
Abstract
Genomics and other large-scale analyses have drawn increasing attention to the potential impacts of transposable elements (TEs) on their host genomes. However, it remains challenging to transition from identifying potential roles to clearly demonstrating the level of impact TEs have on genome evolution and possible functions that they contribute to their host organisms. I summarize TE content and distribution in four well-characterized yeast model systems in this review: the pathogens Candida albicans and Cryptococcus neoformans, and the nonpathogenic species Saccharomyces cerevisiae and Schizosaccharomyces pombe. I compare and contrast their TE landscapes to their lifecycles, genomic features, as well as the presence and nature of RNA interference pathways in each species to highlight the valuable diversity represented by these models for functional studies of TEs. I then review the regulation and impacts of the Ty1 and Ty3 retrotransposons from Saccharomyces cerevisiae and Tf1 and Tf2 retrotransposons from Schizosaccharomyces pombe to emphasize parallels and distinctions between these well-studied elements. I propose that further characterization of TEs in the pathogenic yeasts would enable this set of four yeast species to become an excellent set of models for comparative functional studies to address outstanding questions about TE-host relationships.
Similar content being viewed by others

Background
Transposable elements (TEs) are a diverse set of genetic elements that can move to new sites in genomes and substantially contribute to genotypic and phenotypic variation. They are divided into two main classes, followed by subclasses, superfamilies, and families based on their sequence structures and mechanisms of mobility, or transposition [1]. Mobility of Class 1 elements, known as retrotransposons, occurs through reverse transcription of an RNA into a complementary DNA (cDNA) that is inserted at a new genomic site, which is referred to as a “copy-and-paste” mechanism (Fig. 1). Two example subclasses are long terminal repeat (LTR) retrotransposons, with the four superfamilies Ty1/copia (Pseudoviridae), Ty3/gypsy (Metaviridae), BEL, and endogenous retroviruses, and non-LTR retrotransposons, with approximately 30 superfamilies, such as CRE, I, jockey, L1, and R2 [1]. LTR retrotransposons reverse transcribe cDNA prior to integration by DDE-type integrases, while non-LTR retrotransposons nick target sites using endonuclease domains to initiate reverse transcription (Fig. 1) [1, 2]. Mobility of Class 2 elements, known as DNA transposons, involves DDE transposase proteins that excise DNA copies of elements from donor sites and integrate them at new genomic sites, a “cut-and-paste” mechanism, for approximately 20 superfamilies, such as EnSpm, Harbinger, Mariner/Tc1, and MULE (Mutator-like elements) (Fig. 1) [1, 2]. Mobility of the Crypton superfamily involves tyrosine recombinases and possible circular DNA intermediates, while Helitron mobility involves transposases with HUH nuclease and helicase activities that nick and unwind one transposon strand, which is replicated to form a double-stranded circular DNA that is used to integrate a copy at a new site (“peel-and-paste”, Fig. 1) [2,3,4].

Comparison of transposition cycles of four subclasses of transposable elements (TEs). Simplified representations of major steps in the transposition cycles of two subclasses of DNA transposons (cut-and-paste and peel-and-paste) and two subclasses of retrotransposons (long terminal repeat (LTR) and non-LTR) are shown. Light gray and lavender lines represent donor and target genomic sites, respectively. Boxed arrowheads indicate terminal inverted repeats (cut-and-paste) or LTRs. Curved black arrows indicate repair of the donor site after transposon excision, small purple or blue arrows represent new DNA synthesis, and wavy lines represent RNA. TP- transposase (DDE-type for cut-and-paste or HUH nuclease/helicase-type for peel-and-paste), RT- reverse transcriptase, IN- integrase, EN- endonuclease domain.
TEs are widely known for causing mutations when they are mobile, but they also contribute to chromosomal rearrangements, and have transcriptional control sequences that can affect expression of neighboring genes [2]. Unrestricted activity of a mutagenic agent is typically deleterious, so organisms have many layers of defense that restrict TE mobility at various stages of their complex transposition cycles [2]. It has long been discussed that activation of TEs by stress could produce random genetic variation that gives rise to occasional genotypes better adapted to the relevant stress. Many TEs are activated by particular stresses, and there is some evidence that their transcription or transposition can produce beneficial mutations or changes in gene expression for adaptation to stress [5]. An increasing number of studies, particularly large-scale genomic, transcriptomic, and epigenomic studies, are providing evidence that TEs dispersed throughout genomes provide cis-regulatory sequences and transcriptional start sites for expression of neighboring genes [6]. There are also observations consistent with particular groups of TEs with common cis-regulatory sequences dispersed at various genomic sites contributing to the evolution of gene regulatory networks coordinately controlling large numbers of genes [6]. TEs are also now being analyzed on a massive scale. For instance, a recent study of DNA transposons in 1730 fungal genomes shows that DNA transposon content is correlated with fungal lifestyles (such as soil-living, associated with plants or animals), and increased numbers of DNA transposons but fewer functional copies are present in species with RNA interference defense systems [7]. A second study of total TE content in 625 fungal genomes by the same group reports that the number of TEs in genes is correlated with fungal lifestyles, and differences in clustering of TEs in genomic regions is correlated with their potential for being active elements [8].
The types of studies just discussed are very important and exciting, and are leading to increased interest in studying TEs. However, they must be thoughtfully considered and presented, because they raise exciting possibilities, but in many cases additional functional studies are needed to verify these possibilities [2, 5, 6]. For instance, TEs differ in whether they are activated or inhibited by particular stresses, how they affect expression of neighboring genes, and evidence of negative impacts of TEs during stress have been reported [5]. Biochemical activities consistent with cellular functions for TEs do not guarantee that TEs have those relevant functions, pointing to the need for follow-up functional studies. For example, functional analyses of TE enhancer sequences expected to regulate gene expression in mouse embryonic stem cells based on transcriptomic and epigenomic data show that only a small subset of the enhancers tested had substantial effects on gene expression [9]. Furthermore, one of the large-scale studies of fungi mentioned earlier shows that nearly all TEs analyzed are likely to be experiencing neutral evolution [8]. Overall, there is a growing discussion of the need to experimentally examine the potentially significant impacts of TEs on their hosts [2, 5, 6].
I propose that the model yeast species discussed in this review represent excellent systems for comparative functional analyses to develop a more sophisticated understanding of the regulation and impacts of TEs. These four yeasts – the human pathogens Candida albicans and Cryptococcus neoformans, and the nonpathogenic model organisms Saccharomyces cerevisiae and Schizosaccharomyces pombe – are all well-developed model species with diverse lifestyles and genomic features that may have influenced the evolution of TE-host relationships. I will summarize TE content and distribution, genomic features, lifestyles, and presence of RNA interference pathways in each species to highlight similarities and differences for considering comparative studies. Such studies comparing most/all TEs in a genome, as well as differences in TE regulation/impact between multiple strains of each species, or between the four diverse species has great potential to address many questions about TE evolution and impact on host organisms.
Genomic features of the four yeasts
Yeasts in general have small gene-dense genomes with relatively low TE content that can facilitate manipulation and evaluation of a large proportion of the total TEs in a genome. Genomic features data are shown for reference genomes for two major varieties of Cryptococcus neoformans, var. grubii, also called serotype A, and var. neoformans, also called serotype D (Table 1). It was proposed that these varieties be given different species names [10], but a large international group more recently proposed using the term “Cryptococcus neoformans species complex” until variations between a large number of strains are more thoroughly analyzed [11]. I will refer to the species complex as simply C. neoformans and indicate if particular information is relevant to only one serotype in this review. Cryptococcus neoformans has the largest and most metazoan-like haploid genome of the four yeasts, with introns in virtually all genes (typically multiple per gene) and relatively large regional centromeres that all include retrotransposon sequences (Table 1) [12,13,14]. Candida albicans has the next largest haploid reference genome, with several hundred fewer open reading frames (ORFs), very few introns, and relatively small regional centromeres compared to C. neoformans [15,16,17,18]. An additional noteworthy aspect of that genome is that the CUG codon typically decoded as leucine is decoded as serine in Candida albicans [19]. The Saccharomyces cerevisiae and Schizosaccharomyces pombe reference genomes are the smallest genomes, and despite their similar sizes, S. cerevisiae has substantially more ORFs (Table 1) [20,21,22,23]. S. cerevisiae also has very few introns (similar to C. albicans) and small point centromeres, while S. pombe has many introns and large regional centromeres (more similar to C. neoformans) [21,22,23,24,25].
TE content & distribution in the four yeasts
Candida albicans
Candida albicans is a diploid budding yeast commonly found in the human digestive tract that can switch between distinct cell types and undergo a parasexual cycle, rather than a true sexual cycle [26, 27]. It is an opportunistic pathogen that can cause mucosal infections and more rarely systemic infections, the latter of which are more common in immunocompromised people [28, 29]. The parasexual cycle involves mating of diploid cells to form tetraploid cells, followed by random concerted chromosome loss that reduces DNA content to approximately a diploid state [30,31,32,33]. Mating is regulated by a mating type locus (MTL) that has a and α alleles [27], though same-sex mating has been observed in certain contexts [34]. MTL also regulates switching between white and more elongated opaque cell types, and opaque cells mate much more readily than white cells (about 1 million-fold better) [35]. C. albicans can grow as yeast, pseudohyphae, and hyphae. Environmental cues, such as growth temperatures of 37 °C, neutral pH, or the presence of serum trigger hyphae formation, and hyphal growth is associated with virulence [26].
The C. albicans reference genome (strain SC5314) harbors multiple families of DNA transposons and retrotransposons [16]. Sequences annotated as DNA transposons or transposase genes and retrotransposons or solo LTRs account for approximately 0.8% of the reference genome [16, 36], with retrotransposons and solo LTRs making up the bulk of the elements (Table 1). Solo LTRs result from recombination between LTRs at retrotransposon termini. These recombination events delete the internal sequences and one LTR copy from the genome, and this loss of sequences restricts the accumulation of LTR retrotransposons in genomes. DNA transposons include members of the Mariner/Tc1 (Cirt elements) and MULE superfamilies [37,38,39], but these elements have not been characterized in detail. Thirty-four LTR retrotransposon families and three non-LTR retrotransposon families were identified in early studies of individual elements or surveys of earlier assemblies of the SC5314 reference genome [37, 40,41,42,43]. All 34 LTR retrotransposon families and two of the three non-LTR retrotransposon families are represented in Assembly 22 of the reference genome [16], but in fewer copies than reported in the original studies [37, 43]. An analysis of an early draft of the reference genome reported 16 LTR retrotransposon families, Tca1–16, with internal sequences (sequences between two LTRs) that include multiple members of the Ty1/copia (Pseudoviridae) and the Ty3/gypsy (Metaviridae) superfamilies [37]. However, only nine LTR retrotransposon families are annotated as having internal sequences in the current assembly (Assembly 22) of the reference genome [16].
Potential genomic insertion/distribution biases have not been characterized for C. albicans DNA transposons, but some biases have been identified for retrotransposons (Fig. 2). The Zorro1 and Zorro2 non-LTR retrotransposons and some families of LTRs are present in subtelomeric regions [37, 43]. The beta LTRs of Tca8 elements are biased for sequences upstream of tRNA genes [37, 44]. C. albicans centromeric DNA is approximately 3–4.5 kb and no shared or common repeat sequences have been identified [17, 18]. Approximately half of centromeres were observed to have one or two LTRs in a pericentric region or within the centromeric DNA [18]. Zorro3 non-LTR retrotransposons are present at sites of poly(A) sequences without any known preference for specific chromosomal regions [43], and newly integrated copies identified by a retrotransposition assay are also targeted to poly(A) sites [45]. Use of a retrotransposition assay also revealed that the Tca2 LTR retrotransposon can insert into ORFs, but prefers to insert in a 300 bp window upstream of start codons [46].

Distributions of TEs in four model yeasts. Gray lines and filled boxes are schematic representations of major DNA sequence features present on chromosomes without any intention of indicating relative size, position, or presence of specific sequences on the same chromosome. Sequence features include: pA- poly(A) sites; CEN- centromeres; HML/R or mat2/3- hidden/silent mating loci; MAT, mat1, or MTL- mating locus; Pol2 or Pol3- RNA Polymerase II or III-transcribed genes; rDNA- ribosomal DNA; TEL- telomeres; ? - unknown/no bias for sequence features. Vertical arrows indicate sites of TE insertions and the relative proportions of total insertions as shown in the key. Names above arrows indicate specific TEs or groups of TEs: DNA- DNA transposons, DNA/RT- DNA transposons or retrotransposons, LTR- LTR retrotransposons or solo LTRs. Blue horizontal arrows indicate the direction of gene transcription.
Cryptococcus neoformans
C. neoformans is a encapsulated budding yeast found in soil, particularly soil contaminated with pigeon guano, that can cause infections in humans by inhalation of spores or cells [47]. Cells can then spread from the lungs to cause infection elsewhere, most commonly cryptococcal meningoencephalitis, and this is particularly likely in immunocompromised people [47]. C. neoformans has one mating type locus, MAT, with both a and α alleles (a heterothallic or self-sterile yeast), but the great majority of clinical and environmental isolates are haploid α cells [48]. Mating between a and α cells causes a switch to dikaryotic filamentous growth that can then lead to diploid cells, meiosis, and spore formation [47]. However, same-sex mating between serotype D α cells can produce diploid cells that undergo monokaryotic fruiting, including meiosis and spore formation [49], and diploid α/α cells of serotype A are occasionally observed in environmental and clinical isolates [50]. In addition, cells can undergo phenotypic switching events that change characteristics such as melanin production and capsule size that relate to virulence [47, 51].
The haploid genomes of serotypes A (strain H99) and D (strain JEC21) have multiple families of DNA transposons and retrotransposons [12, 13]. TEs were found to comprise about 5% of the serotype D genome in the original genome sequence report [12]. A more recent study identified 5.9 and 6.6% of the serotype A and D genomes as TEs [52], which is several-fold more than the TE content of the Candida albicans reference strain. DNA transposons represent 0.5 and 0.8%, and retrotransposons represent 3.4 and 4.5% of the serotype A and D genomes, respectively (a portion of TEs in each genome was unclassified, Table 1) [52]. Members of the Crypton, EnSpm, Harbinger, Helitron, Mariner/Tc1, and MULE superfamilies are present, though Helitrons are only present in strain H99 (serotype A) [52]. Cryptons were originally identified in C. neoformans as mobile elements consisting of tyrosine recombinase ORFs that lacked any flanking long terminal or terminal inverted repeats – they were flanked only by a 4 bp repeat that appeared to represent a duplication of the insertion site [53].
Retrotransposons in C. neoformans have been analyzed in more detail. Both the serotype A and D genomes contain CRE-type non-LTR retrotransposons and multiple families of LTR retrotransposons [52, 54]. LTR retrotransposons, particularly members of the Ty3/gypsy superfamily, account for more than half of the total TE content (Table 1) [52]. An early study of the JEC21 genome identified 10 families of LTR retrotransposons (Tcn1–10), five families of solo LTRs, 10 families of retrotransposon fragments, and the Cnl1 CRE-type non-LTR retrotransposon [54]. Members of the 10 LTR retrotransposon and 10 retrotransposon fragment families are evenly split between the Ty1/copia and Ty3/gypsy superfamilies. Full LTR retrotransposon sequences were only reported for Tcn1–6, of which Tcn6 is the only member of the Ty1/copia group [54]. Tcn1 was proposed to use a self-priming mechanism for reverse transcription similar to Tf1/sushi retrotransposons (Tf1 is discussed later for Schizosaccharomyces pombe), and Tcn2–5 were proposed to potentially have novel mechanisms of priming [54].
Distribution biases have been noted for many C. neoformans TEs (Fig. 2), but no studies have characterized insertion preferences from datasets of newly integrated TEs. Disruption of the FRR1 gene (encoding the FKBP12 protein) provides resistance to the drug FK506, and selection for FK506 resistance has been used as a transposon trap assay demonstrating that MULE (T1, T2, and T3) and Harbinger DNA transposons can insert into ORFs [55,56,57]. TEs are also enriched at centromeres, at the MAT locus, flanking rDNA, and the non-LTR retrotransposon Cnl1 is present in telomeres [12, 54]. C. neoformans centromeres span approximately 20–65 kb and contain multiple copies of Tcn1–6 retrotransposons [12,13,14]. Over 95% of Tcn1–6 copies are present at centromeres [14], and the presence of multiple retrotransposons distinguishes C. neoformans centromeres from the other three yeasts (Fig. 2), though a different Schizosaccharomyces species does have centromeric retrotransposons [58].
Saccharomyces cerevisiae
S. cerevisiae has been used for baking and brewing for thousands of years, and many decades ago was developed into an excellent model research organism particularly for biochemistry, cell biology, and genetics. Strikingly, relatively little is known about the ecology and lifecycle of S. cerevisiae in the wild, in part due to the focus on practical, commercial, and laboratory research uses of this yeast [59]. While not generally considered a pathogen, there have been rare reports of S. cerevisiae infections in humans [60]. S. cerevisiae has a MAT locus with a and α alleles, and can stably grow as haploid a or α cells or diploid a/α cells [61]. S. cerevisiae is a homothallic yeast (self-fertile), because haploid cells can switch mating type by expressing the HO endonuclease to cleave a specific site at the MAT locus and repairing the double-strand break using one of two hidden mating loci, HML (α) or HMR (a) [61]. These hidden mating loci are silent copies of the a or α mating-type alleles present at distinct genomic sites from MAT that can be used to change the allele present and expressed at the MAT locus. Many laboratory strains have mutations in the HO gene that prevent switching from occurring. Mating-type switching in a clonal population can create haploid cells of both mating types that readily mate with each other (without nutritional cues) to form diploid cells with two copies of the same haploid genome. Natural populations tend to consist of diploid cells [62], and can be found on tree bark (particularly oak trees), various fruits, and notably have also been found in habitats far from human activity [59, 63, 64].
The only TEs in the S. cerevisiae haploid reference genome (strain S288c) are LTR retrotransposons [20, 22]. An initial analysis identified 3.1% of the genome as LTR retrotransposons and solo LTRs, reporting 331 total insertions consisting of 280 solo LTRs or LTR fragments and 51 retrotransposons [65]. More recent analyses reported an overall retrotransposon content of 3.4% or 3.5% [66, 67]. The annotation of the current version of the reference genome includes 383 total LTRs and 50 retrotransposons [22], though 51 full-length retrotransposons have been previously reported [65, 67] as a result of identifying 32 full-length Ty1 elements, as opposed to 31 in the annotated genome. These retrotransposons include five families, Ty1-Ty5, with Ty3 being the only gypsy-type (Metaviridae) and the other four copia-type (Pseudoviridae) retrotransposons [65]. The relative abundance of these elements in the reference genome is (including solo LTRs): Ty1 > Ty2 > Ty3 > Ty4 > Ty5 [22, 65]. In particular, Ty1 and Ty2 account for 32 (31) and 13 of the 51 (50) full retrotransposon sequences, and Ty1 LTRs account for over half of all LTRs [22, 65, 67]. Ty1 sequences are separated into three subfamilies: Ty1, Ty1′ with divergent gag ORF sequences, and Ty1/2 hybrid elements [65, 67]. Ty2 and a subfamily of Ty3 LTRs referred to as Ty3p appear to have been horizontally transferred to S. cerevisiae from S. mikatae and S. paradoxus, respectively [67].
The genomic distribution of all five families is very biased. About 90% of Ty1-Ty4 elements are found in a 750 bp region upstream of genes transcribed by RNA Polymerase III, and all but three such insertions are upstream of tRNA genes, while Ty5 is found at sites of repressive chromatin, including subtelomeres and the hidden mating loci (Fig. 2) [65, 68]. Newly integrated copies of Ty1 preferentially target an approximately 1 kb region upstream of tRNAs and other genes transcribed by RNA Polymerase III [69,70,71,72]. About 90% of Ty1 insertions occur within 2 kb upstream of RNA Polymerase III-transcribed genes, ≤ 5% of insertions occur in ORFs, and a similarly low percentage of insertions occur in promoters or other flanking regions of RNA Polymerase II-transcribed genes in both haploid and diploid cells, based on analyses of thousands to over a million sequencing reads for new insertions (Fig. 2) [71, 72]. Nearly all newly integrated copies of Ty3 also insert upstream of RNA Polymerase III-transcribed genes, with the great majority occurring 0–20 bp upstream of the 5′ ends of tRNA genes in the vicinity of the transcription start site [73,74,75,76]. There are no functional copies of Ty5 in the reference S. cerevisiae genome, but a functional Ty5 element from S. paradoxus has been shown to preferentially target heterochromatin in subtelomeric regions and near the HML and HMR silent mating loci (Fig. 2) [77, 78]. About 76% of newly integrated Ty5 elements target sites of heterochromatin, defined as the region from chromosome ends to 10 kb centromere-proximal of subtelomeric X repeats or HML and HMR [79]. Integration frequently occurs in intergenic regions upstream of ORFs, peaking around 100 bp and falling to background levels by 1000 bp upstream [79].
Schizosaccharomyces pombe
S. pombe is a nonpathogenic fission yeast found throughout the world that was developed into an excellent model organism for basic cell biology and genetics research many decades ago [80]. The ecology of S. pombe is not well studied, but strains have been isolated from a variety of sources, including cocoa pulp, coffee fruits, grapes, molasses, as well as fermentations of tea (kombucha), sugar cane (cachaca), sorghum (baijiu), and palm sap [81]. The genome has a mating locus, mat1, with plus (P) and minus (M) alleles, and two silent mating loci, mat2-P and mat3-M [61]. Wild-type S. pombe strains are homothallic, and can switch mating type by production of a protected single-strand break at mat1 that results in a double-strand break in one of two daughter cells after another round of DNA replication [80, 82]. This break can be repaired using mat2-P or mat-3 M [82]. However, mutations can give rise to heterothallic strains, h+ and h-, such as in the 972 h- strain used for the reference genome [21, 61]. Strains from natural sources are typically haploid [62], and haploid cells grow stably until they encounter nutrient starvation, which induces mating [80]. Diploid cells typically undergo meiosis soon after they form, unless they are transferred to nutrient-rich conditions [80].
Similar to Saccharomyces cerevisiae, the only TEs in the S. pombe reference genome (strain 972 h-) are LTR retrotransposons [21, 83]. LTR retrotransposons comprise 1.1% of the reference genome [23, 83]. There are 249 LTRs or LTR fragments, 13 full-length retrotransposons, and five retrotransposon fragments containing only portions of internal sequences and sometimes parts of LTRs [83, 84]. Eight of nine families of LTR-retrotransposons are represented only by LTRs or LTR fragments, all full-length retrotransposons are members of the Tf2 family, and the five fragments are Tf1 or Tf2 sequences [83]. S. pombe Tf1 elements have been most intensively studied in terms of their replication cycle and integration biases, and full-length Tf1 elements were identified in other wild-type S. pombe strains [85]. Tf1 is represented by 28 solo LTRs in the reference genome, compared to 35 solo LTRs for Tf2 [83]. Tf1 and Tf2 have similar sequences and are both members of the Ty3/gypsy superfamily of LTR retrotransposons [84, 85]. However, Tf1 and Tf2 are both members of a distinct group of elements that uses self-priming to initiate reverse transcription [86]. There is also a family of 25 ORFs flanked on one or both sides by Tf1 or Tf2 LTRs [21, 83]. There is no evidence that these wtf elements are mobile DNA elements, and the ~ 1 kb ORFs have multiple predicted introns, are predicted to encode membrane proteins, and are transcriptionally induced during meiosis [21, 87]. Recent work shows that at least some members of the wtf gene family encode poison and antidote proteins that kill spores lacking the particular wtf gene, while protecting spores with the gene [88, 89]. These poison-antidote systems allow these genes to act in a selfish manner to increase their transmission to future generations, contributing to the hybrid sterility and reproductive isolation observed for many S. pombe isolates [88, 89].
All Tf1 and Tf2 insertions (solo LTRs and full retrotransposons) in the reference genome are present in intergenic regions [83]. They show a strong bias for promoters of genes transcribed by RNA polymerase II, with 83% of insertions closer to the 5′ end of an ORF than the 3′ end, and the bulk of insertions clustered within 400 bp of a start codon (Fig. 2). In contrast to Tf1, new insertions of Tf2 are primarily incorporated into the genome through homologous recombination between Tf2 cDNA and pre-existing Tf2 sequences [90], and large-scale analyses of the minority of Tf2 integration events at nonhomologous sites have not been reported. Nearly all newly inserted copies of Tf1 integrate into intergenic regions, with a strong bias for a 100–400 bp window upstream of RNA Polymerase II-transcribed genes [91, 92]. More recent analyses of tens of thousands or approximately 10 million new insertions show that > 95% of Tf1 insertions are targeted to intergenic regions, particularly within 500 bp of Pol II promoters, and only ~ 3–4% of insertions occur in ORFs (Fig. 2) [93, 94]. Similar distributions are observed in haploid and diploid cells [93], 93% of insertions occur at intergenic sites between tandemly or divergently transcribed ORFs [93], and 80% of insertions are closer to the 5′ end of an ORF than the 3′ end [94].
In summary, these four yeasts have diverse lifestyles, sexual cycles, and genomic features that could result in different constraints and opportunities for the evolution of TEs in these species. Comparative studies of TEs based on these differences could provide important insights into TE biology. For instance, Candida albicans has a parasexual cycle that is distinct from the sexual cycles of the other three yeasts. Characterizing TE regulation during the C. albicans parasexual cycle in comparison to TE regulation during sexual cycles in the other yeasts could distinguish whether distinct reproductive cycles are characterized by very distinct mechanisms of regulating TEs. The C. albicans parasexual cycle also has the potential to generate high levels of genetic diversity, which could include dramatic changes in TE content and diversity. TE content and distribution changes during experimental evolution of C. albicans through many rounds of the parasexual cycle could be compared to TE content and distribution in the other three species after evolution through many rounds of their sexual cycles. Such studies could test whether TE content and distribution changes much more rapidly in C. albicans or if TEs in this species have any special relationships with their host to protect them from dramatic copy number changes during parasexual reproduction.
The types of TEs present and their abundances also vary between the four species. The Cryptococcus neoformans genome has the greatest TE content and diversity, Candida albicans has moderate TE diversity, but low TE content, while Saccharomyces cerevisiae and Schizosaccharomyces pombe have very low TE diversity. Comparative studies could explore whether changes in TE content and diversity have similar impacts on different species. For instance, S. cerevisiae strains have been engineered that have up to 10 times the normal content of Ty1 retrotransposons, increasing the total DNA content of the strains by up to 15% [95]. These strains are hypersensitive to DNA-damaging agents, but they exhibit remarkably few other phenotypes, including only modest changes in growth rates [95]. Whether the ability of S. cerevisiae cells to tolerate such a large change in TE content depends on specific characteristics of this species or specific aspects of Ty1 regulation and targeting is unknown. Carrying out similar experiments in other species with different types of TEs could address these issues.
Distributions of TEs are also dramatically different in each species (Fig. 2). These distributions reflect both insertion preferences of the TEs themselves and selection processes that act on insertions after integration [96]. TEs are frequently targeted to particular genomic regions through interactions with specific host proteins that have roles in transcription and chromatin regulation, and the three-dimensional arrangement of DNA sequences in the nucleus can also influence targeting [96]. TE distributions in these yeasts likely reflect the evolution of specific TE-host relationships that could impact cellular phenotypes and adaptability [96], and these relationships could be further explored through comparative studies. Expressing TEs from one species in a different species could help address the contribution of TEs themselves, interactions between TEs and conserved proteins or chromatin features, and specialized TE-host interactions to TE distribution biases. For instance, the introduction of the Candida albicans non-LTR retrotransposon Zorro3 into Saccharomyces cerevisiae resulted in targeted retrotransposition with subtle differences in the outcome of reverse transcription between hosts [97]. This indicates that Zorro3 targeting likely depends on element-encoded proteins or interactions with conserved features of DNA/chromatin.
Expressing TEs from one species in different species could also address other aspects of TE biology. Growing cells engineered to harbor types of TEs not normally present in their genomes for long time periods could provide a means to characterize TE copy number and distribution changes prior to the evolution of specific host-TE interactions. Alternatively, specific TE-host relationships identified in one species could be engineered in a diverse species to further explore relevant mechanisms. TE regulation/activity in the pathogenic yeasts would need to be better characterized for many such studies, though.
Informative comparative studies could also involve analyzing multiple wild strains of one species, or comparing any of the four species to more closely related species. Substantial TE content variation in geographically diverse isolates of S. cerevisiae (from a few-fold to approximately 10-fold) provides an opportunity to identify alleles in these strains or changes in the TE-host relationship that may have contributed to these differences, in addition to providing diverse TE landscapes for functional studies [66, 98]. Interestingly, the reference S. cerevisiae genome has a high TE content (3.5%) compared with genomes of many other isolates that have ~ 1–2% TE content [66]. The identification of horizontal transfer of Ty2 from S. mikatae to S. cerevisiae provides the opportunity to compare TE regulation of the same element in these two different host species [67]. Schizosaccharomyces japonicus harbors 10 retrotransposon families present near centromeres and telomeres, while three other Schizosaccharomyces species (including S. pombe) harbor zero to two retrotransposon families and lack retrotransposon sequences near centromeres and telomeres [58]. The reduced presence of retrotransposons in these latter three species is correlated with the evolution of the Cbp1/Abp1 protein family in these species after they diverged from S. japonicus, and this protein family regulates centromeric heterochromatin and retrotransposon silencing [58]. The Tj1 S. japonicus retrotransposon frequently integrates upstream of tRNA genes at centromeres when expressed in S. pombe, consistent with its location at centromeres in S. japonicus, but in contrast to S. pombe Tf1 and Tf2 [99]. This indicates that Tj1 targets these sites based on conserved chromosomal features or protein interactions. These last two studies provide a good example of how comparative approaches that are followed through to functional assays can begin to provide valuable information about host-TE relationships. In the next section, I highlight differences in the presence and nature of RNA interference in the four yeasts that could contribute to the diversity of their TE landscapes.
Diversity in RNA interference pathways
There is a wide diversity in the presence and nature of homology-dependent silencing mechanisms among fungi [100]. RNA interference (RNAi) is one such mechanism that minimally depends on production of short-interfering RNA molecules (siRNA) by Dicer proteins (Dcr), followed by loading of small RNAs onto Argonaute proteins (Ago) and into RNA-induced silencing complexes (RISC), resulting in targeted cleavage (slicing) of homologous RNA molecules or other forms of gene silencing (inhibition of transcription or translation) [100, 101]. RNAi in fungi also frequently involves an RNA-dependent RNA polymerase (RdRP or Rdp), similar to what is observed in plants [100, 101]. Endogenously produced siRNAs (18–30 nucleotides) frequently target TEs to repress mobility of these elements (though in animals piwi-interacting RNA, or piRNA, pathways frequently contribute to TE repression) [101]. Not all fungi have RNAi pathways, and the relative importance of RNAi for controlling TEs can also vary [100]. Here, I summarize the main aspects of RNAi (or note its absence) for the four yeast species as an example comparison of TE regulation in these species.
Candida albicans
One Ago protein (Ago1), a noncanonical Dcr protein (Dcr1) and a Dicer-like protein that does not appear to be catalytically active (Cdl1) are present in C. albicans, but no Rdp homolog has been identified [102,103,104]. Cell extracts or purified Dcr1 have in vitro Dcr-like cleavage activity [103, 104], and small RNAs corresponding to TEs, including the Zorro elements discussed earlier, have been cloned [103]. However, expression of an inverted repeat construct to generate hairpin RNA as a substrate for siRNA production failed to trigger gene silencing, even though cell extracts could produce small RNAs from the hairpin RNA in vitro [105]. It has been reported that introduction of a synthetic siRNA silenced the EFG1 gene, but the mechanism of the silencing and the requirement for RNAi proteins was not investigated [106]. Regulation of the relatively low level of TEs in the C. albicans genome through RNAi has not been reported.
In contrast, homozygous dcr1∆/dcr1∆ mutants are defective for processing rRNA and snRNA transcripts in C. albicans, a function that appears to be independent of Ago1 [104]. Homozygous dcr1∆/dcr1∆ mutants were only recovered when an inducible copy of DCR1 was integrated into the genome [104]. In the absence of DCR1 induction, mutants exhibited poor growth and accumulated lower levels of siRNA, but these phenotypes were not seen in ago1∆/ago1∆ mutants [104]. These authors suggest that the role of Dcr1 in rRNA and snRNA processing is responsible for the growth defect of mutants and compare C. albicans Dcr1 to the Saccharomyces cerevisiae Rnt1 protein, an RNase III enzyme that also contributes to rRNA and snRNA processing.
Cryptococcus neoformans
RNAi has been shown to regulate TEs in C. neoformans [56, 107]. The C. neoformans serotype A genome harbors a single AGO1 gene, while the serotype D genome harbors two – AGO1 and AGO2, and both genomes have two DCR genes (DCR1, DCR2) and a single RDP1 gene [12, 102, 107]. Sequences of these RNAi genes are distinctive from homologs in other basidiomycetes, including the absence of a DEAD/DEAH box helicase domain in DCR1 and DCR2 [102]. Expression of plasmid copies of inverted repeat constructs designed to produce hairpin RNA to two different genes causes gene silencing, demonstrating the existence of an RNAi pathway in C. neoformans [108]. Ago1, Dcr2, and Rdp1 in particular are important for gene silencing by RNAi, and transcriptome analysis shows that multiple DNA transposons and retrotransposons are upregulated in rdp1∆ mutants [107]. Many small RNAs from mating or vegetative cells correspond to DNA transposons, retrotransposons, and centromeres [56, 109], and centromeres are the major sites of Tcn1–6 LTR retrotransposons [14].
A sex-induced silencing system represses TEs in C. neoformans during opposite-sex and same-sex mating, which depends on Ago1, Dcr2, Rdp1, and to a lesser extent Dcr1 [56, 57]. Expression of all four of these proteins is increased at the translational level during mating. Substantial increases in RNA for a Harbinger DNA transposon and the Tcn1, Tcn3, and Tcn4 retrotransposons are observed in cells placed under mating conditions for ago1∆ and rdp1∆ single mutants, and dcr1∆dcr2∆ double mutants [56]. RNA levels for T2 and T3 DNA transposons are also increased in rdp1∆ mutants under mating conditions [57]. Tcn1 H3K9 methylation is reduced in wild type and rdp1∆ cells during mating, indicating that chromatin changes may lead to increased transcription of retrotransposons during mating [56]. Use of the FRR1 gene as a transposon trap shows evidence for increased frequencies of gene disruption by DNA transposons during both opposite-sex and same-sex mating [56, 57]. Overall, RNAi protects the C. neoformans genome from TE insertions during mating.
While Ago1, Dcr1, and Dcr2 were reported to localize with P-bodies in the cytoplasm, Rdp1 localizes to nuclei [56]. A subsequent study identified a nuclear Rdp1 and Ago1 complex that associates with the spliceosome, the Spliceosomal-Coupled and Nuclear RNAi (SCANR) complex [109]. This complex produces siRNA from transcripts that spend a long time associated with the spliceosome due to inefficiently spliced introns. A number of the transcripts most strongly regulated by this pathway include transcripts for DNA transposase genes [109]. These results indicate that C. neoformans has an additional RNAi pathway that represses intron-containing DNA transposons.
Saccharomyces cerevisiae
There is no RNAi pathway in S. cerevisiae and there are no AGO, DCR, or RDP homologs present in the genome [102, 103]. This loss of RNAi in S. cerevisiae and related yeasts has been proposed to have resulted from the presence of double-stranded RNA (dsRNA) killer viruses in these yeasts [110]. In S. cerevisiae, cytoplasmic dsRNA M viruses and L-A viruses together result in stable maintenance of killer toxin-producing and toxin-immunity phenotypes [111]. Yeast cells that produce killer toxins and are themselves immune to the toxin outcompete sensitive yeast cells, providing a selective advantage [111]. RNAi is incompatible with maintenance of the dsRNA killer virus system [110], so the presence of an RNAi pathway can create a disadvantage for cells. An RNAi pathway capable of gene silencing can be engineered in S. cerevisiae cells by expressing AGO1 and DCR1 from the related yeast S. castellii. Interestingly, the engineered S. cerevisiae cells produce small RNAs that match Ty LTR retrotransposons and repress Ty1 mobility (only Ty1 was tested) [103, 110].
Schizosaccharomyces pombe
RNAi has an important role in regulating heterochromatin formation in S. pombe [112], but also contributes to silencing of retrotransposons [113]. S. pombe has one homolog for each type of RNAi protein – Ago1, Dcr1, and Rdp1 [21, 114]. RNAi proteins repress dg and dh centromeric repeats that produce sense and antisense transcripts by degrading their transcripts and promoting H3K9 methylation at those sites, a histone mark associated with heterochromatin [112]. A substantial proportion of siRNA corresponds to dg and dh centromeric sequences, and formation of double-stranded hairpin regions in transcripts from these elements appears to contribute to siRNA formation [21, 115, 116]. Single RNAi component mutants are compromised for centromere function, exhibiting lagging chromosomes during anaphase and reduced cohesin binding at centromeres [117, 118].
In contrast to their significant effects on centromeric repeats, RNAi proteins have only a modest effect on Tf2 LTR retrotransposon expression. Tf2 RNA is reduced only 1.5 to 2-fold in ago1∆, dcr1∆, and rdp1∆ single mutants [119]. Dcr1 and Rdp1 bind to LTRs, based on an assay that detects transient binding to chromosomal sites, and LTR RNA levels are increased 6 to 8-fold in single-gene RNAi mutants [120]. However, Tf2 ORF RNA levels are increased by 2-fold or less, again supporting a modest influence on retrotransposon expression [120]. Multiple studies did not detect retrotransposon-derived siRNAs or detected very low levels of retrotransposon-derived siRNAs by Northern blotting and RNA sequencing [115, 116, 120].
A more recent study demonstrated that very low levels of retrotransposon-derived siRNAs are due to exosome-mediated degradation of Tf2 transcripts [113]. Small RNA sequencing in rrp6∆ mutants compromised for exosome-mediated RNA degradation identified siRNAs corresponding to a number of genomic loci including Tf2 [113]. Ago1 and Dcr1-dependent H3K9 methylation of Tf2 ORFs occurs in rrp6∆ mutants, and siRNA production depends on Ago1, Dcr1, and Rdp1. Furthermore, Tf2 siRNA production and H3K9 methylation in rrp6∆ mutants require the Clr4 H3K9 methyltransferase, the Red1 RNA surveillance protein, the poly(A) polymerase Pla1, and the poly(A)-binding protein Pab2. Tf2 RNA is much more abundant when the rrp6∆ mutation is combined with ago1∆, dcr1∆, rdp1∆, clr4∆, or pla1 mutations [113]. Ago1-dependent Tf2 siRNAs and H3K9 methylation are also observed in wild type cells grown in low glucose [113]. Intriguingly, Tf2 siRNA and H3K9 methylation in rrp6∆ mutants depends on Nrl1-dependent splicing of a cryptic intron in Tf2 elements [121]. This is reminiscent of the SCANR pathway in Cryptococcus neoformans that targets inefficiently spliced RNAs for RNAi [109].
Diversity in the presence and nature of RNAi in these species could form the basis for comparative studies of TE regulation. Only two of the four yeast species regulate TEs through RNAi. In S. pombe, RNAi is redundant with other TE restriction mechanisms but also plays an important role in establishing centromeric heterochromatin. In C. neoformans, RNAi appears to be a major pathway regulating TEs, and this yeast has the greatest TE content and diversity of the four species. Comparative studies could determine whether species lacking RNAi have a greater diversity of host factors that regulate TEs to make up for the absence of RNAi. Studies could also test whether TEs in species with and without RNAi have evolved to have different relative levels of activity or types of impacts on their hosts.
Retrotransposition and its regulation in yeasts
Retrotransposons are the only TEs in the four yeast species that have been studied in great detail, and LTR retrotransposons are the only type of TEs common to all four genomes. Non-LTR and LTR retrotransposons encode distinct types of proteins and carry out reverse transcription through distinct mechanisms for their mobility (Fig. 3) [122]. Elements in each retrotransposon subclass frequently have two ORFs, though some elements have a single ORF. Transcription of non-LTR and LTR retrotransposons is initiated utilizing internal promoters in 5′ untranslated regions or LTR sequences and terminates at the very 3′ end or in the 3′ LTR, respectively. Translation of non-LTR element ORF1 and ORF2 proteins (or a single protein) is followed by binding of these proteins to the mRNA and movement of the ribonucleoprotein complex to the vicinity of potential chromosomal target sites (Fig.3b). Target-primed reverse transcription takes place when an endonuclease domain (EN) in ORF2p (or in an element’s single ORF protein) nicks a genomic site, providing a DNA end with a 3′-OH that can be extended by the ORF2p reverse transcriptase (RT) using the mRNA as a template. Reverse transcription begins at the poly(A) tail of the mRNA and continues towards the 5′ end of the element, but does not always proceed to the very 5′ end of the mRNA template, which produces 5′ truncated elements. Nicking of the other DNA strand at the target site followed by second strand synthesis results in a new copy of the retrotransposon at the target site, but the details of second strand synthesis have not been characterized (Fig. 3b) [122].

Structure and replication of retrotransposons. a Schematic representations of long terminal repeat (LTR) and non-LTR retrotransposons not drawn to scale. Blue boxes with white arrowheads indicate LTRs, and U3-R-U5 indicate regions of LTRs. EN- endonuclease, IN- integrase, pA- poly(A) sequence, PR- protease, RT- reverse transcriptase, RT-RH- reverse transcriptase/RNase H, UTR- untranslated region. b Graphic representation of major steps of retrotransposition through target-primed reverse transcription for non-LTR retrotransposons. c Graphic representation of major steps of retrotransposition for LTR retrotransposons. Colors of ellipses correspond to proteins from panel a, wavy lines represent RNA, boxed arrowheads represent LTRs, and thin blue arrows represent DNA strands newly synthesized by reverse transcriptase.
Terminally redundant LTR retrotransposon mRNA starts with repeat (R) and unique 5′ (U5) LTR sequences from the 5′ LTR and ends with unique 3′ (U3) and R sequences from the 3′ LTR (Figs. 3a and 4). The mRNA is often translated to produce only Gag protein or less often a Gag-Pol fusion protein by frameshifting between the gag and pol ORFs (Fig. 3c), though some elements have a stop codon between gag and pol. Inefficiency of the frameshifting or other mechanisms result in an excess of Gag protein relative to Pol. Gag protein association with the mRNA and Gag-Pol leads to formation of virus-like particles (VLPs) that encapsidate a dimer of RNA and Gag-Pol (Fig. 3c). A protease domain (PR) of Pol cleaves Gag-Pol into Gag, PR, integrase (IN), and reverse transcriptase/RNase H (RT) domains. Reverse transcription occurs in the VLP using a primer binding site (PBS) adjacent to the 5′ LTR that is often complementary to part of a tRNA molecule (extrachromosomal priming, Fig. 4). The initial complementary DNA (cDNA) molecule is a short molecule including U5 and R LTR sequences that can anneal to R sequences at the 3′ end of the mRNA to continue reverse transcription of the minus strand cDNA. RNase H degradation of the mRNA template is incomplete, leaving behind a short purine-rich RNA sequence, the polypurine tract (PPT), that is typically just upstream of the 3′ LTR (Fig. 4). Reverse transcription starting at this point generates a short plus strand cDNA ending in PBS sequences that can anneal to the PBS sequences at the other end of the minus strand cDNA to produce a full plus strand cDNA (Fig. 4 and see [123] for a more detailed discussion of the steps of reverse transcription). IN binds to the double-stranded cDNA to integrate it at a chromosomal site (Fig. 3c) [122].

Majors steps of reverse transcription for LTR retrotransposons. The gray box represents retrotransposon RNA with major sequence features not drawn to scale: LTR sequences U3, R, and U5; gag and pol ORFs; PBS- primer binding site; PPT- polypurine tract. The lavender shape indicates the tRNA primer, dashed blue arrows indicate newly synthesized DNA, and solid blue lines indicate DNA synthesized in a previous step.
The mechanism, regulation, and impact of retrotransposition has been much more intensively studied in Saccharomyces cerevisiae and Schizosaccharomyces pombe than in the other two yeasts. Two retrotransposons (Tca2 and Zorro3) have been studied in some detail in Candida albicans, but transposition of particular retrotransposons (or other TEs) in Cryptococcus neoformans has not been studied in detail. Further study of TEs in the pathogenic yeast species will facilitate comparative studies of these elements.
Candida albicans
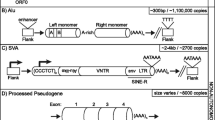
Retrotransposition of Tca2 LTR retrotransposons and Zorro3 non-LTR retrotransposons has been demonstrated using elements harboring retrotransposition-indicator genes [45, 46]. Tca2 was initially discovered because one particular strain of C. albicans accumulates very high levels of extrachromosomal Tca2 DNA [41]. Tca2 is a member of the Pseudoviridae family (Ty1/copia group, IN domain upstream of RT domain in pol) flanked by 280 bp LTRs, is 6426 bp long, and has 972 bp and 4728 bp gag and pol ORFs that are separated by a stop codon (Fig. 5). Tca2 RNA levels are higher at 37 °C than 27 °C, and chromosomal elements harboring a retrotransposition-indicator gene between pol and the 3′ UTR retrotranspose at 37 °C [46]. A small set of marked Tca2 insertions shows a preference for integration in a 300 bp window upstream of start codons [46], but the factors responsible for this bias are unknown.

Characterized retrotransposons from three yeast species. Schematic representations of the indicated retrotransposon families drawn to scale. Boxed arrowheads indicate LTRs. Domains of proteins: EN- endonuclease, IN- integrase, PR- protease, RT- reverse transcriptase, RT-RH- reverse transcriptase-RNase H, ZF- zinc finger. pA- poly(A) sequence. The gag ORF of Ty1-Ty4 is raised relative to pol to indicate a + 1 translational frameshift between gag and pol. A vertical line with “stop” indicates the site of one or more stop codons.
The identification of a stop codon between Tca2 gag and pol led those authors to suggest that translation of Tca2 Pol protein might rely on stop-codon suppression using an 8-bp purine-rich sequence present immediately after the gag stop codon that is followed by a sequence predicted to form a pseudoknot in the RNA [41]. A subsequent study did not find any evidence that a gag-pol junction sequence including 150 bp upstream and downstream of the gag stop codon could allow read-through translation of a reporter gene [124]. In contrast, deletion of the predicted pseudoknot region in the central portion of the junction sequence increased reporter gene expression, and weak promoter activity in the junction sequence was reported [124]. These authors suggested that the weak promoter activity was unlikely to be responsible for appropriate levels of pol expression for retrotransposition, because they found that the promoter was not induced by growth at 37 °C [124], a temperature that induces full-length Tca2 transcription and retrotransposition [46]. The mechanism responsible for expression of appropriate levels of Tca2 pol for retrotransposition therefore remains to be determined.
C. albicans Zorro3 non-LTR retrotransposons (L1 superfamily) have very short 5′ UTRs and are flanked on both sides by poly(A) sequences (Fig. 5) [43, 45]. Poly(A) sequences are expected at the 3′ end of non-LTR elements due to the mechanism of target-primed reverse transcription, but the presence of poly(A) sequences at both flanks is unusual. Zorro3 is 5763 bp long, with a 1608 bp ORF1 and 3450 bp ORF2 separated by multiple stop codons [43]. Comparison of allelic sites lacking Zorro3 insertions in other strains reveals that Zorro3 integrates into poly(A) sequences [45]. New Zorro3 insertions resulting from a chromosomal Zorro3 element harboring a retrotransposition-indicator gene and expressed from a heterologous promoter are targeted to poly(A) sequences in intergenic regions [45]. Insertions into central regions of poly(A) sequences could explain the presence of poly(A) sequences on either side of Zorro3, and target site duplications could potentially increase the length of these poly(A) tracts. However, the total length of the poly(A) tracts on either side of new Zorro3 insertions can be much more than twice the length of the original poly(A) target [45], indicating that other mechanisms likely contribute to the length of these poly(A) tracts. Many newly inserted copies include the full 5′ end. Zorro3 retrotransposition is temperature-sensitive, with no retrotransposition detected at 37 °C, modest levels at 27 °C, and much higher levels at 22 °C [45].
Zorro3 is also active in S. cerevisiae, even though non-LTR elements are not present in that yeast [97]. Expression of a marked Zorro3 element in which all CUG codons were engineered to be UCU (since C. albicans CUG specifies serine [19]) results in detectable retrotransposition events. The frequency of new insertions is dramatically reduced by mutations in the zinc finger region of ORF1, as well as the ORF2 EN or RT domains [97]. New insertions are only rarely full-length and show evidence of RT template-switching to other RNA or DNA sequences, in contrast to results in C. albicans, but all targeted poly(A) sequences. Comparing Zorro3 insertions in the native host to those in S. cerevisiae demonstrates that the Zorro3 integration bias is either independent of cellular factors or relies on factors conserved between C. albicans and S. cerevisiae.
The distinct temperature preferences of Tca2 and Zorro3 elements are intriguing in the context of C. albicans opaque-white cell-type switching. Growth at 37 °C promotes switching to white cells [35] and also promotes Tca2 expression and mobility [46]. Opaque cells are much more mating-competent and are stable at lower temperatures (~ 25 °C) [35], in the range of temperatures permissive for Zorro3 mobility [45]. Also, the Tca1 LTR-retrotransposon is a 5.6 kb nonautonomous element that does not have any ORF sequences, but Tca1 RNA levels are higher at 25 °C than 37 °C [40, 125]. Potential specific connections between retrotransposon regulation, cell-type switching, and mating in C. albicans have not been explored.
Saccharomyces cerevisiae – overview of retrotransposons
Two analyses of the reference genome identified 51 LTR retrotransposons in S. cerevisiae: 32 Ty1, 13 Ty2, two Ty3, three Ty4, and one Ty5 [65, 67]. In addition, two truncated copies each of Ty1 and Ty2, and one truncated copy of Ty4 were identified [67]. The single Ty5 element is not full length, but functional copies of Ty5 from S. paradoxus have been studied in S. cerevisiae [77, 126], and information presented for Ty5 is based on those elements. Ty3 is the only Metaviridae family member (RT upstream of IN in pol), while all other elements belong to the Pseudoviridae family (Fig. 5). LTRs for these elements range from 251 (Ty5) to 371 (Ty4) bp and full-length elements range from 5.4–6.2 kb (Fig. 5) [77, 127,128,129]. Ty1–4 each have gag and pol ORFs, with pol in the + 1 reading frame, and inefficient frameshifting results in the required excess of Gag to Pol protein for retrotransposition [129,130,131,132]. For Ty5, Gag and the functional domains of Pol are processed from a single polypeptide by Ty5 PR, but a Gag to Pol ratio of approximately 5:1 is observed, indicating that the Pol protein domains may be less stable than Gag [126]. Ty1 and Ty3 are the most extensively characterized elements, even though Ty2 is more abundant that Ty3. Ty1 and Ty2 have very similar sequences; relative to Ty1, Ty2 has a 1 bp deletion in its LTR, a divergent gag sequence, and a short divergent region in pol [133]. Ty1 expression and mobility are substantially higher than Ty2, though, and most studies have focused on Ty1 rather than Ty2 [133].
There are excellent recent reviews that discuss the Ty1 and Ty3 retrotransposition cycles in detail [133, 134], which will not be the focus of the following sections. Instead, I will compare and contrast selected aspects of their regulation with some discussion of Ty5 to consider overall regulation of retrotransposons in S. cerevisiae. Multiple large-scale genetic screens for cellular factors (host factors) that regulate Ty1 and Ty3 have identified hundreds of genes that regulate these retrotransposons with roles in cell cycle regulation, chromatin, DNA damage/replication, mRNA degradation/translation regulation, nuclear transport, vesicular trafficking, and other cellular processes [135,136,137,138,139,140,141]. Approximately 15% of these genes overlap for Ty1 and Ty3 [134]. The annotation of the reference genome identifies 175 genes that increase retrotransposon activity when mutated and 311 genes that decrease retrotransposon activity when mutated [142]. This large number of regulators likely reflects the complex replication cycle of retrotransposons [133, 134]. It would also be interesting to determine whether the absence of RNAi and related mechanisms in S. cerevisiae has resulted in the evolution of a greater number of cellular genes regulating retrotransposons, though, or if the presence of RNAi and similar mechanisms in other organisms masks the role of some cellular factors in TE regulation.
Saccharomyces cerevisiae – example host factors regulating Ty1, Ty3, or Ty5
Ty1, Ty3, and Ty5 are all regulated by the pheromone-response pathway (mating-response) in haploid cells, and each is repressed at the transcriptional level by the MATa1/α2 repressor in diploid cells [133, 134, 143]. Ty1 retrotransposition is repressed by treatment with mating pheromone (a or α-factor) at a posttranslational step that involves phosphorylation of Ty1 Gag, changes in VLP structure, and reduced cDNA levels [144]. The pheromone-response pathway shares components with the filamentous-growth pathway, which is triggered by poor nutritional conditions and leads to formation of pseudohyphae, and the Fus3 MAP-kinase for the pheromone-response pathway inhibits cross activation of the filamentous-growth pathway [145]. Fus3 activity inhibits Ty1 retrotransposition at transcriptional and posttranslational steps [146, 147]. Increased levels of Gag and Pol proteins associated with VLPs are observed in fus3∆ mutants, supporting an increase in protein stability or VLP assembly in the absence of Fus3 [146]. The Ste12 transcription factor activates genes in response to both the pheromone-response and filamentous-growth pathways, but Ste12 works together with Tec1 for the filamentous-growth pathway. Ty1 expression and retrotransposition are increased by the filamentous growth pathway and its MAP-kinase Kss1 in both haploid and diploid cells [146, 148], as well as by Ste12 and Tec1 [149,150,151]. These observations indicate that the filamentous-growth pathway positively regulates Ty1, while the pheromone-response pathway negatively regulates Ty1.
In contrast to Ty1 retrotransposition, both Ty3 and Ty5 retrotransposition are activated in mating cells [143, 152]. Transcription and retrotransposition of Ty5 elements located at typical insertion sites in heterochromatin are substantially increased with exposure of haploid cells to mating pheromone, though only modest effects are observed for an element in euchromatin [143]. Ty3 transcription is strongly induced by exposure to mating pheromone [153, 154]. Ty3 VLPs form and mature in cells arrested in G1 with α-factor, but cDNA is not detected and retrotransposition does not occur until cells are released/recover from arrest [155, 156]. Inhibition of retrotransposition depends on cell cycle arrest itself, rather than other aspects of the mating response [156]. Ty3 retrotransposition frequently occurs in haploid cells of opposite mating types allowed to mate and form diploid cells [152]. Ty3 proteins and RNA form cytoplasmic foci in α-factor-treated cells and mating cells that likely represent sites of VLP assembly [155, 157].
Proteins and mRNA for both Ty1 and Ty3 form cytoplasmic foci, retrosomes, that appear to be precursors to VLP formation and associate with cytoplasmic processing bodies (P-bodies) to differing degrees [155, 157,158,159]. P-bodies are cytoplasmic protein-RNA granules that can regulate mRNA degradation and translation [160]. Ty3 retrosomes formed in response to mating pheromone or expression from an inducible heterologous promoter frequently localize with or are found very near to foci of P-body factors Dcp2, Dhh1, Lsm1, Pat, and Xrn1 [155, 157], which have roles in either mRNA decapping/degradation or translational repression [160]. Expression of Ty3 from an inducible promoter also increases the size or leads to formation of P-body protein foci [157]. Fewer retrosomes are observed in dhh1∆, lsm1∆, and to a lesser extent xrn1∆ and dcp2∆ mutants [155]. Moreover, dhh1∆ and xrn1∆ mutants have reduced Ty3 retrotransposition, and dhh1∆, lsm1∆, and xrn1∆ mutants affect RNA levels or packaging in VLPs to reduce cDNA levels [138, 155]. Ty1 cDNA and retrotransposition are also reduced in P-body component mutants, such as dcp2∆, dhh1∆, lsm1∆, pat1∆, and xrn1∆, without substantial effects on Ty1 RNA and protein levels, though the absence of Xrn1 severely reduces RNA packaging in VLPs [158, 159]. Retrosome formation is also greatly reduced in mutants lacking P-body proteins [158]. Unlike Ty3, though, Ty1 retrosomes show only limited overlap with foci of P-body proteins [159], and retrosomes do not form when P-body formation is triggered by glucose deprivation [158, 161]. Despite regulation by common factors, Ty1 and Ty3 again show distinct interactions with the cellular environment.
The positive role of P-body components in Ty1 and Ty3 retrotransposition is intriguing in comparison to TE regulation in other organisms. For instance, Ago1, Dcr1, and Dcr2 proteins localize to P-bodies in Cryptococcus neoformans [56], indicating that P-bodies could be sites of TE repression in this yeast. The situation in C. neoformans is more similar to the many other organisms in which Ago and Dcr proteins repress TEs [101], because Ago proteins, siRNAs, and microRNAs can be found at P-bodies in animal cells [162]. Mammalian LINE-1 (L1) retrotransposon protein-mRNA foci only sometimes have been localized to P-bodies but have also been detected in stress granules [163,164,165], related protein-RNA granules that regulate translation of sequestered RNAs [160]. L1 association with P-bodies can lead to degradation of L1 ribonucleoproteins through autophagy [165]. Studies that contrast mechanisms responsible for the positive influence of P-body factors on TEs in S. cerevisiae with potential negative regulatory mechanisms in Cryptococcus neoformans would provide a more complete understanding of the roles of P-body factors in TE regulation.
The nuclear envelope does not break down during cell division in yeasts, which means that retrotransposon cDNA and integrase protein (at minimum) must be able to enter the nucleus through nuclear pore complexes (NPC) consisting of nucleoporin proteins (Nups) [166]. Ty1 and Ty3 each have basic bipartite nuclear localization signals in C-terminal regions of their IN proteins that are required for IN nuclear localization and retrotransposition (Ty1) [167, 168] or IN nucleolar localization and in vitro integration (Ty3) [169]. Ty1 IN nuclear localization is known to rely on Ran, the karyopherin alpha protein Srp1, the karyopherin beta protein Kap95, and IN interacts directly with Srp1 [170]. Certain Nup mutants decrease Ty1 retrotransposition [136, 140, 141], while some Nup mutants increase and others decrease Ty3 retrotransposition [137, 138]. A more systematic analysis of Ty1 retrotransposition in 19 NPC mutants identified two Nups that restrict retrotransposition and nine Nups that contribute to retrotransposition [171]. Ty3 Gag in VLPs and recombinant Gag interact with Nups containing GLFG (glycine-leucine-phenylalanine-glycine) repeats, and the nucleocapsid domain of Gag enters the nucleus when expressed on its own in a manner that depends on GLFG proteins [172]. The interaction of Gag with Nups was proposed to lead to structural changes in VLPs at nuclear pores, promoting nuclear entry of cDNA, IN, and possibly other VLP components [172].
The roles of many NPC proteins in Ty1 retrotransposition have been studied in greater detail. As just noted, 11 of 19 Nups tested (of about 30 total NPC proteins [166]) regulate Ty1 retrotransposition frequencies, but different Nups have variable and usually modest effects on Ty1 RNA, Gag, and cDNA levels [171]. Interestingly, none of the Nups is required for the nuclear localization of the Ty1 IN domain when expressed on its own. The common theme identified is that all Nups tested, including those that do not affect retrotransposition of a Ty1 marked with a retrotransposition-indicator gene, contribute to the targeting of Ty1 upstream of tRNA genes [171]. Most Nups are required for high frequencies of insertions upstream of two target tRNA genes, and partial deletions of two nuclear basket Nups (Nup1 and Nup60) affect which sites upstream of tRNA genes are targeted. This is not always simply due to lower Ty1 integration, though, because the absence of Nup1, Nup60, Mlp1, or Mlp2 nuclear basket proteins leads to frequent Ty1 insertions into subtelomeric regions [171].
Another recent study of Ty regulation by nucleoporins provides an excellent model for using a comparative approach to probe the coevolution of retrotransposons and their host cells. Nucleoporins from 29 Saccharomyces species were examined for high rates of nonsynonymous substitutions relative to synonymous substitutions as a way to identify proteins that have been recently evolving to control retrotransposons [173]. NUP84 and NUP82 were among the Nup genes that appear to be evolving under positive selection. NUP84 is known to contribute to Ty1 and Ty3 retrotransposition [136, 138, 140], while NUP82 is an essential gene not previously identified as a Ty regulator, but expression of a dominant-negative Nup82 protein reduces Ty1 retrotransposition [173]. Replacing the S. cerevisiae NUP84 allele with an allele from one of three other Saccharomyces species (S. mikatae, S. kudriavzevii, or S. bayanus) leads to moderate but significant increases or decreases in Ty1 retrotransposition, without affecting Ty3 mobility [173]. Similar experiments for NUP82 identified two different alleles that specifically increased either Ty1 or Ty3 retrotransposition [173]. These results indicate that the corresponding S. cerevisiae alleles have evolved to be more restrictive or permissive for specific Ty elements. Overall, this type of approach provides a more sophisticated perspective on the evolution of retrotransposon regulation.
Saccharomyces cerevisiae – Ty1, Ty3, and Ty5 insertion biases & regulation by stress.
The insertion biases of Ty1, Ty3, and Ty5 are all due to specific interactions between their IN proteins and cellular factors present at their preferred target sites, which regulate the mutagenic potential of these elements. Ty5 is the most distinct of the three, with an integration bias for heterochromatin near telomeres and the silent mating type loci [77]. This bias is achieved by an interaction between a targeting domain of six amino acids in Ty5 IN that interacts with the Sir4 heterochromatin protein [174, 175]. This interaction provides the initial localization of IN and cDNA to heterochromatin, but the specific site of insertion then appears to depend on the presence of nucleosome-free regions that are accessible for integration [79]. Ty3 integrates 0–20 bp upstream of tRNA genes, near their transcription start sites [73,74,75,76], which is dependent on the transcriptional competence of the RNA Pol III promoter [74, 75]. Recapitulation of targeted insertions in vitro using VLPs, an RNA Pol III target gene, and purified RNA Pol III transcription factors shows that integration depends on the Pol III transcription factors TFIIIB and TFIIIC [176]. Specifically, TFIIIB components TATA-binding protein (TBP) and Brf1 are the minimum factors needed for targeted insertions, though Bdp1 contributes to integration efficiency, and TFIIIC is required only when needed to load TFIIIB at a promoter [177, 178].
Ty1 most often integrates within ~ 1 kb upstream of RNA Pol III genes [69, 70], which depends on the transcriptional competence of the RNA Pol III-gene promoter and shows a periodicity that depends on nucleosome positioning and structure [70,71,72]. This Ty1 integration bias is due primarily to specific interactions between a C-terminal region of Ty1 IN and the RNA Pol III subunit Rpc40 [179], as well as Rpc53, Rpc34, and Rpc31 [180]. An initial screen that identified the Rpc40-Ty1 IN interaction also identified interactions with Ty2 and Ty4 IN domains, but this was not further investigated [179]. Temperature-sensitive rpc40 and rpc34 alleles reduce integration upstream of a tRNA gene and retrotransposition of a Ty1 marked with an indicator gene, without substantially altering overall tRNA gene expression [180]. Replacing the essential Rpc40 protein with the corresponding protein from Schizosaccharomyces pombe very strongly reduces integration upstream of RNA Pol III-transcribed genes, without having a strong effect on overall retrotransposition frequencies [179]. Interestingly, expression of the S. pombe Rpc40 protein results in frequent mistargeting of Ty1 to telomeres and subtelomeres [179], similar to the effect of certain Nup mutants [171].
A variety of stress conditions can influence Ty element expression and mobility, particularly for Ty1. For example, Ty1 and Ty3 transcription and Ty1 retrotransposition are increased in response to adenine starvation [181, 182]. Ty3 retrotransposition is inhibited by high growth temperature (37 °C) or ethanol stress by a mechanism that blocks VLP formation and results in degradation of Ty3 proteins [183]. Ty1 retrotransposition is very temperature sensitive, with an optimum temperature around 20 °C [184, 185], which is at least partly due to temperature-sensitivity of the Ty1 protease [185]. Ethanol stress increases Ty1 transcription [186], oxidative stress increases Ty1 retrotransposition [187], and nutritional stress increases Ty1 transcription and retrotransposition through the filamentous-growth pathway previously discussed [146, 148]. Both Ty1 and Ty3 are restricted by factors that contribute to DNA repair or replication [133, 134], and Ty1 retrotransposition is also increased by exposure to various DNA-damaging or replication-stress agents, including ionizing radiation, hydroxyurea, methylmethane sulfonate, and 4-nitroquinoline-1-oxide [133]. DNA damage signaling through the Rad24 and Rad9 proteins increases processing of Ty1 Pol proteins, RT activity, and cDNA formation to increase retrotransposition [188].
It is important to highlight a few additional aspects of Ty1, considering that it is the most active and abundant retrotransposon in many S. cerevisiae strains. Ty1 RNA is extremely abundant in haploid cells, accounting for approximately 0.1–0.8% of total RNA and 5–10% of poly(A) RNA [189, 190], and has a half-life of several hours [191]. Much of the regulation of Ty1 therefore occurs at the posttranscriptional level. Ty1 is also regulated through copy-number-control (CNC), by which increasing copy numbers of Ty1 result in decreasing levels of retrotransposition [192]. Ty1 produces short antisense RNAs that initiate in the gag region, have an inhibitory effect on Ty1 retrotransposition, and were considered as potential mediators of CNC [193, 194]. Antisense RNAs are repressed by the 5′-3′ mRNA decay proteins Dcp1, Dcp2, and Xrn1 [193], consistent with the contribution of these proteins to Ty1 mobility [158, 159]. Antisense RNAs are also repressed by the Tye7 transcription factor when Ty1 is activated during adenine starvation [182]. Antisense RNAs were initially reported to inhibit Ty1 transcription [193], but subsequent work shows that antisense RNAs associate with VLPs, reducing levels of mature Pol proteins and RT activity [194]. More recently, Ty1 CNC was shown to depend on production of a truncated Gag protein produced from an internally initiated transcript [195]. This truncated Gag strongly inhibits Ty1 retrotransposition by interacting with the normal Gag protein to decrease VLP assembly and alter VLP structure [195, 196]. Regulation of Ty1 by some ribosome biogenesis proteins and the Mediator transcriptional co-activator occurs at least in part by altering the expression of this truncated Gag protein [197, 198]. Ty1 self-restriction may have evolved as a means of Ty1 stably remaining in an organism that lacks genome-wide small RNA-based repressive mechanisms to limit potentially deleterious unrestricted retrotransposition.
Schizosaccharomyces pombe
Thirteen Tf2 LTR retrotransposons are the only full-length TEs in the reference S. pombe genome [83], but full-length active Tf1 elements are present in other wild type strains [85]. Tf1 and Tf2 are members of the Metaviridae (Ty3/gypsy) group, approximately 4.9 kb in length, have 358 or 349 bp LTRs, and have single ORFs encoding proteins of 1330 or 1333 amino acids (Fig. 5) [85, 199]. Their RT and IN regions are extremely similar in sequence, they share similarities in regions of their LTR sequences, but their gag regions are divergent [85, 199]. The single ORF of each element is translated into one polypeptide that is proteolytically processed into Gag and Pol domains by the PR activity [90, 200]. While Tf1 processing results in mature Gag (capsid, CA), PR, RT, and IN proteins, as expected, Tf2 processing does not separate PR from RT [90, 201]. Retrotransposition of Tf1 and Tf2 has typically been studied by expression of plasmid copies of the elements from inducible promoters [90, 200, 202]. Tf2 mobility, protein levels, and cDNA are much lower than Tf1, and most Tf2 insertions occur by homologous recombination with pre-existing Tf2 sequences [90]. As a result, studies of retrotransposition in S. pombe have focused on Tf1, though many studies of transcriptional regulation have focused on Tf2 elements in the reference genome. An excellent recent review addresses details of the steps of Tf1 retrotransposition and regulation of Tf1 and Tf2 by cellular factors [84], so only certain aspects of Tf1 and Tf2 retrotransposition and regulation will be highlighted in this review.
Two aspects of Tf1 retrotransposition are unusual compared to many other LTR retrotransposons. First, the excess of Gag relative to Pol does not result from differences in translation, but apparently results from increased Pol degradation relative to Gag. Gag and IN (as a measure of Pol) are present in a 26:1 ratio in purified VLPs and stationary phase cell extracts [201]. During log phase there are similar levels of each protein, but as cells continue into stationary phase, IN levels decrease substantially, as cDNA levels increase [201]. These results support degradation of IN, and presumably other Pol proteins, as cells reach stationary phase, but the mechanism is not known. Maturation of VLPs and increased cDNA synthesis as cells approach stationary phase contrasts with Ty1 elements in Saccharomyces cerevisiae, because Ty1 retrotransposition decreases as cells approach stationary phase, which is correlated with more unprocessed or posttranslationally modified Gag [203]. Second, Tf1 is the founding member of a group of retrotransposons that initiate reverse transcription through a self-priming mechanism [204], rather than using a tRNA primer. The first 11 bases of the Tf1 RNA are complementary to the PBS, and RNA structures form that allow base pairing of these two regions, followed by RNase H cleavage between bases 11 and 12 to generate a short RNA primer for reverse transcription [86, 204,205,206].
Many cellular factors regulate retrotransposon RNA levels in S. pombe in addition to the RNAi and exosome pathways previously discussed. Tf2 transcription is repressed by many proteins, including the chromodomain protein Swi6, the HIRA histone chaperone complex, the histone methyltransferases Clr4 and Set1, the RSC chromatin remodeling complex subunit Sfh1, the histone deacetylases Clr3, Clr6, and Hst4, as well as the Nts1 protein present in one specific Clr6 complex [115, 119, 120, 207,208,209,210,211,212,213]. Furthermore, the CENP-B homologs Cbp1/Abp1, Cbh1, and Cbh2 that are involved in establishing centromeric heterochromatin also negatively regulate Tf2 transcription [208]. CENP-B proteins are homologous to transposases of DNA transposons [214, 215], and regulation of Tf2 by CENP-B proteins has been noted as an example of cells co-opting sequences from one type of TE to regulate a different type of TE [208]. Cbp1/Abp1 reduces insertion of Tf1 cDNA into the genome through homologous recombination, recruits Clr3 and Clr6 to Tf2 sequences, and organizes Tf2 retrotransposon sequences into 1–3 nuclear foci, named Tf bodies [208]. Additional factors, such as Set1, Clr3, Clr6, Hst2, Hst4, and the Ku heterodimer are also required for Tf body formation [211, 216]. Ku however does not repress Tf2 transcription, but Ku binding to Tf2 and Tf body formation are reduced by H3K56 acetylation that occurs during S-phase and in response to DNA damage [216].
Incorporation of Tf2 into Tf bodies represses Tf2 mobility [217]. Loss of Cbp1/Abp1 or Set1 each disrupt Tf body formation and results in high mobility of an endogenous Tf2 marked with a retrotransposition-indicator gene [217]. In contrast, absence of individual subunits of the HIRA complex increases Tf2 transcription, but does not prevent Tf body formation, and results in almost no change in Tf2 mobility [217]. Domains of Set1 have different contributions to Tf2 repression and Tf body formation [218]. Use of mutant Set1 proteins compromised only for repression or for Tf body formation shows that loss of Tf body formation increases Tf2 mobility even when repression is maintained, but loss of repression when Tf body formation is maintained does not increase Tf2 mobility [217]. Overall, these results strongly support a repressive role of Tf body formation that is distinct from transcriptional repression. Tf2 transcription is activated by the Sre1 transcription factor in low oxygen conditions [219], and mobility of the marked endogenous Tf2 also increases in response to constitutive Sre1 activity [217].
These observations together with those regarding RNAi and exosome-mediated repression of Tf2 highlight the many layers of cellular defense against retrotransposons. As a further example, the nuclear poly(A)-binding protein Pab2 moderately represses Tf2 RNA levels, but the absence of either Pab2 or Rrp6 (exosome component) can partly suppress the increase in Tf2 RNA observed in the absence of Cbp1/Abp1 [220]. Double mutants lacking Cbp1/Abp1 and Pab2 or Cbp1/Abp1 and Rrp6 accumulate higher levels of Tf2 antisense RNAs than mutants lacking only one of these proteins, which was proposed to trigger increased RNAi-mediated repression of Tf2 [220]. Tf2 antisense RNA increases during middle stages of meiosis, at which point Tf2 mRNA is low, and decreases at later stages, when Tf2 mRNA is high [220]. Antisense RNA may therefore restrict Tf2 retrotransposition during certain stages of meiosis. The Red1 protein that contributes to degradation of meiotic mRNAs also represses Tf2 [221], indicating either that Red1 could contribute to antisense mediated regulation of Tf2 or that multiple lines of defense restrict Tf2 during meiosis.
In contrast to S. cerevisiae Ty1 and Ty3, Tf1 Gag protein exclusively localizes to nuclei in the great majority of stationary phase cells [222], a phase in which retrotransposition is observed [200, 202]. Cosedimentation of Gag, IN, and cDNA supports the interpretation that nuclear Gag signal represents VLPs [222]. Curiously, the Pst1 protein that is present in one specific Clr6 histone deacetylase complex is required for this nuclear localization, with many cells lacking Pst1 showing only cytoplasmic or a mix of cytoplasmic and nuclear Gag [222]. Furthermore, Pst1 positively contributes to Tf1 retrotransposition and cDNA recombination [222], despite the repressive effect of Clr6 noted earlier [119]. The nucleoporin Nup124 interacts with Gag and is also required for Gag nuclear localization [223]. The absence of Nup124 substantially reduces Tf1 retrotransposition without affecting Tf1 protein or cDNA levels [223]. The first ten amino acids of Gag is critical for nuclear import and retrotransposition, but amino acids 20–30 cause nuclear import to specifically depend on the presence of Nup124 [224]. As previously discussed, Ty3 Gag also interacts with nucleoporins, though Ty3 Gag does not localize to the nucleus in the context of VLPs [172], as for Tf1.
Several stress conditions are known to regulate Tf1 expression and retrotransposition, but Tf1 does not show a general response to all stress conditions. Treatments with DNA-damaging agents (bleomycin and 4-nitroquinoline-1-oxide), hydroxyurea, osmotic stress, microtubule depolymerizing agents, and nitrogen starvation do not regulate retrotransposition of Tf1 expressed from an inducible promoter [91]. Retrotransposition frequencies are also not changed over a wide range of growth temperatures (22 °C to 36 °C) [202], which is different from what was discussed for Ty1 and Ty3. Furthermore, Tf1 expression is not activated by Sre1 in response to low oxygen, as Tf2 expression is [219]. Tf1 transcription is induced by a short heat stress (39 °C for 15 min) or a low dose of hydrogen peroxide, but not osmotic stress or cadmium [225]. Retrotransposition of at least some individual elements also increases in reponse to cobalt, zinc, caffeine, or phthalate [226]. There is only limited overlap between the stresses that regulate Tf1 and those that regulate Ty1 in S. cerevisiae.
Tf1 shows a strong integration bias for promoters of RNA Pol II-transcribed genes [93, 94] that is determined by interactions between IN and specific DNA-binding proteins. Tf1 IN interacts with the transcriptional activator Atf1, which can direct integration events to promoters containing Atf1 binding sites in plasmid targets [227, 228]. Integration at specific promoter regions does not depend on high levels of transcriptional activity of those promoters, though [227]. The essential Sap1 DNA-binding protein plays a role in mating-type switching and can arrest replication forks [229,230,231]. A hypomorphic sap1 allele strongly reduces Tf1 retrotransposition without reducing Tf1 protein or cDNA levels [232]. Approximately 63% or 73% of Tf1 insertions occur at sites bound by Sap1, but not all Sap1-binding sites appear to be targeted [232, 233]. This could reflect a need for a threshold level of Sap1 binding to target integration [232]. However, Sap1 replication-fork barrier function may also be needed for integration, based on a preference for integration to occur on the blocking side of the barrier [233]. DNA structures, proteins, or chromatin marks at stalled replication forks may therefore contribute to Tf1 integration. Sap1 preferentially binds within LTRs and adjacent to retrotransposons, which is consistent with a role for Sap1 in Tf1 targeting [234]. Sap1 and Tf1 IN interact when tested in a yeast two-hybrid system, supporting a role for Sap1 in targeting integration events through direct or indirect IN binding [232, 233]. Saccharomyces cerevisiae tRNA genes act as replication fork barriers [235], so there are parallels between integration of Tf1 and Ty1–4.
A large-scale screen for factors that specifically contribute to Tf1 integration identified 61 genes that promote integration [236]. The proteins of these genes contribute to diverse cellular functions, including chromatin structure, DNA repair, nuclear and vesicular transport, splicing and mRNA processing, transcription, and translation. While it is not immediately apparent how proteins involved in some of these activities contribute to integration, Tf1 IN interacts with at least two of these proteins in a two-hybrid system: Cwf3, a subunit of a splicing complex, and the Rhp18 DNA repair protein [236]. Some of the proteins identified are homologs of proteins shown to regulate Ty1 and Ty3, supporting common aspects of retrotransposon regulation in Schizosaccharomyces pombe and Saccharomyces cerevisiae [236].
Impact of retrotransposons on yeast genomes
Retrotransposons can have diverse effects at the level of individual genes as well as on chromosome structure that produce genetic and phenotypic variation in their host cells. There are not many examples of specific impacts of most of the TEs in the four yeast species, but many studies illustrate how Ty1 and Tf1 alter their respective host genomes. Both Ty1 and Tf1 can disrupt protein-coding genes on occasion, as seen by screening for mutations in specific target genes for Ty1 [237, 238] and large-scale analyses of each element showing occasional integration into ORFs [71, 72, 93, 94]. These retrotransposon-induced mutations would often be deleterious or neutral, with rare mutations conferring a selective advantage.
Ty1 and Ty3 integration most often occurs upstream of tRNA genes and can have modest positive influences on the expression of neighboring tRNA genes [239, 240]. It is not clear, though, how much of a phenotypic impact those changes could have. Ty1 also occasionally integrates upstream of RNA Poll II-transcribed genes, which can disrupt their promoters or change their pattern of expression based on the presence of Ty1 transcriptional control sequences [129, 133]. The latter outcome more often occurs when Ty1 is in the opposite orientation of the targeted gene. For example, induction of Ty1 by adenine starvation can lead to insertions upstream of RNA Pol II-transcribed genes that cause those genes to be more highly expressed in response to adenine starvation [241].
However, Tf1 integration almost always occurs into promoters of RNA Pol II-transcribed genes [93, 94]. Tf1 insertions in either relative orientation can compensate for disruption of target gene promoters by providing transcriptional control sequences that promote expression of neighboring genes [227]. Tf1 also shows a preference for integrating into promoters of stress-responsive genes [93]. Individual strains harboring newly integrated Tf1 elements frequently exhibit moderately increased expression of neighboring genes, rather than reduced expression [225]. Heat stress (39 °C) increases Tf1 transcription and can further increase expression of neighboring genes or increase expression of genes that were not originally affected by the insertion [225]. It was noted that the specific genes upregulated by Tf1 are themselves regulated by heat stress in the absence of Tf1, indicating that Tf1 integration into stress-responsive promoters could provide a means of amplifying stress-dependent gene expression [225].
Particular Tf1 insertions can also provide selective advantages to cells in specific environmental contexts. A library of > 40,000 strains each harboring a single newly integrated Tf1 grown together for many generations in the presence of a moderate concentration of cobalt (a stress that can activate Tf1) showed that while thousands of strains became underrepresented, a subset of > 100 strains became consistently overrepresented in replicate final populations [226]. These latter strains had Tf1 insertions that were frequently near genes that are induced by cadmium or peroxide, or genes regulated by the TOR pathway involved in nutrient sensing and cell growth. The TOR pathway was shown to provide resistance to cobalt, indicating that changes in expression of TOR-regulated genes due to neighboring Tf1 sequences could account for overrepresentation of those strains [226]. Strains harboring specific individual Tf1 insertions outcompeted wild type strains when grown in cobalt, but showed a growth disadvantage in the absence of the stress [226]. Overall, these observations support a role for Tf1 in adaptive gene expression changes conferring increased resistance to particular stresses.
Ty1 is known to have several other types of impacts on its host genome due to its reverse transcriptase activity and its repetitive nature. Ty1 occasionally reverse transcribes cellular RNAs to produce processed pseudogenes or retrosequences that are incorporated into the genome [242, 243]. In particular, subtelomeric Y′ element RNA is preferentially packaged into VLPs and Y′ is mobilized at high frequencies by Ty1 in telomerase-negative mutants [244]. Besides duplicating genomic sequences, this activity can replace intron-containing genes with intron-lacking cDNA copies and potentially put coding sequences under the control of nearby heterologous promoters. Ty1 cDNA can also be captured at sites of double-stranded breaks during DNA repair, which may facilitate DNA repair [245, 246]. Ty1-dependent retrosequences can be incorporated at sites of chromosomal rearrangements, raising the possibility that their incorporation could also contribute to DNA repair [247].
The presence of many Ty1 sequences throughout the genome provides substrates for non-allelic homologous recombination to repair DNA damage. Chromosomal rearrangements that occur spontaneously or in response to DNA damage frequently have Ty1 sequences at their breakpoint junctions [248,249,250]. Experimental evolution of cells in nutrient-limited conditions can result in adaptive chromosomal rearrangements that frequently form by recombination between Ty1 sequences [251, 252]. Such rearrangements can delete or amplify genes, for instance, producing adaptive phenotypes in response to various stresses or selective conditions [253, 254]. The presence of Ty1 sequences at intergenic sites prevents gene disruptions during these events, and some events are readily reversible in the absence of selective pressure [254]. The introduction of Ty1 sequences at particular chromosome sites can greatly increase the frequency of chromosomal rearrangements at those sites [255]. The presence of adjacent inverted Ty1 sequences or multiple Ty LTRs in different orientations can promote DNA breaks and create hotspots for chromosomal rearrangements (fragile sites), especially under conditions of replication stress [256, 257]. Ty1 sequences (and Ty2) are genomic sites of R-loops [258], in which one strand of DNA at a chromosomal site is hybridized to RNA, and R-loops cause DNA breaks and replication stress [259]. The most frequent target sites of Ty1, tRNA genes, act as replication fork barriers/pause sites based on formation of transcription initiation complexes at their promoters [235]. The presence of Ty1 elements at these sites could be advantageous by allowing for recombination-mediated restart/repair of stalled/collapsed forks [133].
The frequent involvement of Ty1 in chromosomal rearrangements could simply reflect the abundance of Ty1 sequences in the genome, or it could reflect an active role for Ty1 in these processes. DNA-damage and replication-stress signaling activate Ty1 retrotransposition [188], which could promote chromosomal rearrangements involving Ty1. A new Ty1 retrotransposition event at the site of a translocation formed during adaptation to nutrient-limited conditions provides some support for an active role in formation of chromosomal rearrangements [252]. This perspective has led to the suggestion that Ty1 can act as a genome guardian through its various impacts on gene expression, DNA repair, and adaptive genome rearrangements [260]. Comparative experiments manipulating copy numbers and activities of different Ty family members, as well as experiments in other species could help resolve how much of Ty1’s impact on these aspects of genome dynamics are passive effects of a dispersed repeat, general effects of retrotransposon activity, or specific effects due to the particular regulation/activity of Ty1.
There is less literature on the role of Tf1 and Tf2 in chromosomal rearrangements and DNA repair. The Sap1 protein binds LTR sequences and acts as a replication fork barrier [234], as previously noted. The S. pombe CENP-B homologs Cbp1/Abp1 and Cbh1 also bind to LTRs and can compete to some extent with Sap1 for binding LTRs [208, 234]. In the absence of CENP-B homologs, replication forks often collapse at LTRs, leading to double-stranded breaks and triggering homologous recombination, indicating that Cbp1/Abp1 and Cbh1 normally stabilize replication forks and restrict recombination at these sites [234]. Whether or not the presence of retrotransposon sequences near replication fork barriers provides an advantage for repair/restart of replication forks, as considered for Ty1, is not clear. While the observations reviewed in this section support some similarities between the impacts of Ty1 and Tf1, each element appears to have distinct relationships with its host.
Conclusions
Many studies of functional aspects of TEs will be required to develop a sophisticated understanding of their impacts on host organisms. Comparative functional studies offer advantages for isolating and defining crucial mechanisms underlying the impacts of TEs by examining host-TE interactions in diverse contexts. I think that additional characterization of TEs in the two pathogenic yeasts would develop this set of four diverse yeast species into an outstanding model for comparative functional studies of TEs. Candida albicans has been recently noted as an emerging model for studies of genome dynamics, partly because of the substantial genetic variation that can be present in this species due to its parasexual cycle [261]. I suggest that TEs be included as an important part of these future genome dynamics studies. Parallel and collaborative studies hold great promise for unraveling the complex layers of TE biology.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the preparation of this review article.
Abbreviations
- C. albicans :
-
Candida albicans
- CNC:
-
Copy number control
- C. neoformans :
-
Cryptococcus neoformans
- EN:
-
Endonuclease
- IN:
-
Integrase
- LTR:
-
Long terminal repeat
- NPC:
-
Nuclear pore complex
- ORF:
-
Open reading frame
- PBS:
-
Primer binding site
- PPT:
-
Polypurine tract
- PR:
-
Protease
- RH:
-
RNase H
- RNAi:
-
RNA interference
- RT:
-
Reverse transcriptase
- S. cerevisiae :
-
Saccharomyces cerevisiae
- S. pombe :
-
Schizosaccharomyces pombe
- siRNA:
-
Short interfering RNA
- TE:
-
Transposable element
- UTR:
-
Untranslated region
- VLP:
-
Virus-like particle
References
Kojima KK. Structural and sequence diversity of eukaryotic transposable elements. Genes Genet Syst. 2020;94:233–52.
Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, et al. Ten things you should know about transposable elements. Genome Biol. 2018;19:199.
Grabundzija I, Messing SA, Thomas J, Cosby RL, Bilic I, Miskey C, et al. A Helitron transposon reconstructed from bats reveals a novel mechanism of genome shuffling in eukaryotes. Nat Commun. 2016;7:10716.
Grabundzija I, Hickman AB, Dyda F. Helraiser intermediates provide insight into the mechanism of eukaryotic replicative transposition. Nat Commun. 2018;9:1278.
Horváth V, Merenciano M, González J. Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends Genet. 2017;33:832–41.
Chuong EB, Elde NC, Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 2017;18:71–86.
Muszewska A, Steczkiewicz K, Stepniewska-Dziubinska M, Ginalski K. Cut-and-paste transposons in fungi with diverse lifestyles. Genome Biol Evol. 2017;9:3463–77.
Muszewska A, Steczkiewicz K, Stepniewska-Dziubinska M, Ginalski K. Transposable elements contribute to fungal genes and impact fungal lifestyle. Sci Rep. 2019;9:1–10.
Todd CD, Deniz Ö, Taylor D, Branco MR. Functional evaluation of transposable elements as enhancers in mouse embryonic and trophoblast stem cells. eLife. 2019;8:e44344.
Hagen F, Khayhan K, Theelen B, Kolecka A, Polacheck I, Sionov E, et al. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet Biol. 2015;78:16–48.
Kwon-Chung KJ, Bennett JE, Wickes BL, Meyer W, Cuomo CA, Wollenburg KR, et al. The case for adopting the “species complex” nomenclature for the etiologic agents of Cryptococcosis. mSphere. 2017;2:e00357–16.
Loftus BJ, Fung E, Roncaglia P, Rowley D, Amedeo P, Bruno D, et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science. 2005;307:1321–4.
Janbon G, Ormerod KL, Paulet D, Iii EJB, Yadav V, Chatterjee G, et al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLoS Genet. 2014;10:e1004261.
Yadav V, Sun S, Billmyre RB, Thimmappa BC, Shea T, Lintner R, et al. RNAi is a critical determinant of centromere evolution in closely related fungi. PNAS. 2018;115:3108–13.
Braun BR, van het HM, d’Enfert C, Martchenko M, Dungan J, Kuo A, et al. A human-curated annotation of the Candida albicans genome. PLoS Genet. 2005;1:e1.
Skrzypek MS, Binkley J, Binkley G, Miyasato SR, Simison M, Sherlock G. The Candida Genome Database (CGD): incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res. 2017;45:D592–6.
Sanyal K, Baum M, Carbon J. Centromeric DNA sequences in the pathogenic yeast Candida albicans are all different and unique. PNAS. 2004;101:11374–9.
Mishra PK, Baum M, Carbon J. Centromere size and position in Candida albicans are evolutionarily conserved independent of DNA sequence heterogeneity. Mol Genet Genomics. 2007;278:455–65.
Santos MA, Tuite MF. The CUG codon is decoded in vivo as serine and not leucine in Candida albicans. Nucleic Acids Res. 1995;23:1481–6.
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, et al. Life with 6000 genes. Science. 1996;274:546–67.
Wood V, Gwilliam R, Rajandream M-A, Lyne M, Lyne R, Stewart A, et al. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415:871–80.
Engel SR, Dietrich FS, Fisk DG, Binkley G, Balakrishnan R, Costanzo MC, et al. The reference genome sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda). 2014;4:389–98.
Lock A, Rutherford K, Harris MA, Hayles J, Oliver SG, Bähler J, et al. PomBase 2018: user-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res. 2019;47:D821–7.
Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–54.
Roy B, Sanyal K. Diversity in requirement of genetic and epigenetic factors for centromere function in fungi. Eukaryot Cell. 2011;10:1384–95.
Sudbery P, Gow N, Berman J. The distinct morphogenic states of Candida albicans. Trends Microbiol. 2004;12:317–24.
Bennett RJ, Johnson AD. Mating in Candida albicans and the search for a sexual cycle. Annu Rev Microbiol. 2005;59:233–55.
Maschmeyer G, Ruhnke M. Update on antifungal treatment of invasive Candida and Aspergillus infections. Mycoses. 2004;47:263–76.
Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: Analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–17.
Hull CM, Raisner RM, Johnson AD. Evidence for mating of the “asexual” yeast Candida albicans in a mammalian host. Science. 2000;289:307–10.
Magee BB, Magee PT. Induction of mating in Candida albicans by construction of MTLa and MTLα strains. Science. 2000;289:310–3.
Bennett RJ, Johnson AD. Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. EMBO J. 2003;22:2505–15.
Forche A, Alby K, Schaefer D, Johnson AD, Berman J, Bennett RJ. The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS Biol. 2008;6:e110.
Alby K, Schaefer D, Bennett RJ. Homothallic and heterothallic mating in the opportunistic pathogen Candida albicans. Nature. 2009;460:890–3.
Miller MG, Johnson AD. White-opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell. 2002;110:293–302.
Basenko EY, Pulman JA, Shanmugasundram A, Harb OS, Crouch K, Starns D, et al. FungiDB: an integrated bioinformatic resource for fungi and oomycetes. J Fungi. 2018;4:39.
Goodwin TJD, Poulter RTM. Multiple LTR-retrotransposon families in the asexual yeast Candida albicans. Genome Res. 2000;10:174–91.
Feschotte C, Wessler SR. Mariner-like transposases are widespread and diverse in flowering plants. PNAS. 2002;99:280–5.
Lopes FR, Silva JC, Benchimol M, Costa GG, Pereira GA, Carareto CM. The protist Trichomonas vaginalis harbors multiple lineages of transcriptionally active Mutator-like elements. BMC Genomics. 2009;10:330.
Chen JY, Fonzi WA. A temperature-regulated, retrotransposon-like element from Candida albicans. J Bacteriol. 1992;174:5624–32.
Matthews GD, Goodwin TJ, Butler MI, Berryman TA, Poulter RT. pCal, a highly unusual Ty1/copia retrotransposon from the pathogenic yeast Candida albicans. J Bacteriol. 1997;179:7118–28.
Plant EP, Goodwin TJD, Poulter RTM. Tca5, a Ty5-like retrotransposon from Candida albicans. Yeast. 2000;16:1509–18.
Goodwin TJD, Ormandy JE, Poulter RTM. L1-like non-LTR retrotransposons in the yeast Candida albicans. Curr Genet. 2001;39:83–91.
Perreau VM, Santos MAS, Tuite MF. beta, a novel repetitive DNA element associated with tRNA genes in the pathogenic yeast Candida albicans. Mol Microbiol. 1997;25:229–36.
Goodwin TJ, Busby JN, Poulter RT. A yeast model for target-primed (non-LTR) retrotransposition. BMC Genomics. 2007;8:263.
Holton NJ, Goodwin TJD, Butler MI, Poulter RTM. An active retrotransposon in Candida albicans. Nucleic Acids Res. 2001;29:4014–24.
Lin X, Heitman J. The biology of the Cryptococcus neoformans species complex. Annu Rev Microbiol. 2006;60:69–105.
Kwon-Chung KJ, Bennett JE. Distribution of alpha and alpha mating types of Cryptococcus neoformans among natural and clinical isolates. Am J Epidemiol. 1978;108:337–40.
Lin X, Hull CM, Heitman J. Sexual reproduction between partners of the same mating type in Cryptococcus neoformans. Nature. 2005;434:1017–21.
Lin X, Patel S, Litvintseva AP, Floyd A, Mitchell TG, Heitman J. Diploids in the Cryptococcus neoformans serotype A population homozygous for the α mating type originate via unisexual mating. PLoS Pathogens. 2009;5:e1000283.
Leopold Wager CM, Hole CR, Wozniak KL, Wormley FLJ. Cryptococcus and phagocytes: Complex interactions that influence disease outcome. Front Microbiol. 2016;7:105.
Castanera R, López-Varas L, Borgognone A, LaButti K, Lapidus A, Schmutz J, et al. Transposable elements versus the fungal genome: impact on whole-genome architecture and transcriptional profiles. PLoS Genet. 2016;12:e1006108.
Goodwin TJD, Butler MI, Poulter RTM. Cryptons: a group of tyrosine-recombinase-encoding DNA transposons from pathogenic fungi. Microbiology. 2003;149:3099–109.
Goodwin TJD, Poulter RTM. The diversity of retrotransposons in the yeast Cryptococcus neoformans. Yeast. 2001;18:865–80.
Cruz MC, Cavallo LM, Görlach JM, Cox G, Perfect JR, Cardenas ME, et al. Rapamycin antifungal action is mediated via conserved complexes with FKBP12 and TOR kinase homologs in Cryptococcus neoformans. Mol Cell Biol. 1999;19:4101–12.
Wang X, Hsueh Y-P, Li W, Floyd A, Skalsky R, Heitman J. Sex-induced silencing defends the genome of Cryptococcus neoformans via RNAi. Genes Dev. 2010;24:2566–82.
Wang X, Darwiche S, Heitman J. Sex-induced silencing operates during opposite-sex and unisexual reproduction in Cryptococcus neoformans. Genetics. 2013;193:1163–74.
Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ, Habib N, et al. Comparative functional genomics of the fission yeasts. Science. 2011;332:930–6.
Liti G. The fascinating and secret wild life of the budding yeast S. cerevisiae. eLife. 2015;4:e05835.
Enache-Angoulvant A, Hennequin C. Invasive Saccharomyces infection: a comprehensive review. Clin Infect Dis. 2005;41:1559–68.
Hanson SJ, Wolfe KH. An evolutionary perspective on yeast mating-type switching. Genetics. 2017;206:9–32.
Dujon B. Yeast evolutionary genomics. Nat Rev Genet. 2010;11:512–24.
Sampaio JP, Gonçalves P. Natural populations of Saccharomyces kudriavzevii in portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl Environ Microbiol. 2008;74:2144–52.
Wang Q-M, Liu W-Q, Liti G, Wang S-A, Bai F-Y. Surprisingly diverged populations of Saccharomyces cerevisiae in natural environments remote from human activity. Mol Ecol. 2012;21:5404–17.
Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF. Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998;8:464–78.
Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, et al. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–41.
Carr M, Bensasson D, Bergman CM. Evolutionary genomics of transposable elements in Saccharomyces cerevisiae. PLoS One. 2012;7:e50978.
Zou S, Wright DA, Voytas DF. The Saccharomyces Ty5 retrotransposon family is associated with origins of DNA replication at the telomeres and the silent mating locus HMR. PNAS. 1995;92:920–4.
Ji H, Moore DP, Blomberg MA, Braiterman LT, Voytas DF, Natsoulis G, et al. Hotspots for unselected Ty1 transposition events on yeast chromosome III are near tRNA genes and LTR sequences. Cell. 1993;73:1007–18.
Devine SE, Boeke JD. Integration of the yeast retrotransposon Ty1 is targeted to regions upstream of genes transcribed by RNA polymerase III. Genes Dev. 1996;10:620–33.
Baller JA, Gao J, Stamenova R, Curcio MJ, Voytas DF. A nucleosomal surface defines an integration hotspot for the Saccharomyces cerevisiae Ty1 retrotransposon. Genome Res. 2012;22:704–13.
Mularoni L, Zhou Y, Bowen T, Gangadharan S, Wheelan SJ, Boeke JD. Retrotransposon Ty1 integration targets specifically positioned asymmetric nucleosomal DNA segments in tRNA hotspots. Genome Res. 2012;22:693–703.
Chalker DL, Sandmeyer SB. Transfer RNA genes are genomic targets for de novo transposition of the yeast retrotransposon Ty3. Genetics. 1990;126:837–50.
Chalker DL, Sandmeyer SB. Ty3 integrates within the region of RNA polymerase III transcription initiation. Genes Dev. 1992;6:117–28.
Qi X, Daily K, Nguyen K, Wang H, Mayhew D, Rigor P, et al. Retrotransposon profiling of RNA polymerase III initiation sites. Genome Res. 2012;22:681–92.
Patterson K, Shavarebi F, Magnan C, Chang I, Qi X, Baldi P, et al. Local features determine Ty3 targeting frequency at RNA polymerase III transcription start sites. Genome Res. 2019;29:1298–309.
Zou S, Ke N, Kim JM, Voytas DF. The Saccharomyces retrotransposon Ty5 integrates preferentially into regions of silent chromatin at the telomeres and mating loci. Genes Dev. 1996;10:634–45.
Zou S, Voytas DF. Silent chromatin determines target preference of the Saccharomyces retrotransposon Ty5. PNAS. 1997;94:7412–6.
Baller JA, Gao J, Voytas DF. Access to DNA establishes a secondary target site bias for the yeast retrotransposon Ty5. PNAS. 2011;108:20351–6.
Hoffman CS, Wood V, Fantes PA. An ancient yeast for young geneticists: a primer on the Schizosaccharomyces pombe model system. Genetics. 2015;201:403–23.
Jeffares DC. The natural diversity and ecology of fission yeast. Yeast. 2018;35:253–60.
Egel R. Fission yeast mating-type switching: programmed damage and repair. DNA Repair. 2005;4:525–36.
Bowen NJ, Jordan IK, Epstein JA, Wood V, Levin HL. Retrotransposons and their recognition of pol II promoters: a comprehensive survey of the transposable elements from the complete genome sequence of Schizosaccharomyces pombe. Genome Res. 2003;13:1984–97.
Esnault C, Levin HL. The long terminal repeat retrotransposons Tf1 and Tf2 of Schizosaccharomyces pombe. Microbiol Spectr. 2015;3:MDNA3–0040-2014.
Levin HL, Weaver DC, Boeke JD. Two related families of retrotransposons from Schizosaccharomyces pombe. Mol Cell Biol. 1990;10:6791–8.
Lin JH, Levin HL. Self-primed reverse transcription is a mechanism shared by several LTR-containing retrotransposons. RNA. 1997;3:952–3.
Watanabe T, Miyashita K, Saito TT, Yoneki T, Kakihara Y, Nabeshima K, et al. Comprehensive isolation of meiosis-specific genes identifies novel proteins and unusual non-coding transcripts in Schizosaccharomyces pombe. Nucleic Acids Res. 2001;29:2327–37.
Hu W, Jiang Z-D, Suo F, Zheng J-X, He W-Z, Du L-L. A large gene family in fission yeast encodes spore killers that subvert Mendel’s law. eLife. 2017;6:e26057.
Nuckolls NL, Bravo Núñez MA, Eickbush MT, Young JM, Lange JJ, Yu JS, et al. Wtf genes are prolific dual poison-antidote meiotic drivers. eLife. 2017;6:e26033.
Hoff EF, Levin HL, Boeke JD. Schizosaccharomyces pombe retrotransposon Tf2 mobilizes primarily through homologous cDNA recombination. Mol Cell Biol. 1998;18:6839–52.
Behrens R, Hayles J, Nurse P. Fission yeast retrotransposon Tf1 integration is targeted to 5′ ends of open reading frames. Nucleic Acids Res. 2000;28:4709–16.
Singleton TL, Levin HL. A long terminal repeat retrotransposon of fission yeast has strong preferences for specific sites of insertion. Eukaryot Cell. 2002;1:44–55.
Guo Y, Levin HL. High-throughput sequencing of retrotransposon integration provides a saturated profile of target activity in Schizosaccharomyces pombe. Genome Res. 2010;20:239–48.
Chatterjee AG, Esnault C, Guo Y, Hung S, McQueen PG, Levin HL. Serial number tagging reveals a prominent sequence preference of retrotransposon integration. Nucleic Acids Res. 2014;42:8449–60.
Scheifele LZ, Cost GJ, Zupancic ML, Caputo EM, Boeke JD. Retrotransposon overdose and genome integrity. PNAS. 2009;106:13927–32.
Sultana T, Zamborlini A, Cristofari G, Lesage P. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat Rev Genet. 2017;18:292–308.
Dong C, Poulter RT, Han JS. LINE-like retrotransposition in Saccharomyces cerevisiae. Genetics. 2009;181:301–11.
Peter J, Chiara MD, Friedrich A, Yue J-X, Pflieger D, Bergström A, et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature. 2018;556:339–44.
Guo Y, Singh PK, Levin HL. A long terminal repeat retrotransposon of Schizosaccharomyces japonicus integrates upstream of RNA pol III transcribed genes. Mob DNA. 2015;6:19.
Billmyre RB, Calo S, Feretzaki M, Wang X, Heitman J. RNAi function, diversity, and loss in the fungal kingdom. Chromosome Res. 2013;21:561–72.
Claycomb JM. Ancient endo-siRNA pathways reveal new tricks. Curr Biol. 2014;24:R703–15.
Nakayashiki H, Kadotani N, Mayama S. Evolution and diversification of RNA silencing proteins in fungi. J Mol Evol. 2006;63:127–35.
Drinnenberg IA, Weinberg DE, Xie KT, Mower JP, Wolfe KH, Fink GR, et al. RNAi in budding yeast. Science. 2009;326:544–50.
Bernstein DA, Vyas VK, Weinberg DE, Drinnenberg IA, Bartel DP, Fink GR. Candida albicans Dicer (CaDcr1) is required for efficient ribosomal and spliceosomal RNA maturation. PNAS. 2012;109:523–8.
Staab JF, White TC, Marr KA. Hairpin dsRNA does not trigger RNA interference in Candida albicans cells. Yeast. 2011;28:1–8.
Moazeni M, Khoramizadeh MR, Kordbacheh P, Sepehrizadeh Z, Zeraati H, Noorbakhsh F, et al. RNA-mediated gene silencing in Candidaalbicans: inhibition of hyphae formation by use of RNAi technology. Mycopathologia. 2012;174:177–85.
Janbon G, Maeng S, Yang D-H, Ko Y-J, Jung K-W, Moyrand F, et al. Characterizing the role of RNA silencing components in Cryptococcus neoformans. Fungal Genet Biol. 2010;47:1070–80.
Liu H, Cottrell TR, Pierini LM, Goldman WE, Doering TL. RNA interference in the pathogenic fungus Cryptococcus neoformans. Genetics. 2002;160:463–70.
Dumesic PA, Natarajan P, Chen C, Drinnenberg IA, Schiller BJ, Thompson J, et al. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell. 2013;152:957–68.
Drinnenberg IA, Fink GR, Bartel DP. Compatibility with killer explains the rise of RNAi-deficient fungi. Science. 2011;333:1592–2.
Schmitt MJ, Breinig F. The viral killer system in yeast: from molecular biology to application. FEMS Microbiol Rev. 2002;26:257–76.
Volpe TA, Kidner C, Hall IM, Teng G, Grewal SIS, Martienssen RA. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–7.
Yamanaka S, Mehta S, Reyes-Turcu FE, Zhuang F, Fuchs RT, Rong Y, et al. RNAi triggered by specialized machinery silences developmental genes and retrotransposons. Nature. 2013;493:557–60.
Aravind L, Watanabe H, Lipman DJ, Koonin EV. Lineage-specific loss and divergence of functionally linked genes in eukaryotes. PNAS. 2000;97:11319–24.
Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SIS. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat Genet. 2005;37:809–19.
Djupedal I, Kos-Braun IC, Mosher RA, Söderholm N, Simmer F, Hardcastle TJ, et al. Analysis of small RNA in fission yeast; centromeric siRNAs are potentially generated through a structured RNA. EMBO J. 2009;28:3832–44.
Provost P, Silverstein RA, Dishart D, Walfridsson J, Djupedal I, Kniola B, et al. Dicer is required for chromosome segregation and gene silencing in fission yeast cells. PNAS. 2002;99:16648–53.
Volpe T, Schramke V, Hamilton GL, White SA, Teng G, Martienssen RA, et al. RNA interference is required for normal centromere function in fission yeast. Chromosome Res. 2003;11:137–46.
Hansen KR, Burns G, Mata J, Volpe TA, Martienssen RA, Bähler J, et al. Global effects on gene expression in fission yeast by silencing and RNA interference machineries. Mol Cell Biol. 2005;25:590–601.
Woolcock KJ, Gaidatzis D, Punga T, Bühler M. Dicer associates with chromatin to repress genome activity in Schizosaccharomyces pombe. Nat Struct Mol Biol. 2011;18:94–9.
Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, et al. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell. 2013;155:1061–74.
Beauregard A, Curcio MJ, Belfort M. The take and give between retrotransposable elements and their hosts. Annu Rev Genet. 2008;42:587–617.
Hughes SH. Reverse transcription of retroviruses and LTR retrotransposons. Microbiol Spectr. 2015;3:MDNA3–0027–2014.
Forbes EM, Nieduszynska SR, Brunton FK, Gibson J, Glover LA, Stansfield I. Control of gag-pol gene expression in the Candida albicans retrotransposon Tca2. BMC Mol Biol. 2007;8:94.
Chen J, Wang Q, Fu Z, Zhou S, Fonzi WA. Tca1, the retrotransposon-like Element of Candida albicans, is a degenerate and inactive element. J Bacteriol. 1998;180:3657–62.
Irwin PA, Voytas DF. Expression and processing of proteins encoded by the Saccharomyces retrotransposon Ty5. J Virol. 2001;75:1790–7.
Hansen LJ, Chalker DL, Sandmeyer SB. Ty3, a yeast retrotransposon associated with tRNA genes, has homology to animal retroviruses. Mol Cell Biol. 1988;8:5245–56.
Janetzky B, Lehle L. Ty4, a new retrotransposon from Saccharomyces cerevisiae, flanked by tau-elements. J Biol Chem. 1992;267:19798–805.
Lesage P, Todeschini AL. Happy together: the life and times of Ty retrotransposons and their hosts. Cytogenet Genome Res. 2005;110:70–90.
Clare JJ, Belcourt M, Farabaugh PJ. Efficient translational frameshifting occurs within a conserved sequence of the overlap between the two genes of a yeast Ty1 transposon. PNAS. 1988;85:6816–20.
Farabaugh PJ, Zhao H, Vimaladithan A. A novel programed frameshift expresses the POL3 gene of retrotransposon Ty3 of yeast: frameshifting without tRNA slippage. Cell. 1993;74:93–103.
Kawakami K, Pande S, Faiola B, Moore DP, Boeke JD, Farabaugh PJ, et al. A rare tRNA-Arg (ccu) that regulates Ty1 element ribosomal frameshifting is essential for Ty1 retrotransposition in Saccharomyces cerevisiae. Genetics. 1993;135:309–20.
Curcio MJ, Lutz S, Lesage P. The Ty1 LTR-Retrotransposon of Budding Yeast, Saccharomyces cerevisiae. Microbiol Spectr. 2015;3:MDNA3–0053-2014.
Sandmeyer S, Patterson K, Bilanchone V. Ty3, a position-specific retrotransposon in budding yeast. Microbiol Spectr. 2015;3:MDNA3–0057-2014.
Scholes DT, Banerjee M, Bowen B, Curcio MJ. Multiple regulators of Ty1 transposition in Saccharomyces cerevisiae have conserved roles in genome maintenance. Genetics. 2001;159:1449–65.
Griffith JL, Coleman LE, Raymond AS, Goodson SG, Pittard WS, Tsui C, et al. Functional genomics reveals relationships between the retrovirus-like Ty1 element and its host Saccharomyces cerevisiae. Genetics. 2003;164:867–79.
Aye M, Irwin B, Beliakova-Bethell N, Chen E, Garrus J, Sandmeyer S. Host factors that affect Ty3 retrotransposition in Saccharomyces cerevisiae. Genetics. 2004;168:1159–76.
Irwin B, Aye M, Baldi P, Beliakova-Bethell N, Cheng H, Dou Y, et al. Retroviruses and yeast retrotransposons use overlapping sets of host genes. Genome Res. 2005;15:641–54.
Nyswaner KM, Checkley MA, Yi M, Stephens RM, Garfinkel DJ. Chromatin-associated genes protect the yeast genome from Ty1 insertional mutagenesis. Genetics. 2008;178:197–214.
Dakshinamurthy A, Nyswaner KM, Farabaugh PJ, Garfinkel DJ. BUD22 affects Ty1 retrotransposition and ribosome biogenesis in Saccharomyces cerevisiae. Genetics. 2010;185:1193–205.
Risler JK, Kenny AE, Palumbo RJ, Gamache ER, Curcio MJ. Host co-factors of the retrovirus-like transposon Ty1. Mob DNA. 2012;3:12.
Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–5.
Ke N, Irwin PA, Voytas DF. The pheromone response pathway activates transcription of Ty5 retrotransposons located within silent chromatin of Saccharomyces cerevisiae. EMBO J. 1997;16:6272–80.
Xu H, Boeke JD. Inhibition of Ty1 transposition by mating pheromones in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:2736–43.
Saito H. Regulation of cross-talk in yeast MAPK signaling pathways. Curr Opin Microbiol. 2010;13:677–83.
Conte D, Barber E, Banerjee M, Garfinkel DJ, Curcio MJ. Posttranslational regulation of Ty1 retrotransposition by mitogen-activated protein kinase Fus3. Mol Cell Biol. 1998;18:2502–13.
Conte D, Curcio MJ. Fus3 controls Ty1 transpositional dormancy through the invasive growth MAPK pathway. Mol Microbiol. 2000;35:415–27.
Morillon A, Springer M, Lesage P. Activation of the Kss1 invasive-filamentous growth pathway induces Ty1 transcription and retrotransposition in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:5766–76.
Company M, Adler C, Errede B. Identification of a Ty1 regulatory sequence responsive to STE7 and STE12. Mol Cell Biol. 1988;8:2545–54.
Laloux I, Dubois E, Dewerchin M, Jacobs E. TEC1, a gene involved in the activation of Ty1 and Ty1-mediated gene expression in Saccharomyces cerevisiae: cloning and molecular analysis. Mol Cell Biol. 1990;10:3541–50.
Laloux I, Jacobs E, Dubois E. Involvement of SRE element of Ty1 transposon in TEC1-dependent transcriptional activation. Nucleic Acids Res. 1994;22:999–1005.
Kinsey PT, Sandmeyer SB. Ty3 transposes in mating populations of yeast: a novel transposition assay for Ty3. Genetics. 1995;139:81–94.
Arsdell SWV, Stetler GL, Thorner J. The yeast repeated element sigma contains a hormone-inducible promoter. Mol Cell Biol. 1987;7:749–59.
Bilanchone VW, Claypool JA, Kinsey PT, Sandmeyer SB. Positive and negative regulatory elements control expression of the yeast retrotransposon Ty3. Genetics. 1993;134:685–700.
Bilanchone V, Clemens K, Kaake R, Dawson AR, Matheos D, Nagashima K, et al. Ty3 retrotransposon hijacks mating yeast RNA processing bodies to infect new genomes. PLoS Genet. 2015;11:e1005528.
Menees TM, Sandmeyer SB. Transposition of the yeast retroviruslike element Ty3 is dependent on the cell cycle. Mol Cell Biol. 1994;14:8229–40.
Beliakova-Bethell N, Beckham C, Giddings TH, Winey M, Parker R, Sandmeyer S. Virus-like particles of the Ty3 retrotransposon assemble in association with P-body components. RNA. 2006;12:94–101.
Checkley MA, Nagashima K, Lockett SJ, Nyswaner KM, Garfinkel DJ. P-body components are required for Ty1 retrotransposition during assembly of retrotransposition-competent virus-like particles. Mol Cell Biol. 2010;30:382–98.
Dutko JA, Kenny AE, Gamache ER, Curcio MJ. 5′ to 3′ mRNA decay factors colocalize with Ty1 Gag and human APOBEC3G and promote Ty1 retrotransposition. J Virol. 2010;84:5052–66.
Decker CJ, Parker R. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol. 2012;4:a012286.
Malagon F, Jensen TH. The T Body, a new cytoplasmic RNA granule in Saccharomyces cerevisiae. Mol Cell Biol. 2008;28:6022–32.
Leung AKL, Sharp PA. Quantifying Argonaute proteins in and out of GW/P-bodies: implications in microRNA activities. Adv Exp Med Biol. 2013;768:165–82.
Doucet AJ, Hulme AE, Sahinovic E, Kulpa DA, Moldovan JB, Kopera HC, et al. Characterization of LINE-1 ribonucleoprotein particles. PLoS Genet. 2010;6:e1001150.
Goodier JL, Mandal PK, Zhang L, Kazazian HH. Discrete subcellular partitioning of human retrotransposon RNAs despite a common mechanism of genome insertion. Hum Mol Genet. 2010;19:1712–25.
Guo H, Chitiprolu M, Gagnon D, Meng L, Perez-Iratxeta C, Lagace D, et al. Autophagy supports genomic stability by degrading retrotransposon RNA. Nature Comm. 2014;5:1–11.
Hoelz A, Debler EW, Blobel G. The structure of the nuclear pore complex. Annu Rev Biochem. 2011;80:613–43.
Kenna MA, Brachmann CB, Devine SE, Boeke JD. Invading the yeast nucleus: a nuclear localization signal at the C terminus of Ty1 integrase is required for transposition in vivo. Mol Cell Biol. 1998;18:1115–24.
Moore SP, Rinckel LA, Garfinkel DJ. A Ty1 integrase nuclear localization signal required for retrotransposition. Mol Cell Biol. 1998;18:1105–14.
Lin SS, Nymark-McMahon MH, Yieh L, Sandmeyer SB. Integrase mediates nuclear localization of Ty3. Mol Cell Biol. 2001;21:7826–38.
McLane LM, Pulliam KF, Devine SE, Corbett AH. The Ty1 integrase protein can exploit the classical nuclear protein import machinery for entry into the nucleus. Nucleic Acids Res. 2008;36:4317–26.
Manhas S, Ma L, Measday V. The yeast Ty1 retrotransposon requires components of the nuclear pore complex for transcription and genomic integration. Nucleic Acids Res. 2018;46:3552–78.
Beliakova-Bethell N, Terry LJ, Bilanchone V, DaSilva R, Nagashima K, Wente SR, et al. Ty3 nuclear entry is initiated by viruslike particle docking on GLFG nucleoporins. J Virol. 2009;83:11914–25.
Rowley PA, Patterson K, Sandmeyer SB, Sawyer SL. Control of yeast retrotransposons mediated through nucleoporin evolution. PLoS Genet. 2018;14:e1007325.
Zhu Y, Zou S, Wright DA, Voytas DF. Tagging chromatin with retrotransposons: target specificity of the Saccharomyces Ty5 retrotransposon changes with the chromosomal localization of Sir3p and Sir4p. Genes Dev. 1999;13:2738–49.
Xie W, Gai X, Zhu Y, Zappulla DC, Sternglanz R, Voytas DF. Targeting of the yeast Ty5 retrotransposon to silent chromatin is mediated by interactions between Integrase and Sir4p. Mol Cell Biol. 2001;21:6606–14.
Kirchner J, Connolly CM, Sandmeyer SB. Requirement of RNA polymerase III transcription factors for in vitro position-specific integration of a retroviruslike element. Science. 1995;267:1488–91.
Yieh L, Kassavetis G, Geiduschek EP, Sandmeyer SB. The Brf and TATA-binding protein subunits of the RNA Polymerase III Transcription Factor IIIB mediate position-specific integration of the gypsy-like element, Ty3. J Biol Chem. 2000;275:29800–7.
Yieh L, Hatzis H, Kassavetis G, Sandmeyer SB. Mutational analysis of the Transcription Factor IIIB-DNA target of Ty3 retroelement integration. J Biol Chem. 2002;277:25920–8.
Bridier-Nahmias A, Tchalikian-Cosson A, Baller JA, Menouni R, Fayol H, Flores A, et al. An RNA polymerase III subunit determines sites of retrotransposon integration. Science. 2015;348:585–8.
Cheung S, Ma L, Chan PHW, Hu H-L, Mayor T, Chen H-T, et al. Ty1 integrase interacts with RNA Polymerase III-specific subcomplexes to promote insertion of Ty1 elements upstream of Polymerase (Pol) III-transcribed genes. J Biol Chem. 2016;291:6396–411.
Todeschini A-L, Morillon A, Springer M, Lesage P. Severe adenine starvation activates Ty1 transcription and retrotransposition in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:7459–72.
Servant G, Pinson B, Tchalikian-Cosson A, Coulpier F, Lemoine S, Pennetier C, et al. Tye7 regulates yeast Ty1 retrotransposon sense and antisense transcription in response to adenylic nucleotides stress. Nucleic Acids Res. 2012;40:5271–82.
Menees TM, Sandmeyer SB. Cellular stress inhibits transposition of the yeast retrovirus-like element Ty3 by a ubiquitin-dependent block of virus-like particle formation. PNAS. 1996;93:5629–34.
Paquin CE, Williamson VM. Temperature effects on the rate of ty transposition. Science. 1984;226:53–5.
Lawler JF, Haeusser DP, Dull A, Boeke JD, Keeney JB. Ty1 defect in proteolysis at high temperature. J Virol. 2002;76:4233–40.
Stanley D, Fraser S, Stanley GA, Chambers PJ. Retrotransposon expression in ethanol-stressed Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2010;87:1447–54.
Stoycheva T, Pesheva M, Venkov P. The role of reactive oxygen species in the induction of Ty1 retrotransposition in Saccharomyces cerevisiae. Yeast. 2010;27:259–67.
Curcio MJ, Kenny AE, Moore S, Garfinkel DJ, Weintraub M, Gamache ER, et al. S-phase checkpoint pathways stimulate the mobility of the retrovirus-like transposon Ty1. Mol Cell Biol. 2007;27:8874–85.
Elder RT, St John TP, Stinchcomb DT, Davis RW, Scherer S, Davis RW. Studies on the transposable element Ty1 of yeast. I. RNA homologous to Ty1. II. Recombination and expression of Ty1 and adjacent sequences. Cold Spring Harb Symp Quant Biol. 1981;45(Pt 2):581–91.
Curcio MJ, Hedge AM, Boeke JD, Garfinkel DJ. Ty RNA levels determine the spectrum of retrotransposition events that activate gene expression in Saccharomyces cerevisiae. Mol Gen Genet. 1990;220:213–21.
Huang Q, Purzycka KJ, Lusvarghi S, Li D, LeGrice SFJ, Boeke JD. Retrotransposon Ty1 RNA contains a 5′-terminal long-range pseudoknot required for efficient reverse transcription. RNA. 2013;19:320–32.
Garfinkel DJ, Nyswaner K, Wang J, Cho J-Y. Post-transcriptional cosuppression of Ty1 retrotransposition. Genetics. 2003;165:83–99.
Berretta J, Pinskaya M, Morillon A. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev. 2008;22:615–26.
Matsuda E, Garfinkel DJ. Posttranslational interference of Ty1 retrotransposition by antisense RNAs. PNAS. 2009;106:15657–62.
Saha A, Mitchell JA, Nishida Y, Hildreth JE, Ariberre JA, Gilbert WV, et al. A trans-dominant form of Gag restricts Ty1 retrotransposition and mediates copy number control. J Virol. 2015;89:3922–38.
Tucker JM, Larango ME, Wachsmuth LP, Kannan N, Garfinkel DJ. The Ty1 retrotransposon restriction factor p22 targets Gag. PLoS Genet. 2015;11:e1005571.
Ahn HW, Tucker JM, Arribere JA, Garfinkel DJ. Ribosome biogenesis modulates Ty1 copy number control in Saccharomyces cerevisiae. Genetics. 2017;207:1441–56.
Salinero AC, Knoll ER, Zhu ZI, Landsman D, Curcio MJ, Morse RH. The Mediator co-activator complex regulates Ty1 retromobility by controlling the balance between Ty1i and Ty1 promoters. PLoS Genet. 2018;14:e1007232.
Weaver DC, Shpakovski GV, Caputo E, Levin HL, Bocke JD. Sequence analysis of closely related retrotransposon families from fission yeast. Gene. 1993;131:135–9.
Levin HL, Weaver DC, Boeke JD. Novel gene expression mechanism in a fission yeast retroelement: Tf1 proteins are derived from a single primary translation product. EMBO J. 1993;12:4885–95.
Atwood A, Lin JH, Levin HL. The retrotransposon Tf1 assembles virus-like particles that contain excess Gag relative to integrase because of a regulated degradation process. Mol Cell Biol. 1996;16:338–46.
Levin H l., Boeke J d. Demonstration of retrotransposition of the Tf1 element in fission yeast. EMBO J. 1992;11:1145–1153.
Peifer AC, Maxwell PH. Preferential Ty1 retromobility in mother cells and nonquiescent stationary phase cells is associated with increased concentrations of total Gag or processed Gag and is inhibited by exposure to a high concentration of calcium. Aging. 2018;10:402–24.
Levin HL. A novel mechanism of self-primed reverse transcription defines a new family of retroelements. Mol Cell Biol. 1995;15:3310–7.
Levin HL. An unusual mechanism of self-primed reverse transcription requires the RNase H domain of reverse transcriptase to cleave an RNA duplex. Mol Cell Biol. 1996;16:5645–54.
Lin J-H, Levin HL. Reverse transcription of a self-primed retrotransposon requires an RNA structure similar to the U5-IR stem-loop of retroviruses. Mol Cell Biol. 1998;18:6859–69.
Greenall A, Williams ES, Martin KA, Palmer JM, Gray J, Liu C, et al. Hip3 interacts with the HIRA proteins Hip1 and Slm9 and is required for transcriptional silencing and accurate chromosome segregation. J Biol Chem. 2006;281:8732–9.
Cam HP, Noma K, Ebina H, Levin HL, Grewal SIS. Host genome surveillance for retrotransposons by transposon-derived proteins. Nature. 2008;451:431–6.
Anderson HE, Wardle J, Korkut ŞV, Murton HE, López-Maury L, Bähler J, et al. The fission yeast HIRA histone chaperone is required for promoter silencing and the suppression of cryptic antisense transcripts. Mol Cell Biol. 2009;29:5158–67.
Durand-Dubief M, Sinha I, Fagerström-Billai F, Bonilla C, Wright A, Grunstein M, et al. Specific functions for the fission yeast Sirtuins Hst2 and Hst4 in gene regulation and retrotransposon silencing. EMBO J. 2007;26:2477–88.
Lorenz DR, Mikheyeva IV, Johansen P, Meyer L, Berg A, Grewal SIS, et al. CENP-B cooperates with Set1 in bidirectional transcriptional silencing and genome organization of retrotransposons. Mol Cell Biol. 2012;32:4215–25.
Zilio N, Codlin S, Vashisht AA, Bitton DA, Head SR, Wohlschlegel JA, et al. A novel histone deacetylase complex in the control of transcription and genome stability. Mol Cell Biol. 2014;34:3500–14.
Kotomura N, Tsunemine S, Kuragano M, Asanuma T, Nakagawa H, Tanaka K, et al. Sfh1, an essential component of the RSC chromatin remodeling complex, maintains genome integrity in fission yeast. Genes Cells. 2018;23:738–52.
Smit AF, Riggs AD. Tiggers and DNA transposon fossils in the human genome. PNAS. 1996;93:1443–8.
Tudor M, Lobocka M, Goodell M, Pettitt J, O’Hare K. The pogo transposable element family of Drosophila melanogaster. Mol Gen Genet. 1992;232:126–34.
Tanaka A, Tanizawa H, Sriswasdi S, Iwasaki O, Chatterjee AG, Speicher DW, et al. Epigenetic regulation of condensin-mediated genome organization during the cell cycle and upon DNA damage through histone H3 lysine 56 acetylation. Mol Cell. 2012;48:532–46.
Murton HE, Grady PJR, Chan TH, Cam HP, Whitehall SK. Restriction of retrotransposon mobilization in Schizosaccharomyces pombe by transcriptional silencing and higher-order chromatin organization. Genetics. 2016;203:1669–78.
Mikheyeva IV, Grady PJR, Tamburini FB, Lorenz DR, Cam HP. Multifaceted genome control by Set1 dependent and independent of H3K4 methylation and the Set1C/COMPASS complex. PLoS Genet. 2014;10:e1004740.
Sehgal A, Lee C-YS, Espenshade PJ. SREBP controls oxygen-dependent mobilization of retrotransposons in fission yeast. PLoS Genet. 2007;3:e131.
Mallet P-L, Larochelle M, Bachand F. Multiple transcriptional and post-transcriptional pathways collaborate to control sense and antisense RNAs of Tf2 retroelements in fission yeast. Genetics. 2017;205:621–32.
Sugiyama T, Sugioka-Sugiyama R. Red1 promotes the elimination of meiosis-specific mRNAs in vegetatively growing fission yeast. EMBO J. 2011;30:1027–39.
Dang VD, Benedik MJ, Ekwall K, Choi J, Allshire RC, Levin HL. A new member of the Sin3 family of corepressors is essential for cell viability and required for retroelement propagation in fission yeast. Mol Cell Biol. 1999;19:2351–65.
Balasundaram D, Benedik MJ, Morphew M, Dang V-D, Levin HL. Nup124p Is a nuclear pore factor of Schizosaccharomyces pombe that is important for nuclear import and activity of retrotransposon Tf1. Mol Cell Biol. 1999;19:5768–84.
Dang V-D, Levin HL. Nuclear import of the retrotransposon Tf1 is governed by a nuclear localization signal that possesses a unique requirement for the FXFG nuclear pore factor Nup124p. Mol Cell Biol. 2000;20:7798–812.
Feng G, Leem Y-E, Levin HL. Transposon integration enhances expression of stress response genes. Nucleic Acids Res. 2013;41:775–89.
Esnault C, Lee M, Ham C, Levin HL. Transposable element insertions in fission yeast drive adaptation to environmental stress. Genome Res. 2019;29:85–95.
Leem Y-E, Ripmaster TL, Kelly FD, Ebina H, Heincelman ME, Zhang K, et al. Retrotransposon Tf1 Is targeted to Pol II promoters by transcription activators. Mol Cell. 2008;30:98–107.
Majumdar A, Chatterjee AG, Ripmaster TL, Levin HL. Determinants that specify the integration pattern of retrotransposon Tf1 in the fbp1 promoter of Schizosaccharomyces pombe. J Virol. 2011;85:519–29.
Arcangioli B, Klar AJ. A novel switch-activating site (SAS1) and its cognate binding factor (SAP1) required for efficient mat1 switching in Schizosaccharomyces pombe. EMBO J. 1991;10:3025–32.
Arcangioli B, Copeland TD, Klar AJ. Sap1, a protein that binds to sequences required for mating-type switching, is essential for viability in Schizosaccharomyces pombe. Mol Cell Biol. 1994;14:2058–65.
Krings G, Bastia D. Sap1p binds to Ter1 at the ribosomal DNA of Schizosaccharomyces pombe and causes polar replication fork arrest. J Biol Chem. 2005;280:39135–42.
Hickey A, Esnault C, Majumdar A, Chatterjee AG, Iben JR, McQueen PG, et al. Single-nucleotide-specific targeting of the Tf1 retrotransposon promoted by the DNA-binding protein Sap1 of Schizosaccharomyces pombe. Genetics. 2015;201:905–24.
Jacobs JZ, Rosado-Lugo JD, Cranz-Mileva S, Ciccaglione KM, Tournier V, Zaratiegui M. Arrested replication forks guide retrotransposon integration. Science. 2015;349:1549–53.
Zaratiegui M, Vaughn MW, Irvine DV, Goto D, Watt S, Bähler J, et al. CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature. 2011;469:112–5.
Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272:1030–3.
Rai SK, Sangesland M, Lee M Jr, Esnault C, Cui Y, Chatterjee AG, et al. Host factors that promote retrotransposon integration are similar in distantly related eukaryotes. PLoS Genet. 2017;13:e1006775..
Paquin CE, Williamson VM. Ty insertions at two loci account for most of the spontaneous antimycin A resistance mutations during growth at 15 degrees C of Saccharomyces cerevisiae strains lacking ADH1. Mol Cell Biol. 1986;6:70–9.
Natsoulis G, Thomas W, Roghmann MC, Winston F, Boeke JD. Ty1 transposition in Saccharomyces cerevisiae is nonrandom. Genetics. 1989;123:269–79.
Bolton EC, Boeke JD. Transcriptional interactions between yeast tRNA genes, flanking genes and Ty elements: a genomic point of view. Genome Res. 2003;13:254–63.
Kinsey PT, Sandmeyer SB. Adjacent pol II and pol III promoters: transcription of the yeast retrotransposon Ty3 and a target tRNA gene. Nucleic Acids Res. 1991;19:1317–24.
Servant G, Pennetier C, Lesage P. Remodeling yeast gene transcription by activating the Ty1 long terminal repeat retrotransposon under severe adenine deficiency. Mol Cell Biol. 2008;28:5543–54.
Derr LK, Strathern JN, Garfinkel DJ. RNA-mediated recombination in S. cerevisiae. Cell. 1991;67:355–64.
Schacherer J, Tourrette Y, Souciet J-L, Potier S, de Montigny J. Recovery of a function involving gene duplication by retroposition in Saccharomyces cerevisiae. Genome Res. 2004;14:1291–7.
Maxwell PH, Coombes C, Kenny AE, Lawler JF, Boeke JD, Curcio MJ. Ty1 mobilizes subtelomeric Y′ elements in telomerase-negative Saccharomyces cerevisiae survivors. Mol Cell Biol. 2004;24:9887–98.
Moore JK, Haber JE. Capture of retrotransposon DNA at the sites of chromosomal double-strand breaks. Nature. 1996;383:644–6.
Teng SC, Kim B, Gabriel A. Retrotransposon reverse-transcriptase-mediated repair of chromosomal breaks. Nature. 1996;383:641–4.
Maxwell PH, Curcio MJ. Retrosequence formation restructures the yeast genome. Genes Dev. 2007;21:3308–18.
Umezu K, Hiraoka M, Mori M, Maki H. Structural analysis of aberrant chromosomes that occur spontaneously in diploid Saccharomyces cerevisiae: retrotransposon Ty1 plays a crucial role in chromosomal rearrangements. Genetics. 2002;160:97–110.
Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA. Double-strand breaks associated with repetitive DNA can reshape the genome. PNAS. 2008;105:11845–50.
Hoang ML, Tan FJ, Lai DC, Celniker SE, Hoskins RA, Dunham MJ, et al. Competitive repair by naturally dispersed repetitive DNA during non-allelic homologous recombination. PLoS Genet. 2010;6:e1001228.
Dunham MJ, Badrane H, Ferea T, Adams J, Brown PO, Rosenzweig F, et al. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. PNAS. 2002;99:16144–9.
Gresham D, Desai MM, Tucker CM, Jenq HT, Pai DA, Ward A, et al. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008;4:e1000303.
Zhang H, Zeidler AFB, Song W, Puccia CM, Malc E, Greenwell PW, et al. Gene copy-number variation in haploid and diploid strains of the yeast Saccharomyces cerevisiae. Genetics. 2013;193:785–801.
Chang S-L, Lai H-Y, Tung S-Y, Leu J-Y. Dynamic large-scale chromosomal rearrangements fuel rapid adaptation in yeast populations. PLoS Genet. 2013;9:e1003232.
Chan JE, Kolodner RD. A genetic and structural study of genome rearrangements mediated by high copy repeat Ty1 elements. PLoS Genet. 2011;7:e1002089.
Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell. 2005;120:587–98.
Admire A, Shanks L, Danzl N, Wang M, Weier U, Stevens W, et al. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006;20:159–73.
Chan YA, Aristizabal MJ, Lu PYT, Luo Z, Hamza A, Kobor MS, et al. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-Chip. PLoS Genet. 2014;10:e1004288.
García-Muse T, Aguilera A. R Loops: from physiological to pathological roles. Cell. 2019;179:604–18.
Curcio MJ. Border collies of the genome: domestication of an autonomous retrovirus-like transposon. Curr Genet. 2019;65:71–8.
Legrand M, Jaitly P, Feri A, d’Enfert C, Sanyal K. Candida albicans: an emerging yeast model to study eukaryotic genome plasticity. Trends Genet. 2019;35:292–307.
Acknowledgements
I thank the members of my department for their support and collegiality.
Funding
The author’s work is supported by his institution, Siena College.
Author information
Authors and Affiliations
Contributions
PM wrote the manuscript and prepared the figures and table.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares he has no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Maxwell, P.H. Diverse transposable element landscapes in pathogenic and nonpathogenic yeast models: the value of a comparative perspective. Mobile DNA 11, 16 (2020). https://doi.org/10.1186/s13100-020-00215-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13100-020-00215-x