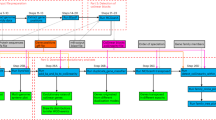
BMX is implemented in Perl, BioPerl and R, and includes a bash script wrapper. The user supplies an orthologous map input file of interest in OrthoMCL format for analysis, and the number of genomes under study. OrthoMCL format is used for convenience and uniformity, and orthologous maps in other database formats could be easily converted to this input format. However, updates to BMX are anticipated soon after the ortholog database community fully embraces and implements a standardised XML database format. Although various ortholog databases have signed-up for and agreed to use XML, there currently is still community efforts towards adoption of standard file formats and benchmarking [17]. Detailed step-by-step documentation that includes input files and how to run the software is also provided. Figure 1 portrays the entire BMX pipeline in pseudo-code. Ortholog clusters are first organized into a matrix of genome content, while taking into account absent genes. In this matrix the matrix columns represent different strain genomes, while the rows represent the orthologous clusters across the genomes. The composition of core (genes common to all strain genomes) and accessory (genes not common to all strain genomes) gene clusters is determined using randomly sampled genomes (the matrix columns) iteratively, in an arithmetic progression fashion using the mathematical formula:

A flow-chart describing the pipeline implemented in BMX. (A) The pipeline consists of three mains steps that involve (i) reorganisation and quality assessment of the data, (ii) random sampling of genomes for each computation event, and (iii) iterative computation of the core and accessory genome size and composition. These are marked as step 1 through 3 respectively. (B) A quality assessment heatmap is generated at step 1, with genes represented in red and the absence of genes represented in blue. The core genome is represented by solid red columns of genes. Corrupted or poor quality genomes can be visually identified, for instance as having missing genes in the core genomes. (C) During the core genome analysis, genomes in the dataset are selected at random from the dataset and each genome (represented by a number) is selected only once. The calculation of the core genome size of N genomes is performed N times, starting with determination of the size determination of one genome up to N genomes, with an increment of 1 genome is each subsequent calculation. This is known as an event and is implemented as an arithmetic progression. 100 such events are performed and the average value is used to establish the core genome size of N genomes. (D) A graph of the core genome size is generated. The files showing the core genome sizes (“CG.txt”) and accessory clusters (“not_CG.txt”), which do not form part of the core genome, are also generated.
The number of random sampling events when determining the core genome size given N number of genomes, S N , is established using the least number of genomes under consideration, which is set to 1 genome by default, a 1 , the total number of genomes under consideration (i.e. the dataset size), N, and the common difference of successive genomes, d, (i.e. 1) used during sampling. During random sampling, the total number of genomes, N, is initialized as 1 genome for the first event, and increased by one unit for each of the subsequent events, until it is equivalent to the total number of genomes in the dataset in the final event. The random genomes are only sampled once during each event and the total number of orthologs shared by all genomes counted once for each cluster, thereby excluding paralogous counts. The default setting uses 100 iterations resulting in 100 different input orders per event, and the more advanced users may alter this value depending on their preference, in the BMX script L.pl. However, it’s important to note previous tests show that 100 iterations give the optimal results. The average core genome size per event is computed enabling correlation with the number of genomes sampled. Poor quality genomes can be identified for exclusion, by visual examination of a quality control diagram generated using R scripts. Core and accessory genome size and composition comparisons between different datasets can be achieved by this approach, thereby allowing annotations to be easily transferred to genes of draft genomes. A plot of the core genome size, which only consists of clusters that have a gene in every taxon, is generated using R scripts.
Results and discussion
BMX has already been used on highly divergent bacterial genomes of Streptococcus pneumoniae (pneumococcus) (Additional file 1: Table S1). Pneumococci are clinically important and have highly recombigenic genomes from multiple diverse lineages, which make it challenging to study their evolutionary history [18]. BMX was used to assess the composition of 140 invasive disease pneumococcal genomes. Datasets of 16, 70 and 140 Streptococcus genome maps are included in the software download itself, and can be found at this url: http://sourceforge.net/projects/bmexplorer/files/latest/download. The estimated run times for analysis of these datasets using BMX are: 16 genomes in less than 3 mins, 70 genomes in 15–20 mins, and 140 genomes in 45 mins on a 2.7 GHz Intel Core i7 processor and 8Gb 1333 MHz DDR3 memory. Paralogs, core genes and other accessory genes were clearly distinguished, allowing the subsequent robust reconstruction of phylogenetic trees. BMX can be exploited to identify relevant functional sets of core genes, which encode important virulence complements always present in the genome that can be exploited as therapeutic and diagnostic targets.
Conclusion
BMX is a scalable, reference-free framework for computing gene-by-gene core and accessory genome composition and making comparisons between datasets. We believe that BMX will facilitate hypothesis generation and design of new experiments that explore the genomic diversity of bacteria, and its use can be extended to other prokaryotic organisms.
Availability and requirements
Project name: BMX.
Project home page: http://sourceforge.net/projects/bmexplorer/
Operating system(s): Multiple platforms.
Programming language: Perl, BioPERL and R.
Other requirements: The following modules must be installed in perl: Bio::Perl, IO::String, Bio::SeqIO, List::Util, List::Util ‘max’, Text::CSV, integer.
The following libraries/packages must be installed in R: RColorBrewer, gplots.
License: GNU GPL.
Any restrictions to use by non-academics: none.
Availability of supporting data
The data set of the orthologous map from 140 invasive disease pneumococcal genomes supporting the results of this article is available in the publically accessible source forge repository, http://sourceforge.net/projects/bmexplorer/.
References
Jensen RA. Orthologs and paralogs - we need to get it right. Genome Biol. 2001;2:INTERACTIONS1002.1–1002.3.
Fitch WM. Homology a personal view on some of the problems. Trends Genet. 2000;16:227–31.
Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–113.
Studer RA, Robinson-Rechavi M. How confident can we be that orthologs are similar, but paralogs differ? Trends Genet. 2009;25:210–16.
Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. The bacterial species challenge: making sense of genetic and ecological diversity. Science. 2009;323:741–6.
Maiden MC, van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013;11:728–36.
Parkhill J. What has high-throughput sequencing ever done for us? Nat Rev Microbiol. 2013;11:664–5.
Altenhoff AM, Dessimoz C. Inferring orthology and paralogy. Methods Mol Biol. 2012;855:259–79.
Kuzniar A, van Ham RC, Pongor S, Leunissen JA. The quest for orthologs: finding the corresponding gene across genomes. Trends Genet. 2008;24:539–51.
Donati C, Hiller NL, Tettelin H, Muzzi A, Croucher NJ, Angiuoli SV, et al. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol. 2010;11:R107.
Dessimoz C, Gabaldon T, Roos DS, Sonnhammer EL, Herrero J. Toward community standards in the quest for orthologs. Bioinformatics. 2012;28:900–4.
Parsons M, Myler PJ, Berriman M, Roos DS, Stuart KD. Identity crisis? The need for systematic gene IDs. Trends Parasitol. 2011;27:183–4.
Serruto D, Rappuoli R. Post-genomic vaccine development. FEBS Lett. 2006;580:2985–92.
Barocchi MA, Censini S, Rappuoli R. Vaccines in the era of genomics: the pneumococcal challenge. Vaccine. 2007;25:2963–73.
Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102:13950–5.
Li L, Stoeckert Jr CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Sonnhammer EL, Gabaldon T, da Silva AW S, Martin M, Robinson-Rechavi M, Boeckmann B, et al. Big data and other challenges in the quest for orthologs. Bioinformatics. 2014;30:2993–8.
Hanage WP, Fraser C, Tang J, Connor TR, Corander J. Hyper-recombination, diversity, and antibiotic resistance in pneumococcus. Science. 2009;324:1454–7.
Acknowledgements
The author thanks Dean Everett, Rob Heyderman, Aras Kadioglu, Martin Maiden and Chisomo Msefula for their constructive comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that he has no competing interests.
Author’s contributions
BK conceived the study and planned experiments, performed the analysis and wrote the manuscript. The author read and approved the final manuscript.
Additional file
Additional file 1: Table S1.
The 213 publically available Streptococcus pneumoniae genomes used a test datasets for the BMX program. OrthoMCL was used to generate orthologous maps from these dataset.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kulohoma, B.W. BMX: a tool for computing bacterial phyletic composition from orthologous maps. BMC Res Notes 8, 51 (2015). https://doi.org/10.1186/s13104-015-1017-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-015-1017-z