
Abstract
Background
Metabolic syndrome (MetS) is a cluster of conditions associated with glucose intolerance, hypertension, abdominal obesity, dyslipidemia, and insulin resistance that increase the risk of cardiovascular diseases (CVD) and type 2 diabetes (T2D). Since MetS is known as a complex symptom with a high incidence of genetic factors, it is important to identify genetic variants for each clinical characteristic of MetS.
Methods
We performed targeted next-generation sequencing (NGS) to identify genetic variants related to obesity, blood glucose, triacylglycerol (TG), and high-density lipoprotein (HDL)-cholesterol level, and hypertension in 48 subjects with MetS and in 48 healthy subjects.
Results
NGS analysis revealed that 26 of 48 subjects (54.2%) with MetS had putative non-synonymous variants related to the clinical features of MetS. Of the subjects with MetS, 8 (16.7%) had variants in 4 genes (COL6A2, FTO, SPARC, and MTHFR) related to central obesity, 17 (35.4%) had variants in 6 genes (APOB, SLC2A2, LPA, ABCG5, ABCG8, and GCKR) related to hyperglycemia, 3 (6.3%) had variants in 4 genes (APOA1, APOC2, APOA4, and LMF1) related to hypertriglyceridemia, 8 (16.7%) had variants in 4 genes (ABCA1, CETP, SCARB1, and LDLR) related to low HDL-cholesterolemia, and 5 (10.4%) had variants in ADD1 related to hypertension.
Conclusions
Our findings may contribute to broadening the genetic spectrum of risk variants related to the development of MetS.
Similar content being viewed by others

Introduction
MetS, known as syndrome X, Deadly quartet, or insulin resistance syndrome, is defined by a cluster of five risk factors that increase the likelihood of developing CVD, stroke, and T2D [1]. The 5 risk factors for MetS include hypertension, obesity, hypertriglyceridemia, hyperlipidemia, and hyperglycemia. MetS is diagnosed when someone has three or more of these risk factors. There are also other factors that are likely increase the risk for MetS. These include age, genetic susceptibility, and not getting enough exercise. MetS is known to have different prevalence depending on geographic, racial, and ethnic origins, where its prevalence is estimated to be about 35% in North America [2], 11–26% in Europe [3], and 12–37% in Asia–Pacific region [4].
These clinical features of MetS are also caused by genetic factors, and up to 50% of all MetS cases are reported to be inherited [5]. However, as MetS includes multiple combinations of the effects of more than 3 risk factors, pinpointing a causative genotype is difficult and most MetS cases are considered polygenic. Therefore, inheritance of a single specific risk variant may be less important than the additive effects of many alleles, and thus, determination of multiple risk variants is necessary. Considerable progress has been made in the identification of causes that influence the development of MetS. Especially, population-specific genetic risk factors may help early diagnosis of individuals with high susceptibility to MetS.
In addition, as the major important pathogenic single nucleotide polymorphisms (SNPs) for specific target diseases vary among different populations, it is important to explore the genetic variants for clinical factors of MetS in specific populations.
Currently, comprehensive multiplex genomic sequencing technology approaches including NGS, are accelerating the identification of new molecular biomarker targets [6]. Advances in NGS have provided an unprecedented opportunity for identifying rare variants with moderate-to-large effects. These have increased our understanding of the molecular mechanisms underlying many inherited diseases in outlier populations. Targeted NGS is useful for rapidly identifying common and rare genetic variations [7]. Therefore, it can be an interesting and important work to identify rare variants affecting the development of MetS in specific populations with a single NGS panel in which genes identified for individual factors related to the clinical features of MetS are integrated.
In this study, we aimed to identify relevant genetic variants according to the clinical status of MetS using NGS.
Materials and methods
Study population, diagnosis of MetS, and ethical approval
Forty-eight participants were classified into the MetS group through clinical diagnoses based on the results of basic blood tests and health examinations at Health Checkup Center of HANARO Medical Foundation, Seoul, Korea, and 48 participants with no clinical features of MetS were included as the healthy control group (Table 1).The 48 subjects with MetS were diagnosed and enrolled according to a harmonized definition of International Diabetes Federation/National Heart, Lung, and Blood Institute/American Heart Association/International Association for the Study of Obesity [8]. All the subjects with MetS had at least three clinical features among hypertension (≥ 130 mmHg systolic and/or ≥ 85 mmHg diastolic), hyperglycemia (fasting glucose: ≥ 100 mg/dl), elevated TG (≥ 150 mg/dl), decreased HDL-cholesterol (< 40 mg/dl in men; < 50 mg/dl in women), and central obesity (waist circumference > 90 cm in men; > 85 cm in women), which followed a definition of the Korean Academy of Family Medicine [9]. Participants with two or more combination medications, chemotherapy, or anticancer drugs on special medications were excluded from the study. Written informed consent was obtained from all study participants. The study protocol was approved by the Institutional Review Board of Seoul Clinical Laboratory (2018-31-02F).
DNA preparation and selection of target NGS panel genes
Whole blood from all participants was collected and DNA was extracted using MagNa Pure 96 System (Roche Life Science, USA). NGS analysis was performed with NextSeq550 NGS system (Illumina, USA). NGS data were obtained with custom made NGS panel (Agilent Technologies, USA) on 28 selected genes related to the 5 clinical features for target capture sequencing. Gene selection was based on a review of research literature related to the 5 clinical features (central obesity, hyperglycemia, hypertriglyceridemia, low HDL-cholesterolemia, and hypertension) used for diagnosing MetS. A list of target genes that were sequenced is shown in Table 2.
Target capture and NGS
DNA extracted from whole blood of study participants was sheared into approximately 180 bp fragments with QSonica Sonicator (QSonica, USA). Sheared DNA was purified with AMPure XP beads (Beckman Coulter, USA) and the NGS library was prepared to target hybridization capture with the NGS panel using an Agilent SureSelectXT Custom Panel with SureSelectXT reagent kit (Agilent Technologies, USA) following the manufacturers’ instructions. Target capture libraries were sequenced on the NextSeq550 platform (Illumina, USA) using 2 × 150 bp paired-end runs.
Data analyses
All sequenced reads were aligned to the human reference genome National Center for Biotechnology Information build 37 (GRCh37/hg19) using the Burrows-Wheeler Aligner (Ver. 0.7.12). Local re-alignment around the indels and pair-end fixing was performed using Genome Analysis Tool Kit (GATK) Lite (Ver. 2.3-9), and PCR duplicates were removed using Picard (Ver. 1.128). The GATK Unified Genotyper was used to call the genomic variants. Mutated loci were annotated using snpEff (Ver. 4.3q). Non-synonymous single nucleotide variants (SNVs) and indels in the coding exons and splicing sites of target genes were included in the analysis. Known SNP with minor allelic frequency > 5% in the 1000 Genome Project Phase I East Asian (April 2012) and Genome Aggregation Database (gnomAD) (v2.1.1 release) were annotated and removed as the common non-disease-associated SNPs. Variant allele frequency ≤ 35%, total read depth ≤ 10X, and reads supporting a variant allele count ≤ 3 were also rejected as non-significant variants. False-positive indels were removed manually using the Integrated Genome Viewer (IGV Ver. 1.8.0.). In addition, all variants found in the 48 control subjects were rejected as non-disease-associated SNPs. After removing non-significant variants, in silico prediction was performed using SIFT, PolyPhen-2, PROVEAN, and MutationTaster.
According to American College of Medical Genetics (ACMG) guideline, variants identified in this study were classified as pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign. Predicted benign or likely benign variants in more than half of the programs were regarded as benign or likely benign variants and were not regarded as significant variants associated with MetS.
Results
Target region of NGS panel and targeted sequencing depth and coverage of each subject
Target capture genomic regions of interest included 359 exons and 109,173 bases. As shown in Additional file 1: Table S1, NGS probes were designed to capture exons in target genes and were used for target capture sequencing on subjects with MetS. The calculated mean depth and on-target ratio were 1099X and 30.8%, respectively. The coverage in the target region was 96.5%, with an average of ≥ 20X non-duplicated reads.
Variant spectrum from target capture sequencing
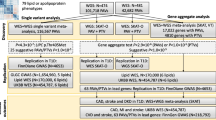
After variant calling, synonymous and off-target variants, and those that did not reach the cut-off value were rejected. Variants classified as benign and likely benign by in silico prediction tools were also rejected. We then identified non-synonymous variants associated with the clinical features of MetS, including 74 missense, 9 nonsense, and 1 frameshift indel after common variant removal based on the 1000 Genomes Project (East Asian frequency ≥ 5%) and GenomAD (East Asian frequency ≥ 5%) and compared with the control group data (Table 3). By grouping SNVs according to the nature of the nucleotide changes, we observed enrichment of C:G → T:A transition variants (40.3% on average) followed by A:T → G:C transition variants (33.1% on average) (Fig. 1A). Clinical information for each sample according to variant spectrum is described in Fig. 1B with subjects classified as normal and risk group. Validated loci were further analyzed for the functional prediction of amino acid changes using 4 different prediction algorithms (SIFT, PolyPhen-2, PROVEAN, and Mutation Taster). Overall, 26 of 48 subjects (54.2%) had putative non-synonymous variants associated with the clinical features of MetS (Fig. 1C).

Variant spectrum from target capture sequencing: A Overall variant profiles. B Characteristics of MetS subjects. Normal and risk subjects were classified according to the definition of MetS. C Genes with putative variants (East Asian frequency < 5%) associated with clinical feature of MetS and the number of subjects with following genetic variants (rightmost graph). Each block shown in Fig. 1 represents each individual participant with MetS
Variant related to central obesity
CAV1, COL6A2, FTO, LEP, IGF1, SPARC, GCKR, and MTHFR genes were included for the NGS analysis for detecting variants related to central obesity. Eleven types of variants in 4 genes (COL6A2, FTO, SPARC, and MTHFR) were found to be related to central obesity, and 8 of 48 subjects with MetS (16.7%) showed putative non-synonymous variants in these genes (Fig. 1C). Six missense variants in COL6A2 were found in 6 subjects with MetS, 1 subject had a missense variant in FTO, 3 subjects had 2 missense variants and 1 frameshift indel in SPARC, and 1 subject had a missense variant in MTHFR.
Variant related to hyperglycemia
ADIPOQ, APOB, SLC2A2, LPA, ABCG5, ABCG8, and GCKR genes were included in the NGS analysis for detecting variants related to fasting blood glucose level. Seventeen of 48 subjects (35.4%) had putative non-synonymous variants in APOB, SLC2A2, LPA, ABCG5, ABCG8, and GCKR (Fig. 1C and Table 3). Eight subjects with MetS had 17 missense variants and 3 nonsense variants in APOB. One subject had a missense variant in SLC2A2, 3 subjects had 3 missense variants in ABCG5, 1 subject had a missense variant in ABCG8, 2 subjects had 2 missense variants in GCKR, and 10 subjects with MetS had 11 missense variants and 1 nonsense variant in LPA.
Variant related to hypertriglyceridemia
APOA1, APOA4, APOA5, APOC2, APOC3, ZHX3, GPIHBP1, and LMF1 genes were included for the NGS analysis for detecting variants related to hypertriglyceridemia. Overall, 3 of 48 subjects (6.3%) had putative non-synonymous variants in APOA1, APOC2, APOA4, and LMF1 (Fig. 1C and Table 3). One subject with MetS had 10 non-synonymous variants in APOA1, APOC2, APOA4, and LMF1, including 7 missense variants and 3 nonsense variants. The other 2 subjects had 2 missense variants in LMF1.
Variant related to low HDL-cholesterolemia
Among the low HDL-cholesterolemia-related genes (ABCA1, CETP, SCARB1, LPL, and LDLR) included in the NGS panel, ABCA1, CETP, SCARB1, and LDLR showed putative non-synonymous variants in subjects with MetS. Eight of 48 subjects with MetS (16.7%) showed variants in ABCA1, CETP, SCARB1, and LDLR (Fig. 1C and Table 3). Four subjects with MetS had non-synonymous variants in ABCA1 with 6 missense variants and 1 nonsense variant. Two subjects with MetS had 2 missense variants in SCARB1. One subject with MetS had a missense variant in CEPT, 5 subjects with MetS had 7 missense variants and 1 nonsense variant in LDLR.
Variant related to hypertension
ADD1, ADM, and ADRB2 were included for the NGS analysis for detecting variants related to hypertension. Overall, 5 of 48 subjects (10.4%) had putative non-synonymous variants in ADD1. Five subjects with MetS had 6 missense variants in ADD1 (Fig. 1C and Table 3). Four subjects with MetS had the same c.16C > T p.(Arg6Cys) variant and 1 had c.1003G > C p.(Ala335Pro) and c.1051G > C p.(Ala351Pro) variants.
Discussion
Our NGS analyses identified 84 non-synonymous variants related to the 5 clinical features of MetS in the 19 genes, including 74 missense and 9 nonsense SNPs, and 1 frameshift indel variant. The 8, 17, 3, 8, and 5 subjects with MetS had the 11 variants in 4 genes (COL6A2, FTO, SPARC, and MTHFR) related to obesity, 39 in 6 genes (APOB, SLC2A2, LPA, ABCG5, ABCG8, and GCKR) related to hyperglycemia, 10 in 4 genes (APOA1, APOC2, APOA4, and LMF1) related to hypertriglyceridemia, 18 in 4 genes (ABCA1, CETP, SCARB1, and LDLR) related to low HDL-cholesterolemia, and 6 in 1 gene (ADD1) related to hypertension, respectively. To our knowledge, 50 variants identified in our NGS analysis are novel ones that may be related to the clinical features of MetS.
The contribution of genetic factors to the inter-individual variation in obesity accounts for 40–70% [10]. Previous studies have shown that COL6A2 is a putative preadipocyte marker gene [11] and is also involved in the formation of adipocyte extracellular matrix [12]. Although many genetic loci related to obesity have been reported through genome-wide association studies (GWAS) in large cohorts of European [13] or East Asian populations [14] and in Genetics of Noninsulin dependent Diabetes Mellitus (GENNID) multiethnic family study [15], no studies have reported rare variants in COL6A2 in subjects with obesity. Notably, the c.2524G > T p.(Ala842Ser) and c.1327G > A p.(Glu443Lys) variants in COL6A2 were absent in the 1000 Genome and gnomAD databases, indicating they may be novel variants. Based on these results, it is presumed that the 6 variants in COL6A2 may influence the development of obesity. FTO, a well-known gene associated with obesity, has been identified as a major genetic contributor to polygenic obesity in a cohort study of European populations [16, 17]. A large cohort study of Korean populations has also shown that FTO SNP rs9939609 is significantly associated with body mass index (BMI), a common measure of obesity [14]. However, we identified FTO c400G > A p.(Ala134Thr) variant in only 1 obese subject with MetS, suggesting that this variant may be detected with very low frequency in obese Koreans. A previous study has shown that the SPARC gene is associated with human obesity and its expression is increased in adipose tissue of obese individuals [18]. A recent study has revealed that subnetworks of key genes such as SPARC play roles in regulating known genes for obesity, CVD, and T2D [19]. However, any variants in SPARC related to MetS have not been identified. The 3 variants (c.1024_1025delTA p.(Ter342fs), c253C > A p.(Leu85Met), and c.230G > T p.(Cys77Phe) in SPARC related to obesity in our study were classified as pathogenic or likely pathogenic according to the ACMG guidelines, suggesting that these variants may be closely related to obesity. Interestingly, the SPARC c.253C > A p.(Leu85Met) and c.230G> T p.(Cys77phe) were identified as previously unreported novel variants, as they were absent in the 2 public databases. The Cys677Thr polymorphism in MTHFR has been known to be a significant variant associated with an increased risk of obesity [20]. Although the MTHFR c.1498T > G p.(Trp500Gly) variant identified in our study is not Cys677Thr, the variant we identified needs further functional study as it has been classified as pathogenic according to the ACMG guidelines.
A previous study has shown that APOB variants are significantly related to higher blood glucose levels in patients with T2D [21]. However, many studies have focused on investigating an association of APOB variants with metabolic diseases such as familial hypercholesterolemia [22, 23]. Among the APOB variants we identified in this study, c.6655C > T p.(Arg2219Cys), c.12853A > T p.(Lys4285Ter), c.7397T > A p.(Leu2466Ter), c.5409C > A p.(Tyr1803Ter), c.3122G < T p.(Gly1041Val), and c.4364T > A p.(Phe1455Tyr) variants were classified as pathogenic or likely pathogenic according to the ACMG guidelines, suggesting a significant relationship of these variants with higher fasting blood glucose levels in subjects with MetS. Notably, the c.13525T > A p.(Tyr4509Asn), c.13055T > A p.(Leu4352Gln), c.7138G > T p.(Val2380Phe), c.6538C > A p.(Gln2180Lys), c.6480A > T p.(Leu2160Phe), c.2459G > T p.(Gly820Val), c.8266G > T p.(Gly2756Cys), c.12853A > T p.(Lys4285Ter), c.12794T > A p.(Val4265Glu), c.7397T > A p.(Leu2466Ter), c.5409C > A p.(Tyr1803Ter), and c.3122G > T p.(Gly1041Val) in APOB were identified as novel variants that had not been previously reported. Thus, further studies are needed to elucidate the functional associations of these novel variants. Two GWA studies have reported that missense variants including the Thr110Ile encoded by rs5400 SNP in SLC2A2, known as GLUT2 are significantly associated with impaired fasting glucose [24, 25]. Interestingly, another study has suggested that study participants with the missense variant, Thr110Ile, in SLC2A2 show a preference for carbohydrates [26]. However, the missense variant, c1521C > A p.(His507Gln), in the SLC2A2 related to increased fasting glucose levels that we identified in this study has not been previously reported in GWAS or NGS-based study. Recent cohort studies of MetS in Korean populations have not reported the c1521C > A p.(His507Gln) variant in SCL2A2 [27, 28]. Therefore, the SLC2A2 variant we identified may be a novel variant related to hyperglycemia in the Korean population, and additional studies are needed to reveal that the novel variant has a functional association with the phenotypes of MetS. Several studies have also reported that LPA variants that are considered risk factors for cardiovascular disease [29] and GCKR variants are associated with hyperglycemia [24, 30]. Interestingly, the c.5690T > C p.(Phe1897Ser), c.5221T > A p.(Cys1741Ser), c.6068A > T p.(Tyr2023Phe), c.6037G > T p.(Gly2013Cys), c.4880C > A p.(Thr1627Lys), and c.3053C > T p.(Ser1018Leu) variants in LPA and the c.169G > T p.(Asp57Tyr) variants in GCKR were absent in the 2 public databases, indicating their novelty. Notably, the c.5897A > G p.(Glu1966Gly), c.5690T > C p.(Phe1897Ser), and c.6068A > T p.(Tyr2023Phe) variants in LPA and the c.169G > T p.(Asp57Tyr) variant in GCKR were classified as pathogen or likely pathogen according to the ACMG guidelines, suggesting that these novel variants are likely to be significantly related to hyperglycemia in individuals with MetS. In a cohort of patients with T2D, two variants (rs6720173 and rs4148211) in ABC transporter genes, ABCG5 and ABCG8, have been found to increase the risk of T2D in humans [31]. The most significant SNP (rs4299376) in ABCG8 was also found to be associated with increased fasting plasma glucose levels [32]. In our study, c.629A > C p.(Asp210Ala), c.254T > A p.(Leu85Gln), and c.1114C > A p.(Leu372Met) in ABCG5 and c.900G > A p.(Met300Ile) in ABCG8 were found to be related to higher fasting glucose levels. Notably, these 3 variants in ABCG5 were previously unknown, suggesting that these novel variants may be novel genetic determinants of MetS and be ethnic-specific genetic variants under clinical conditions of Mets.
A GWA study of individuals with hypertriglyceridemia has shown that common variants in 4 genetic loci (APOB, LPL, GCKR, and APOA5) are significantly related to increased TG levels [33]. Among genetic variants affecting TG metabolism, a rare APOC3 p.(Gln38Lys), well-known as a gain-of-function (GOF) variant has been found to contribute to increased TG levels [34]. Our NGS analysis revealed 10 rare variants at 4 genes (APOA1, APOA4, APOC2, and LMF1) related to hypertriglyceridemia. Among them, c.76G > T p.(Glu26Ter) in APOA1, c.23T > A p.(Leu8Gln) in APOA4, c.214A > T p.(Arg72Trp) in APOC2, c.1644C > A p.(Ser548Arg), c.1207G > T p.(Val403Phe), c.1177A > T p.(Met393Leu), and c.668T > A p.(Leu223Gln) in LMF1 were identified as novel variants because these variants were not present in the 2 public databases. Notably, the APOA1 c.76G > T p.(Glu26Ter), the APOC2 c.214A > T p.(Arg72Trp), and the LMF1 c.668T > A p.(Leu223Gln) variants were classified as likely pathogenic, pathogenic, and likely pathogenic according to the ACMG guidelines, respectively, suggesting that these rare variants may be significantly related to hypertriglyceridemia in individuals with MetS.
A previous study has shown that a variant (rs9282541, Arg230Cys) in ABCA1, known as ATP-binding cassette transporter A1 gene is significantly associated with low HDL-cholesterolemia in Native Americans, suggesting that the variant is not only exclusive to Native Americans but is also a significant genetic determinant of HDL-cholesterol levels [35]. The 7 variants, c.6406G > T p.(Gly2136Ter), c.6205G > T p.(Asp2069Tyr), c.2940G > T p.(Gln980His), c.4995G > T p.(Met1665Ile), c.1631G > A p.(Gly544Asp), c.6608T > A p.(Ile2203Lys), and c.796G > A p.(Gly266Arg), in ABCA1 identified in this study were previously unreported. Moreover, these variants were classified as pathogenic or likely pathogenic according to the ACMG guidelines. Given these results, the 7 variants may play roles in the pathogenesis of low HDL-cholesterolemia. Interestingly, we identified 2 nonsense variants in ABCA1 and LDLR related to low HDL-cholesterolemia, c.6406G > T p.(Gly2136Ter) and c.2541C > A p.(Tyr847Ter), respectively. The 2 variants were also classified as pathogenic according to the ACMG guidelines, suggesting that the variants may be significantly related to an increased risk of low HDL-cholesterolemia in individuals with MetS as well as play an important role in the pathogenesis of low HDL-cholesterolemia. Thus, further studies are needed to elucidate the functional associations of these variants.
A previous study has shown that ADD1 is a salt-sensitive gene that plays a role in the etiology of hypertension [36]. Recently, a study has shown that a genetic polymorphism (rs4961, Gly460Trp) in ADD1 associated with hypertension is a genetic biomarker of hypertension in Asians [37]. However, the known variant was absent in our study, suggesting that genetic variants affecting the development of MetS may vary across ethnic groups. In our study, since the 4 individuals with MetS had the same c.16C > T p.(Arg6Cys) variant in hypertension-related ADD1, this was considered the most frequent variant. Interestingly, the c.1003G > C p.(Ala335Pro) and c.1051G > C p.(Ala351Pro) variants in ADD were previously unknown, indicating their novelty.
Our study has several strengths. First, study participants were recruited from the same geographical area, and baseline examinations were conducted at the same medical center using stringent phenotype selection approaches. Second, this study was designed to use subjects with non-medication and no symptoms related to the clinical features of MetS as control subjects. The variant identification process is comparable to that used for high quality data, which greatly enhances the significance of the variant data.
As our study is a DNA-mutation-based cohort study, more accurate results could be obtained by additional in vivo analysis accompanied with RNA or protein expression data. Especially, clinical information and the presence or absence of genetic mutations did not show clearly matched in some of cases, which seem to be because genetic mutations are not necessarily associated with clinical symptoms and not all genes related to clinical symptoms have been screened. Thus, to determine more accurate individual cause genes for each clinical feature, individual cellular protein expression needs to be confirmed to validate the function of mutations, but this was difficult to apply, which is a limitation of the present study.
A recent MetS cohort study involving a total of 8,150 participants revealed that the prevalence of MetS in Koreans over 30 years was about 35.2% [38]. With the increasing incidence of MetS, it is necessary to discover additional genetic variants of MetS. In addition, since the novel variants identified in this study were detected in Koreans with MetS, it is necessary to investigate the potential roles of these variants in other ethnic groups with MetS.
Although common 17 SNPs associated with hypertriglyceridemia and low HDL-cholesterolemia in Korean populations with MetS have recently been reported through GWAS [28], these SNPs are different from the rare non-synonymous variants identified by our NGS analysis. Identifying genetic variants, such as mutations with potential molecular predictors, can enable early identification on risk of MetS and therapeutic targets for drugs. In addition, information regarding genetic factors may help to decide the best decisions on MetS treatment.
Conclusion
Our study identified 84 non-synonymous variants related to the 5 clinical features of MetS, which include 74 missense and 9 nonsense SNPs, and 1 frameshift indel variant. Among them, 50 variants identified in our NGS analysis are novel ones that may be related to the clinical features of MetS. Our results suggest that the candidate genes and rare non-synonymous variants related to the 5 clinical features of MetS may be used as potential genetic variants or molecular predictors for MetS. However, additional functional studies are needed to validate these novel variants.
Availability of data and materials
Not applicable.
References
Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C. Definition of metabolic syndrome report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation. 2004;109:433–8.
Hirode G, Wong RJ. Trends in the Prevalence of Metabolic Syndrome in the United States, 2011–2016. JAMA. 2020;323:2526–8.
van Vliet-Ostaptchouk JV, Nuotio M-L, Slagter SN, Doiron D, Fischer K, Foco L, Gaye A, Gögele M, Heier M, Hiekkalinna T, Joensuu A, Newby C, Pang C, Partinen E, Reischl E, Schwienbacher C, Tammesoo M-L, Swertz MA, Burton P, FerrettiV Fortier I, Giepmans L, Harris JR, Hillege HL, Holmen J, Jula A, Kootstra-Ros JE, Kvaløy K, Holmen TL, Männistö S, Metspalu A, Midthjell K, Murtagh MJ, Peters A, Pramstaller PP, Saaristo T, Salomaa V, Stolk RP, Uusitupa M, van der Harst P, van der Klauw MM, Waldenberger M, Perola M, Wolffenbuttel BH. The prevalence of metabolic syndrome and metabolically healthy obesity in Europe: a collaborative analysis of ten large cohort studies. BMC Endocr Disorder. 2014;14:9.
Ranasinghe P, Mathangasinghe Y, Hills AP, Misra A. Prevalence and trends of metabolic syndrome among adults in the Asia-Pacific region: a systematic review. BMC Public Health. 2017;17:101.
Ziki MDA, Mani A. Metabolic syndrome: genetic insights into disease pathogenesis. Curr Opin Lipidol. 2016;27:162–71.
Hu T, Chitnis N, Monos D, Dinh A. Next-generation sequencing technologies: an overview. Human Immunol. 2021;82:801–11.
Gulilat M, Lamb T, Teft WA, Wang J, Dron JS, Robinson JF, Tirona RG, Hegele RA, Kim RB, Schwarz UI. Targeted next-generation sequencing as a tool for precision medicine. BMC Med Genomics. 2019;12:81.
Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart J-C, James WPT, Loria CM, Smith SC Jr. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5.
Kang H-T, Kim S-Y, Kim J, Kim J, Kim J, Park HA, Shin J, Cho SH, Choi Y, Shim JY. Clinical practice guideline of prevention and treatment for metabolic syndrome. Korean J Fam Pract. 2015;5:375–420.
Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human obesity. Behav Genet. 1997;27:325–51.
Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–56.
Mariman ECM, Wang P. Adipocyte extracellular matrix composition, dynamics, and role in obesity. Cell Mol Life Sci. 2010;67:1277–92.
Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM, the GIANT Consortium. Meta-analysis of genome-wide association studies for height and body mass index in ∼700 000 individuals of European ancestry. Hum Mol Genet. 2018;27:3641–9.
Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban H-J, YoonD Lee MH, Kim D-J, Park M, ChaS-H Kim J-W, Han B-G, Min H, Ahn Y, Park MS, Han HR, Jang H-Y, Cho EY, Lee J-E, Cho NH, Shin C, Park T, Park JW, Lee J-K, Cardon L, Clarke G, McCarthy MI, Lee J-Y, Lee J-K, Oh B, Kim H-L. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet. 2009;41:527–34.
Wan JY, Goodman DL, Willems EL, Freedland AR, Norden-Krichmar TM, Santorico SA, Edwards KL, American Diabetes GENNID Study Group. Genome-wide association analysis of metabolic syndrome quantitative traits in the GENNID multiethnic family study. Diabetol Metab Syndr. 2021;13:59.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94.
Babenko V, Babenko R, Markel A. FTO haplotyping underlines high obesity risk for European populations. BMC Med Genomics. 2019;12:46.
Kos K, Wilding JP. SPARC: a key player in the pathologies associated with obesity and diabetes. Nat Rev Endocrinol. 2010;6:225–35.
Shu L, Chan KHK, Zhang G, Huan T, Kurt Z, Zhao Y, Codoni V, Cardiogenics Consortium, Trégouët D-A, Yang J, Wilson JG, Luo X, Levy D, Lusis AJ, Liu S, Yang X. Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet. 2017;13:e1007040.
Lewis SJ, Lawlor DA, Nordestgaard BG, Tybjaerg-Hansen A, Ebrahim S, Zacho J, Ness A, Leary S, Smith GD. The methylenetetrahydrofolate reductase C677T genotype and the risk of obesity in three large population-based cohorts. Eur J Endocrinol. 2008;159:35–40.
Duman BS, Öztürk M, Yilmazer S, Çagatay P, Hatemi H. Apolipoprotein B gene variants are involved in the determination of blood glucose and lipid levels in patients with non-insulin dependent diabetes mellitus. Cell Biochem Funct. 2006;24:261–7.
Xu H, Ryan KA, Jaworek TJ, Southam L, Reid JG, Overton JD, Baras A, Puurunen MK, Zeggini E, Taylor SI, Shuldiner AR, Mitchell BD. Familial hypercholesterolemia and type 2 diabetes in the old order Amish. Diabetes. 2017;66:2054–8.
Sharifi M, Futema M, Nair D, Humphries SE. Genetic archtecture of familial hypercholesterolaemia. Curr Cardiol Rep. 2017;19:44.
Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, Lindgren CM, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–16.
Barker A, Sharp SJ, Timpson NJ, Bouatia-Naji N, WarringtonNM Kanoni S, Beilin LJ, Brage S, Deloukas P, Evans DM, Grontved A, Hassanali N, et al. Association of genetic loci with glucose levels in childhood and adolescence: a meta-analysis of over 6,000 children. Diabetes. 2011;60:1805–12.
Eny KM, Wolever TM, Fontaine-Bisson B, El-Sohemy A. Genetic variant in the glucose transporter type 2 is associated with higher intakes of sugars in two distinct populations. Physiol Genomics. 2008;33:355–60.
Lee H-S, Kim Y, Park T. New common and rare variants influencing metabolic syndrome and its individual components in a Korean population. Sci Rep. 2018;8:5701.
Oh S-H, Lee J-E, ShinE Kwon H, Choe EK, Choi S-Y, Rhee H, Choi SH. Genome-wide association study of metabolic syndrome in Korean populations. PLoS ONE. 2020;15:e0227357.
Ronald J, Rajagopalan R, Cerrato F, Nord AS, Hatsukami T, Kohler T, Marcovina S, Heagerty P, Jarvik GP. Genetic variation in LPAL2, LPA, and PLG predicts plasma lipoprotein(a) level and carotid artery disease risk. Stroke. 2011;42:2–9.
Stancáková A, Civelek M, Saleem NK, Soininen P, Kangas AJ, Cederberg H, Paananen J, Pihlajamäki J, Bonnycastle LL, Morken MA, Boehnke M, Pajukanta P, Lusis AJ, Collins FS, Kuusisto J, Ala-Korpela M, Laakso M. Hyperglycemia and a common variant of GCKR are associated with the levels of eight amino acids in 9,369 Finnish men. Diabetes. 2012;61:1895–902.
Gok O, Karaali ZE, Acar L, Kilic U, Ergen A. ABCG5 and ABCG8 gene polymorphisms in type 2 diabetes mellitus in the Turkish population. Can J Diabetes. 2015;39:405–10.
Cederberg H, Gylling H, Miettinen TA, Paananen J, Vangipurapu J, Pihlajamäki J, Kuulasmaa T, Alena Stancáková A, Smith U, Kuusisto J, Laakso M. Non-cholesterol sterol levels predict hyperglycemia and conversion to type 2 diabetes in Finnish men. PLoS ONE. 2013;8: e67406.
Johansen CT, Wang J, Lanktree MB, Cao H, McIntyre AD, BanMR Martins RA, Kennedy BA, Hassell RG, Visser ME, Schwartz SM, Voight BF, Elosua R, Salomaa V, O’Donnell CJ, Dallinga-Thie GM, Anand SS, Yusuf S, Huff MW, Kathiresan S, Hegele RA. Mutation skew in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42:684–7.
Sundaram M, Curtis KR, Alipour MA, LeBlond ND, Margison KD, Yaworski RA, Parks RJ, McIntyre AD, Hegele RA, Fullerton MD, Yao Z. The apolipoprotein C-III (Gln38Lys) variant associated with human hypertriglyceridemia is a gain-of-function mutation. J Lipid Res. 2017;58:2188–96.
Acunña-Alonzo V, Flores-Dorantes T, Janine K, Kruit JK, Villarreal-Molina T, Arellano-Campos O, Hünemeier T, Moreno-Estrada A, Ortiz-López MG, Villamil-Ramírez H, León-Mimila P, Villalobos-Comparan M, Jacobo-Albavera L, Ramıírez-Jiménez S, Sikora M, Zhang L-H, Pape TD, de Ángeles Granados-Silvestre M, Montufar-Robles I, Tito-Alvarez AM, Zurita-Salinas C, Bustos-Arriaga J, Cedillo-Barrón L, Gómez-Trejo C, Barquera-Lozano R, Vieira-Filho JP, Granados J, Romero-Hidalgo S, Huertas-Vázquez A, González-Martín A, Gorostiza A, Bonatto SL, Rodríguez-Cruz M, Wang L, Tusié-Luna T, Aguilar-Salinas CA, Lisker R, Moises RS, Menjivar M, Salzano FM, Knowler WC, Bortolini MC, Hayden MR, Baier LJ, Canizales-Quinteros S, Acunña-Alonzo V, et al. A functional ABCA1 gene variant is associated with low HDL-cholesterol levels and shows evidence of positive selection in Native Americans. Human Mol Genet. 2021;19:2877–85.
Bianchi G, Ferrari P, Staessen JA. Adducin polymorphism: detection and impact on hypertension and related disorders. Hypertension. 2005;45:331–40.
Jin H, Huang Y, Yang G. Association between a-adducin rs4961 polymorphism and hypertension: a meta-analysis based on 40 432 subects. J Cell Biochem. 2019;120:4613–9.
Lee S-H, Tao S, Kim H-S. The prevalence of metabolic syndrome and its related risk complications among Koreans. Nutrients. 2019;11:1755.
Acknowledgements
We are grateful to all members of HANARO Medical Foundation and SCL Biobank for supporting this study.
Author information
Authors and Affiliations
Contributions
SL and KRL conceived, coordinated, and designed the study. KC and SYB recruited the study participants and diagnosed and classified MetS and control groups. SL, SAK, JH, YK, and GH performed the NGS experiment. JH and MKL annotated the NGS data. SL and KRL wrote the original draft and finally edited the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Written informed consent was obtained from all study participants. The study protocol was approved by the Institutional Review Board of Seoul Clinical Laboratory (2018-31-02F).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Target region of NGS panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lee, S., Kim, SA., Hong, J. et al. Identification of genetic variants related to metabolic syndrome by next-generation sequencing. Diabetol Metab Syndr 14, 119 (2022). https://doi.org/10.1186/s13098-022-00893-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13098-022-00893-y