
Abstract
Fibroblast-like synoviocytes (FLSs) play a central role in RA pathogenesis and are the main cellular component in the inflamed synovium of patients with rheumatoid arthritis (RA). FLSs are emerging as promising new therapeutic targets in RA. However, fibroblasts perform many essential functions that are required for sustaining tissue homeostasis. Direct targeting of general fibroblast markers on FLSs is challenging because fibroblasts in other tissues might be altered and side effects such as reduced wound healing or fibrosis can occur. To date, no FLS-specific targeted therapies have been applied in the clinical management of RA. With the help of high-throughput technologies such as scRNA-seq in recent years, several specific pathogenic FLS subsets in RA have been identified. Understanding the characteristics of these pathogenic FLS clusters and the mechanisms that drive their differentiation can provide new insights into the development of novel FLS-targeting strategies for RA. Here, we discuss the pathogenic FLS subsets in RA that have been elucidated in recent years and potential strategies for targeting pathogenic FLSs.
Similar content being viewed by others

Introduction
Rheumatoid arthritis (RA) is one of the most common rheumatic diseases characterized by persistent synovial inflammation in multiple joints along with bone damage [1] and affects approximately 0.5 ~ 1% of the population worldwide [2]. Current drugs for treating RA including conventional synthetic disease-modifying anti-rheumatic drugs (csDMARD) and biological or targeted DMARDs (b/tsDMARDs) mainly target immune cells and inflammatory cytokines. Although b/tsDMARDs have greatly improved outcomes in patients with RA, approximately 40% of patients with RA do not respond to individual biologic therapies [3], and a significant proportion of patients with RA still have active disease and are considered as “difficult-to-treat RA” [4]. Additionally, general immune suppression by traditional immunosuppressive cell therapies can significantly increase the risk of infection, which remains a major challenge for RA management. A previous study showed molecular signature of synovium was different in patients who responded to IL-6 or CD20 respectively. Moreover, fibroblasts encoding gene signature was substantially increased in patients who showed no response to both IL-6 and CD20 targeted therapies [5]. Patients who do not respond to two biologics presented with a pauci-immune phenotype in the synovium [6]. These findings suggest that stromal cells are an emerging attractive new therapeutic target in RA.
Fibroblast-like synoviocytes (FLSs) are the main cell component in the inflamed synovium of patients with RA [7]. The synovium is composed of two layers: the intimal lining layer and the sublining layer [6]. In homeostatic states, the synovial lining is comprised of FLSs and macrophages (2–3 cell layers in total). In RA, the lining layer expands, and immune cells such as lymphocytes, macrophages and dendritic cells accumulate in the sublining layer [8]. Instead of sustaining homeostasis in the physiological state, FLSs in RA exhibit a “proinflammatory” or “aggressive/tissue damage” phenotype and increase the expression of inflammatory cytokines, chemokines and matrix metalloproteinases, leading to inflammation persistence and bone damage [8]. FLSs have been proven to play a central role in the pathogenesis of RA [9], and therapeutic strategies targeting FLSs might avoid systemic immunosuppressive consequences, in contrast with the immunosuppressive therapies. Thus, FLSs have long been considered as promising new therapeutic targets for RA [10]. Recent published reviews have summarized where do we stand in the era of FLS-targeted therapy [11, 12]. To date, no effective FLS-targeting therapies have been approved for use in the clinical treatment of RA, as fibroblasts are enriched in a wide array of tissues with multiple functions that are important for sustaining tissue homeostasis, and FLSs in the synovium cannot be easily distinguished from fibroblasts in other tissues by specific markers; thus, direct targeting of general fibroblasts markers in FLSs is a challenge for RA treatment.
With the help of high-throughput technologies such as scRNA-seq and CyTOF in recent years, our understanding of functionally distinct subsets of fibroblasts has been largely explored. Several pathological FLS phenotypes were found to be specifically expanded in RA. These novel RA-specific pathogenic FLS clusters might provide new promising therapeutic targets that could significantly decrease the side effects of general fibroblast-targeted therapies. Here, we review recent findings on pathogenic FLS subsets in RA and potential targeting strategies with the aim of providing a better understanding of the heterogeneity of FLSs in RA and new insights into FLS-targeted therapy.
Pathogenic effects of FLSs in RA
In the physiological state, FLSs directly impact the synovial fluid composition by producing hyaluronic acid and other joint lubricants, such as lubricin (also known as proteoglycan 4). FLSs play an important role in helping shape and maintain the synovial extracellular matrix (ECM) by producing matrix components (such as fibronectin, type I and III collagens, vimentin, tenascin, proteoglycans and laminin) and ECM-degrading enzymes (such as proteases, matrix metalloproteinases, hyaluronan synthase, and cathepsins) [6, 13]. Additionally, FLSs under physiological conditions may have an anti-inflammatory/pro-resolving ability that helps sustain immune homeostasis in the local immune environment [14]. In RA, FLSs lose their homeostatic phenotype and acquire “proinflammatory” and “aggressive/tissue-damaging” phenotypes that mediate the persistence of inflammation and cartilage/bone damage. The pathological effects mediated by RA-FLSs include the following: (1) enhanced migration and proliferation with reduced apoptosis, which results in hyperplastic rheumatoid pannus formation and leads to direct cartilage and bone damage; (2) overproduction of matrix metalloproteinases (MMPs) (such as MMP1, MMP3 and MMP13), aggrecanases (ADAMTS4 and ADAMTS5) [9], and RANKL [15], which damage the collagen-rich structures of joint tissues and promote osteoclast differentiation; (3) overproduction of proinflammatory cytokines (such as type 1 interferons [16], IL6 [17]) and chemokines (such as CCL5, CCL8, CXCL5 and CXCL10 [18]) that direct the recruitment of immune cells into joints; (4) promotion of T-cell [19,20,21] and B-cell [22,23,24] activation and differentiation. The various physiological and pathogenic effects of FLSs suggest their functional heterogeneity.
Pathogenic FLS subsets in RA
The FLS subsets in RA reported in recent studies are summarized in Table 1 [14, 25,26,27,28,29,30,31,32,33,34], and the phenotypic characteristics of the pathogenic FLS subsets are described. Among those markers, cadherin-11 (CDH-11), fibroblast activation protein α (FAPα) and podoplanin (PDPN/GP38) are considered as general markers that associated with the pathogenic FLS phenotype in RA [14, 35]; however, FLSs in the physiological state may also express these markers but at relatively low levels. CDH-11 was reported to be a relatively specific marker of FLSs compared with fibroblasts in other tissues [36, 37]. CDH-11 regulates the production of several proinflammatory cytokines, such as IL-6 [38]. CDH-11 knockout mice were resistant to joint inflammation and cartilage erosion, suggesting the vital role of CDH-11+ FLSs in the pathogenesis of RA [36]. PDPN is expressed predominantly on the lining layer, and studies have shown that a small number of FLSs in the sublinling layer also express PDPN [25, 29]. PDPN+ FLSs were found to be expanded in RA but not in OA or healthy synovial tissue [14, 39]. PDPN+ FLSs could migrate and invade cartilage in a mouse model of cartilage destruction, suggesting that PDPD+ FLSs are pathogenic and capable of invasion and destruction [25]. Previous research has demonstrated that PDPN+CD45−CD31− cells, termed PRIME cells, can be found in peripheral blood and are similar to PDPN+ FLSs. The proportion of PRIME cells increased before RA flare-up, thus verifying the important role of PDPN+ FLSs in RA pathogenesis [40]. FAPα is considered as a marker of fibroblast activation. In RA, FAPα colocalized with PDPN in the synovium. Moreover, FAPα plays an important role in remodeling the immune environment by mediating the interaction of FLSs with immune cells, regulating cytokine secretion and initiating the immune response [41], and deletion of FAPα+ FLSs ameliorated both inflammation and bone erosion in a mouse model of arthritis [29].
Pathogenic PDPN+ or FAPα+ FLSs can be further divided into functionally distinct pathogenetic subsets. In 2019, Adam P Croft et al. [29] classified FAPα+PDPN+ FLSs into proinflammatory (CD90+FAPα+PDPN+, sublining) and bone damage (CD90−FAPα+PDPN+, lining) subsets based on the expression of CD90. Injection of CD90+FAPα+PDPN+ FLS into the inflamed ankle joint of mice led to more severe and persistent joint swelling, with greater leukocyte infiltration. In contrast, injection of CD90−FAP+PDPN+ FLSs led to increased osteoclast activity and joint damage but did not affect the severity of joint inflammation. Similarly, several other studies also revealed expanded CD90+ FLSs with proinflammatory features in the sublining area in RA patients [26,27,28]. Thus, expression of CD90 can be used to designate proinflammatory FLS subsets. Furthermore, Fan Zhang et al. [28] classified CD90+ FLSs into three subsets, namely, CD90+CD34+ FLSs, CD90+HLA-DRAhigh FLSs, and CD90+DKK3+ FLSs. Among these subsets, CD90+HLA-DRAhigh FLSs were substantially expanded and correlated with cytokine and chemokine expression in RA. The pathogenic FLS subsets in RA and their characteristic markers are summarized in Fig. 1.

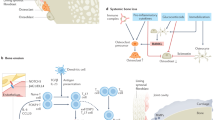
Pathogenic FLS subsets in RA. FLSs in the inflamed synovium can be anatomically distinguished into lining FLSs and sublining FLSs. FAPα, PDPN, and CDH-11 are expressed mainly on lining FLSs and also on sublining FLSs. The expression of FAPα, PDPN, and CDH-11 may resemble the pathogenic FLS phenotype, as targeting these markers can ameliorate arthritis in animal models of RA. Pathogenic FLSs in RA can be further subclassified into immune-interacting FLSs and bone-effector FLSs based on the expression of CD90 and CD55/PRG4, respectively. CD90 is expressed on sublining FLSs and can be used to designate proinflammatory FLS subsets that mediate inflammation persistence. Among CD90+ FLSs, a subset with high HLA-DRA expression that can secrete several proinflammatory cytokines and chemokines is substantially expanded in RA patients
Resolving FLS subsets in RA
FLSs play various physiological functions in the homeostatic state but transform from a friend to a foe in patients with RA [42], indicating that different FLS subsets are involved in active RA and remission/homeostatic states. In 2020, Stefano Alivernini et al. [31]. analyzed FLSs from patients with active RA and those in remission. FLSs expressing MMPs can be classified into a lining-layer FLS cluster and those expressing collagens and immune mediators can be classified into four sublining-layer clusters. Although the relative proportions of these clusters were similar in patients with active RA and those in remission, their transcriptomes differed. FLS clusters in RA patients in remission expressed more mediators related to tissue repair and the resolution of inflammation. Among these subsets, CD90+CXCL14+ cells in RA patients in remission expressed high levels of GAS6, which may contribute to the regulatory functions of lining-layer MerTKpos macrophages to promote the resolution of inflammation. To explore the profiles of normal FLSs and molecular networks controlling the transition from homeostatic to arthritic FLSs, in 2022, Marietta Armaka et al. [14]. performed a combined analysis of single-cell transcriptomes and epigenomes of FLSs derived from naïve and hTNFtg mice (mice that overexpress human TNF, a murine model for RA). FLSs play roles in chondrogenesis and osteogenesis, tissue repair, and immune surveillance in healthy synovium. The presence of arthritis was accompanied by reduction of homeostatic FLSs and the emergence of pathogenic FLS profiles marked by Dkk3 and Lrrc15 expression [14, 33]; these FLSs promote the inflammatory response and matrix catabolic processes. Moreover, a recent study demonstrated for the first time that FLSs have the ability to transform from proinflammatory to pro-resolving phenotypes (CD200+), which can reduce inflammation via interactions with ILC2 in patients with inflammatory arthritis [33]. These studies indicated the presence of resolving FLS subsets in the remission/homeostatic state that function in inflammation resolution instead of promotion; these subsets might be a new therapeutic tool to promote the resolution of inflammation and restore tissue homeostasis in patients with RA.
Strategies for targeting pathogenic FLS subsets in RA
Targeting cell surface markers on pathogenic FLSs
Direct depletion of pathogenic FLS subsets by targeting cell surface markers with antibodies, chimeric antigen receptor (CAR)-T cells or vaccines might be the most effective way to target pathogenic FLSs in RA. Among these FLS cell surface markers, CDH-11 first attracted attention as a promising target for RA treatment. However, a phase II trial of monoclonal antibodies targeting CDH-11 (RG6125) in RA patients was discontinued in 2018 due to a lack of efficacy [43]. Other markers, especially FAPα, are promising potential therapeutic targets for RA.
FAPα is a type II cell surface serine protease with dipeptidylpeptidase and endopeptidase activity [44] that is overexpressed in activated fibroblasts, such as those involved in cancer and fibrosis; the expression of FAPα in healthy tissues is scarce, making FAPα an attractive therapeutic target in disease [45]. Although a study in mice demonstrated that depletion of FAPα+ cells can result in cachexia and anemia and FAPα+ cells were found to reside in most tissues, including muscle and bone marrow [46], studies investigating the potential use of FAPα as a therapeutic target in diseases are ongoing. Depletion of FAPα-expressing cells by antibodies, FAP CAR-T cells and various FAP vaccines has been widely investigated in the treatment of cancer [45] and fibrosis [47, 48] and has shown safety and tolerability in phase I trials [49, 50]. Moreover, small-molecule inhibitors of FAPα (FAPIs) with high affinity and selectivity for FAPα provide new strategies to image FAPα-expressing tissues [51], as well as new treatment strategies based on linking traditional drugs with FAPα-targeted molecules [52,53,54]. In RA, FAPI labeled with gallium 68 (68Ga-FAPI) or aluminum-(18-F)-labeled 1,4,7-triazacyclononane-N, N’,N″-triacetic acid (18F-AIF-NOTA-FAPI) can be used to clearly reveal inflammatory joints and assess disease activity [55, 56], indicating that FAPα is an excellent candidate for RA therapy. Daphne N. Dorst [57] developed a treatment strategy for the selective destruction of FAPα+ cells by coupling an anti-FAP antibody with the photosensitizer IRDye700DX. This compound can accumulate in inflamed joints and induce local FAPα+ cell death, which moderately delayed the development of arthritis in CIA mice. A vaccine with the consensus FAPα mRNA encapsulated in a lipid nanoparticle (cFAP mRNA-LNP) prevented disease onset and arthritis development in a mouse model of RA [58]. Zinc ferrite nanoparticles (ZF-NPs) engineered to target FAPα+ FLSs significantly suppressed synovitis and protected against bone damage in a mouse model of RA [59]. These studies in mice further suggest that FAPα is a promising therapeutic target in RA. FAPα+ cell targeting studies in cancer may help us exploring new strategies for targeting FAPα+ FLSs in RA.
The potential use of other pathogenic FLS surface markers as therapeutic targets in RA has also been investigated in animal studies. PDPN is a mucin type-1 glycoprotein with a molecular weight of 40–43 kDa. Besides on FLS, PDPN is expressed in many tumors and normal cells, especially lymphatic epithelial cells and follicular DCs [60]. PDPN has been studied in cancer as a therapeutic target by using antibodies or antagonistic peptides [61, 62]. In RA, Christopher D Buckley et al. reported in 2018 that anti-PDPN antibodies efficiently protected mice with CIA from arthritis [63]. THY-1 (CD90) is a highly N-glycosylated, glycosylphosphatidylinositol (GPI)-anchored cell surface protein, first identified for the recognition of thymoma cells, and was found to be expressed on various types of cells, such as mesenchymal stem cells (MSCs) [64]. CD90 is associated with the proinflammatory phenotype of FLSs. An anti-CD90 antibody alleviated disease progression in CIA mice by inhibiting FLS proliferation, proinflammatory cytokine release, osteoclast differentiation and angiogenesis [65], suggesting that CD90 is a potential therapeutic target for RA. CD248 is a transmembrane glycoprotein that is expressed on FLSs in the sublining area in RA. A study in CD248-deficient mice demonstrated that CD248 contributes to leukocyte accumulation and synovial hyperplasia in inflammatory arthritis, indicating that CD248 is a potential therapeutic target in RA [66]. However, the function of CD248 as a therapeutic target in RA needs to be further investigated. Potential strategies for targeting pathogenic FLS cell surface markers in RA are summarized in Fig. 2.

CDH-11, FAPα, PDPN, CD90 and CD248 are relatively specific synovial FLS markers. Targeting pathogenic FLS surface markers with specific antibodies, inhibitors, vaccines or CART cell might be potential strategies for the treatment of RA
Targeting signaling pathways that drive pathogenic FLS subsets differentiation
FLSs exhibit high phenotypic plasticity, and cytokines are important factors that can drive the differentiation of FLSs toward specific pathogenic RA-FLS subsets [25]; thus, cytokines can be used to modulate the FLS phenotype. TNF-α and IL-1β have been shown to stimulate fibroblasts to produce proteolytic enzymes that destroy bone and cartilage [67], and TGF-β1 can inhibit the synthesis of metalloproteinases and thereby reduce joint damage [68]. TNF-α or IL-1β can stimulate RA-FLSs to upregulate PDPN expression [25], suggesting that blockade of TNF-α or IL-1β in RA patients can inhibit the differentiation of PDPN+ FLSs. By using paired single-cell RNA and ATAC sequencing, multiplexed imaging, and spatial transcriptomics, along with in vitro modeling of cell-extrinsic factor signaling, a recent study revealed that myeloid and T-cell-derived TNF-α, IFN-γ, and IL-1β were important drivers of pathogenic FLS subset heterogeneity in RA [69]. Shuyang Zhao et al. [70]. showed that NK-derived IFN-γ could induce the differentiation of the inflammatory HLA-DR+CD90+ FLS phenotype, which can induce CD69 expression on CD4+ T cells. JAK inhibition by upadacitinib can prevent HLA-DR induction. These data indicated that JAK1 inhibition could reduce the generation of HLA-DR+CD90+ FLSs. These results may provide new insights into the mechanisms underlying the effects of cytokines and JAK inhibitors in RA.
In 2020, Kevin Wei et al. [30]. investigated the upstream signaling pathway that drives the expansion of the proinflammatory CD90+ FLS subset. They identified the central role of endothelium-derived Notch ligands (DLL4/JAG2) in driving the expansion of CD90+ sublining FLS through inductive Notch3 signaling in RA. The Notch activation signature was more enriched in CD90high FLSs than in CD90low FLSs, and the Notch inhibitor DAPT blocked CD90high FLS differentiation. Genetic deletion or blockade of Notch3 signaling in mice relieved inflammation and protected joints in an arthritis model. LY411575 (which inhibits NOTCH-1 and the NOTCH-3 intracellular domain) suppressed inflammation and bone damage in CIA [71]. These results indicated that Notch3 is a potential therapeutic target for inhibiting the differentiation of proinflammatory FLSs in RA. Potential strategies for targeting pathogenic FLS subsets differentiation are summarized in Fig. 3.

Targeting signaling pathways that drive pathogenic FLS differentiation might be a potential strategy for the treatment of RA. FLSs exhibit high phenotypic plasticity, and different cytokines or cells can stimulate the differentiation of different FLS subsets. Targeting NOTCH3 signaling at FLSs might affect CD90 expression in FLSs. IFN-γ-mediated stimulation of JAK-STAT1 signaling might also block HLA-DR expression in CD90+FLSs, which has proinflammatory effects during RA pathogenesis
Restoring FLS homeostasis by promoting subsets with resolving phenotypes
Fibroblasts are complex, functional, heterogeneous cells with a wide range of effects ranging from immunosuppressive to proinflammatory effects as well as tissue repair and tissue damage effects. Thus, a delicate balance between these contradictory functions of fibroblasts may be essential for sustaining tissue homeostasis. Instead of the pathogenic phenotype of FLSs in active RA, resolving FLS clusters in RA patients in remission and immunosuppressive clusters in a homeostatic state have been demonstrated. Thus, restoring FLS homeostasis by promoting the immunosuppressive/pro-resolving and tissue repair phenotypes or switching expanded pathogenic subsets into protective subsets may be an ideal therapeutic strategy for the treatment of RA.
Consistent with the role of resolving CD200+ FLSs in mediating RA remission, a previous study also demonstrated that CD200-Fc, which can target proinflammatory cytokine expression in the joint without any obvious systemic immunosuppressive effects, is an effective therapeutic agent for CIA [72]. Umbilical cord-derived MSCs were reported to decrease CDH-11 expression in RA-FLSs, mainly by producing the anti-inflammatory cytokine IL-10 [73]. In several clinical trials of RA, such as a phase 1/2 trial (NCT03618784), promising results have been demonstrated following the treatment of joint inflammation with MSCs. Although the expanded CD34-CD90+ FLS subset in RA is considered to constitute a pathogenic phenotype, one study showed that the CD34+CD90+ FLS subset has high osteoblastic and chondrogenic potential in vitro [74]. Research on the functions of the CD34+CD90+ subgroup may lead to new treatment strategies for regenerating damaged bone/cartilage in arthritic joints. Skin fibroblasts in a homeostatic state can inhibit the proliferation of T lymphocytes. An intravenous injection of normal skin fibroblasts efficiently suppressed the severity of CIA-related inflammatory arthritis and delayed disease onset [75]. Similarly, our study showed that FLSs stimulated by IFN-γ can upregulate the expression of several inhibitory molecules, such as PD-L1 and galectin-9, on the cell membrane, which might be negative feedback mechanisms of inflammatory cytokines. FLS cell membranes with high expression of inhibitory molecules ameliorated inflammation and bone damage in CIA [76]. These data suggest that the introduction of fibroblasts with immunosuppressive/pro-resolving effects may help restore the balance of FLS subsets. However, although the driving factors of proinflammatory or tissue damaging FLS clusters in RA are beginning to be understood, the factors that induce resolving/immunosuppressive FLS subset differentiation have not been identified. Potential strategies for restoring FLS subset homeostasis are summarized in Fig. 4.

Restoring FLS homeostasis might be a potential strategy for the treatment of RA. FLS subsets in active RA patients are different from those in healthy individuals or patients in remission. Transforming proinflammatory and aggressive FLSs in RA patients into resolving or tissue repair FLSs via normal fibroblasts or mesenchymal stem cells might restore FLS homeostasis and alleviate RA
Potential FLS-targeting strategies implemented in studies of cancer
In addition to the pathogenic effects of FLSs in RA, fibroblasts are well known for their functions in immune suppression [77] and tissue repair and play important roles in the induction of immune tolerance, inflammation resolution [78] and wound healing [79], indicating that distinct fibroblast subsets can be found in different microenvironments. Studying on fibroblast heterogeneity in other microenvironments may help us understand the mechanisms underlying FLS subset dysregulation in RA and explore new treatment strategies.
In contrast to those in RA, fibroblasts in cancer (cancer-associated fibroblasts, CAFs) exert potent immune suppression effects. CD36+ CAFs were recently identified as a new CAF subset with immunosuppressive effects. CD36 mediates oxidized LDL uptake to promote MIF expression, which promotes immunosuppressive MDSC accumulation and accelerates cancer progression, and CD36 inhibitors enhance the treatment efficacy of immunotherapies [80]. These findings suggest that strategies for enhancing the effects of CD36 on FLSs might be a potential therapeutic approach to reduce inflammation in individuals with RA. Among the CAF subsets in cancer [81], antigen-presenting fibroblasts, which are characterized by high expression of MHCII molecules and CD74 and can present antigens to T cells [82], were also found to be expanded in RA (CD90+HLA-DRAhigh FLS) [70]; these results suggest potential new therapeutic strategies based on targeting CD74 on fibroblasts [83]. However, whether this antigen-presenting fibroblast subset can fully activate T cells or induce T-cell tolerance is still debated [84, 85]. An in-depth study on the function of antigen-presenting fibroblasts may help us better understand peripheral tolerance in RA and design methods to restore autoimmunity. Whether fibroblasts with similar phenotypes across different disease microenvironments share similar functional characteristics remains to be determined.
Conclusions and future perspectives
FLSs play a central role in RA pathogenesis by acting as both drivers and effectors. There has been increasing interest in FLSs as important therapeutic targets in RA. FLSs are quite heterogeneous and widely arranged in different microenvironments. The increase in our understanding of pathogenic FLS clusters specific to RA has provided us with promising novel therapeutic targets. The targeting of pathogenic FLSs subsets by specific cell surface markers or upstream driving pathways may succeed in treating “non-responders” to immunosuppressive therapies, and open new RA targeting treatment era with less adverse effects associated with traditional systemic immunosuppressive therapy including csDMARDs or b/tsDMARDs.
Although high-throughput technologies have revealed several RA-specific pathogenic FLS subsets, along with the newly identified resolving FLSs in RA patients in remission, the mechanisms driving the differentiation of pathogenic or resolving subsets have not been fully elucidated. An ideal strategy for terminating persistent inflammation in individuals with RA may be restoring the homeostasis of FLS subsets by switching the pathogenic FLS phenotype to a pro-resolving/immunosuppressive phenotype. Studies on the detailed mechanisms that drive pathogenic FLS subset differentiation, including local triggers and imprinting changes, as well as the identification of factors that drive resolving/immunosuppressive fibroblast differentiation in other microenvironments, such as cancer, may help us develop treatment strategies that can restore FLS subset homeostasis. Moreover, novel therapeutic strategies that target and deliver drugs to FLSs can facilitate the development of FLS-based treatments.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- RA:
-
Rheumatoid arthritis
- FLS:
-
Fibroblast-like synoviocytes
- DMARDs:
-
Disease-modifying anti-rheumatic drugs
- csDMARDs:
-
Conventional synthetic disease-modifying anti-rheumatic drugs
- b/ts DMARDs:
-
Biological/targeted disease-modifying anti-rheumatic drugs
- ECM:
-
Extracellular matrix
- MMP:
-
Matrix metalloproteinases
- PDGFRα:
-
Platelet-derived growth factor receptor α
- PDPN:
-
Podoplanin
- αSMA:
-
α-smooth muscle actin
- FAPα:
-
Fibroblast activation protein α
- UDPGD:
-
Uridine diphosphoglucose dehydrogenase
- PRG4:
-
Proteoglycan 4
- CIA:
-
Collagen induced arthritis
- CAF:
-
Cancer associated fibroblast
References
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38.
Tobón GJ, Youinou P, Saraux A. The environment, geo-epidemiology, and autoimmune disease: rheumatoid arthritis. Autoimmun Rev. 2010;9(5):A288-292.
Buch MH. Defining refractory rheumatoid arthritis. Ann Rheum Dis. 2018;77(7):966–9.
Nagy G, Roodenrijs NMT, Welsing PM, Kedves M, Hamar A, van der Goes MC, Kent A, Bakkers M, Blaas E, Senolt L, et al. EULAR definition of difficult-to-treat rheumatoid arthritis. Ann Rheum Dis. 2021;80(1):31–5.
Rivellese F, Surace AEA, Goldmann K, Sciacca E, Cubuk C, Giorli G, John CR, Nerviani A, Fossati-Jimack L, Thorborn G, et al. Rituximab versus tocilizumab in rheumatoid arthritis: synovial biopsy-based biomarker analysis of the phase 4 R4RA randomized trial. Nat Med. 2022;28(6):1256–68.
Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16(6):316–33.
Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–55.
Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. 2008;223:252–70.
Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33.
Filer A. The fibroblast as a therapeutic target in rheumatoid arthritis. Curr Opin Pharmacol. 2013;13(3):413–9.
Nemeth T, Nagy G, Pap T. Synovial fibroblasts as potential drug targets in rheumatoid arthritis, where do we stand and where shall we go? Ann Rheum Dis. 2022; 81(8):1055–64.
Tsaltskan V, Firestein GS. Targeting fibroblast-like synoviocytes in rheumatoid arthritis. Curr Opin Pharmacol. 2022;67:102304.
Yoshida M, Sai S, Marumo K, Tanaka T, Itano N, Kimata K, Fujii K. Expression analysis of three isoforms of hyaluronan synthase and hyaluronidase in the synovium of knees in osteoarthritis and rheumatoid arthritis by quantitative real-time reverse transcriptase polymerase chain reaction. Arthritis Res Ther. 2004;6(6):R514–520.
Armaka M, Konstantopoulos D, Tzaferis C, Lavigne MD, Sakkou M, Liakos A, Sfikakis PP, Dimopoulos MA, Fousteri M, Kollias G. Single-cell multimodal analysis identifies common regulatory programs in synovial fibroblasts of rheumatoid arthritis patients and modeled TNF-driven arthritis. Genome Med. 2022;14(1):78.
Shigeyama Y, Pap T, Kunzler P, Simmen BR, Gay RE, Gay S. Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum. 2000;43(11):2523–30.
Crow MK. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res Ther. 2010;12(Suppl 1):S5.
Guerne PA, Zuraw BL, Vaughan JH, Carson DA, Lotz M. Synovium as a source of interleukin 6 in vitro. Contribution to local and systemic manifestations of arthritis. J Clin Invest. 1989;83(2):585–92.
Palmer CD, Mutch BE, Page TH, Horwood NJ, Foxwell BM. Bmx regulates LPS-induced IL-6 and VEGF production via mRNA stability in rheumatoid synovial fibroblasts. Biochem Biophys Res Commun. 2008;370(4):599–602.
Filer A, Parsonage G, Smith E, Osborne C, Thomas AM, Curnow SJ, Rainger GE, Raza K, Nash GB, Lord J, et al. Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils. Arthritis Rheum. 2006;54(7):2096–108.
Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, Liu Y, Bicker KL, Wahamaa H, Hoffmann V, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. 2017;2(10):eaag3358.
Tran CN, Davis MJ, Tesmer LA, Endres JL, Motyl CD, Smuda C, Somers EC, Chung KC, Urquhart AG, Lundy SK, et al. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. 2007;56(5):1497–506.
Bombardieri M, Kam NW, Brentano F, Choi K, Filer A, Kyburz D, McInnes IB, Gay S, Buckley C, Pitzalis C. A BAFF/APRIL-dependent TLR3-stimulated pathway enhances the capacity of rheumatoid synovial fibroblasts to induce AID expression and ig class-switching in B cells. Ann Rheum Dis. 2011;70(10):1857–65.
Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest. 2001;107(3):305–15.
Reparon-Schuijt CC, van Esch WJ, van Kooten C, Rozier BC, Levarht EW, Breedveld FC, Verweij CL. Regulation of synovial B cell survival in rheumatoid arthritis by vascular cell adhesion molecule 1 (CD106) expressed on fibroblast-like synoviocytes. Arthritis Rheum. 2000;43(5):1115–21.
Croft AP, Naylor AJ, Marshall JL, Hardie DL, Zimmermann B, Turner J, Desanti G, Adams H, Yemm AI, Muller-Ladner U, et al. Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage. Arthritis Res Ther. 2016;18(1):270.
Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, Nguyen HN, Noss EH, Turner JD, Earp BE, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun. 2018;9(1):789.
Stephenson W, Donlin LT, Butler A, Rozo C, Bracken B, Rashidfarrokhi A, Goodman SM, Ivashkiv LB, Bykerk VP, Orange DE, et al. Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nat Commun. 2018;9(1):791.
Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, Goodman SM, Tabechian D, Hughes LB, Salomon-Escoto K, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. 2019;20(7):928–42.
Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, Savary L, Wehmeyer C, Naylor AJ, Kemble S, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570(7760):246–51.
Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, Croft AP, Watts J, Blazar PE, Lange JK, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. 2020;582(7811):259–64.
Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, Petricca L, Di Mario C, Bui L, Perniola S, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med. 2020;26(8):1295–306.
Korsunsky I, Wei K, Pohin M, Kim EY, Barone F, Major T, Taylor E, Ravindran R, Kemble S, Watts GFM, et al. Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Med. 2022;3(7):481–e518414.
Rauber S, Mohammadian H, Schmidkonz C, Atzinger A, Soare A, Treutlein C, et al. CD200(+) fibroblasts form a pro-resolving mesenchymal network in arthritis. Nat Immunol. 2024;25(4):682–92.
Micheroli R, Elhai M, Edalat S, Frank-Bertoncelj M, Burki K, Ciurea A, MacDonald L, Kurowska-Stolarska M, Lewis MJ, Goldmann K, et al. Role of synovial fibroblast subsets across synovial pathotypes in rheumatoid arthritis: a deconvolution analysis. RMD Open. 2022;8(1):e001949.
Del Rey MJ, Fare R, Izquierdo E, Usategui A, Rodriguez-Fernandez JL, Suarez-Fueyo A, Canete JD, Pablos JL. Clinicopathological correlations of podoplanin (gp38) expression in rheumatoid synovium and its potential contribution to fibroblast platelet crosstalk. PLoS One. 2014;9(6):e99607.
Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M, Brenner MB. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315(5814):1006–10.
Valencia X, Higgins JM, Kiener HP, Lee DM, Podrebarac TA, Dascher CC, Watts GF, Mizoguchi E, Simmons B, Patel DD, et al. Cadherin-11 provides specific cellular adhesion between fibroblast-like synoviocytes. J Exp Med. 2004;200(12):1673–9.
Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, Leon L, Lee SY, Lee DM, Brenner MB. Cadherin-11 regulates fibroblast inflammation. Proc Natl Acad Sci U S A. 2011;108(20):8402–7.
Ekwall AK, Eisler T, Anderberg C, Jin C, Karlsson N, Brisslert M, Bokarewa MI. The tumour-associated glycoprotein podoplanin is expressed in fibroblast-like synoviocytes of the hyperplastic synovial lining layer in rheumatoid arthritis. Arthritis Res Ther. 2011;13(2):R40.
Orange DE, Yao V, Sawicka K, Fak J, Frank MO, Parveen S, Blachere NE, Hale C, Zhang F, Raychaudhuri S, et al. RNA identification of PRIME cells Predicting Rheumatoid Arthritis flares. N Engl J Med. 2020;383(3):218–28.
Wang Z, Wang J, Lan T, Zhang L, Yan Z, Zhang N, Xu Y, Tao Q. Role and mechanism of fibroblast-activated protein-alpha expression on the surface of fibroblast-like synoviocytes in rheumatoid arthritis. Front Immunol. 2023;14:1135384.
Mousavi MJ, Karami J, Aslani S, Tahmasebi MN, Vaziri AS, Jamshidi A, Farhadi E, Mahmoudi M. Transformation of fibroblast-like synoviocytes in rheumatoid arthritis; from a friend to foe. Auto Immun Highlights. 2021;12(1):3.
Finch R, Sostelly A, Sue-Ling K, Blaeuer A, Duchateau-Nguyen G, Ukarma L, Petry C, Ravva P, Villiger P, Junker U. Op0224 results of a phase 2 study of Rg6125, an anti-cadherin-11 monoclonal antibody, in rheumatoid arthritis patients with an inadequate response to Anti-tnfalpha Therapy. Oral Presentations. 2019;189:181–9.
Aertgeerts K, Levin I, Shi L, Snell GP, Jennings A, Prasad GS, Zhang Y, Kraus ML, Salakian S, Sridhar V, et al. Structural and kinetic analysis of the substrate specificity of human fibroblast activation protein alpha. J Biol Chem. 2005;280(20):19441–4.
Shahvali S, Rahiman N, Jaafari MR, Arabi L. Targeting fibroblast activation protein (FAP): advances in CAR-T cell, antibody, and vaccine in cancer immunotherapy. Drug Deliv Transl Res. 2023;13(7):2041–56.
Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, Espeli M, Kraman M, McKenna B, Wells RJ, et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. 2013;210(6):1137–51.
Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573(7774):430–3.
Rurik JG, Tombacz I, Yadegari A, Mendez Fernandez PO, Shewale SV, Li L, Kimura T, Soliman OY, Papp TE, Tam YK, et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375(6576):91–6.
Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26(1):44–8.
Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, Divgi CR, Hanson LH, Mitchell P, Gansen DN, et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9(5):1639–47.
Mori Y, Dendl K, Cardinale J, Kratochwil C, Giesel FL, Haberkorn U. FAPI PET: fibroblast activation protein inhibitor use in oncologic and nononcologic disease. Radiology. 2023;306(2):e220749.
Millul J, Bassi G, Mock J, Elsayed A, Pellegrino C, Zana A, Dakhel Plaza S, Nadal L, Gloger A, Schmidt E, et al. An ultra-high-affinity small organic ligand of fibroblast activation protein for tumor-targeting applications. Proc Natl Acad Sci U S A. 2021;118(16):e2101852118.
Zana A, Galbiati A, Gilardoni E, Bocci M, Millul J, Sturm T, Stucchi R, Elsayed A, Nadal L, Cirillo M, et al. Fibroblast activation protein triggers release of drug payload from non-internalizing small Molecule Drug Conjugates in Solid tumors. Clin Cancer Res. 2022;28(24):5440–54.
Ruopp M, Zhu S, Worschech R, Haas D, Maschauer S, Prante O, Meinel L, Luhmann T. Bioconjugation of a fibroblast activation protein targeted Interleukin-4. ACS Biomater Sci Eng. 2023;9(10):5580–8.
Luo Y, Pan Q, Zhou Z, Li M, Wei Y, Jiang X, Yang H, Li F. (68)Ga-FAPI PET/CT for rheumatoid arthritis: a prospective study. Radiology. 2023;307(3):e222052.
Ge L, Fu Z, Wei Y, Shi D, Geng Y, Fan H, Zhang R, Zhang Y, Li S, Wang S, et al. Preclinical evaluation and pilot clinical study of [(18)F]AlF-NOTA-FAPI-04 for PET imaging of rheumatoid arthritis. Eur J Nucl Med Mol Imaging. 2022;49(12):4025–36.
Dorst DN, Rijpkema M, Boss M, Walgreen B, Helsen MMA, Bos DL, Brom M, Klein C, Laverman P, van der Kraan PM, et al. Targeted photodynamic therapy selectively kills activated fibroblasts in experimental arthritis. Rheumatology (Oxford). 2020;59(12):3952–60.
Zhang X, Jozic A, Song P, Xu Q, Shi X, Wang H, Bishop L, Struthers HM, Rutledge J, Chen S, et al. mRNA vaccine against fibroblast activation protein ameliorates murine models of inflammatory arthritis. Rheumatol Immunol Res. 2023;4(2):90–7.
Qi W, Jin L, Wu C, Liao H, Zhang M, Zhu Z, Han W, Chen Q, Ding C. Treatment with FAP-targeted zinc ferrite nanoparticles for rheumatoid arthritis by inducing endoplasmic reticulum stress and mitochondrial damage. Mater Today Bio. 2023;21:100702.
Krishnan H, Rayes J, Miyashita T, Ishii G, Retzbach EP, Sheehan SA, Takemoto A, Chang YW, Yoneda K, Asai J, et al. Podoplanin: an emerging cancer biomarker and therapeutic target. Cancer Sci. 2018;109(5):1292–9.
Feng C, Yu A, Wang Z, Wang K, Chen J, Wu Y, Deng T, Chen H, Hou Y, Ma S, et al. A novel PDPN antagonist peptide CY12-RP2 inhibits melanoma growth via Wnt/beta-catenin and modulates the immune cells. J Exp Clin Cancer Res. 2024;43(1):9.
Takemoto A, Takagi S, Ukaji T, Gyobu N, Kakino M, Takami M, Kobayashi A, Lebel M, Kawaguchi T, Sugawara M, et al. Targeting Podoplanin for the Treatment of Osteosarcoma. Clin Cancer Res. 2022;28(12):2633–45.
GE Desanti AS, Naylor AJ, Kemble S, Falconer J, Wehmeyer C, Marshall JL, Nakamura K, Goodall M, Navarro-Núñez L, Watson SP, CD Buckley. Podoplanin (GP38), a marker of synovial inflammation, is an excellent therapeutic target in mouse collagen-induced arthritis. Ann Rheum Dis. 2018;77(Suppl 1):A1-77.
Kumar A, Bhanja A, Bhattacharyya J, Jaganathan BG. Multiple roles of CD90 in cancer. Tumour Biol. 2016;37(9):11611–22.
Hu X, Li M, Zhang Y, Sang K, Zhang Y, Li W, Liu B, Wan L, Du B, Qian J, et al. An innovative immunotherapeutic strategy for rheumatoid arthritis: selectively suppressing angiogenesis and osteoclast differentiation by fully human antibody targeting thymocyte antigen-1. Exp Cell Res. 2023;424(1):113490.
Maia M, de Vriese A, Janssens T, Moons M, van Landuyt K, Tavernier J, Lories RJ, Conway EM. CD248 and its cytoplasmic domain: a therapeutic target for arthritis. Arthritis Rheum. 2010;62(12):3595–606.
Yoshitomi H. Regulation of Immune responses and chronic inflammation by Fibroblast-Like Synoviocytes. Front Immunol. 2019;10:1395.
Muller-Ladner U, Gay S. MMPs and rheumatoid synovial fibroblasts: siamese twins in joint destruction? Ann Rheum Dis. 2002;61(11):957–9.
Smith MH, Gao VR, Periyakoil PK, Kochen A, DiCarlo EF, Goodman SM, Norman TM, Donlin LT, Leslie CS, Rudensky AY. Drivers of heterogeneity in synovial fibroblasts in rheumatoid arthritis. Nat Immunol. 2023;24(7):1200–10.
Zhao S, Grieshaber-Bouyer R, Rao DA, Kolb P, Chen H, Andreeva I, Tretter T, Lorenz HM, Watzl C, Wabnitz G, et al. Effect of JAK inhibition on the induction of Proinflammatory HLA-DR + CD90 + rheumatoid arthritis synovial fibroblasts by Interferon-gamma. Arthritis Rheumatol. 2022;74(3):441–52.
Chen J, Cheng W, Li J, Wang Y, Chen J, Shen X, Su A, Gan D, Ke L, Liu G, et al. Notch-1 and Notch-3 mediate Hypoxia-Induced activation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheumatol. 2021;73(10):1810–9.
Simelyte E, Criado G, Essex D, Uger RA, Feldmann M, Williams RO. CD200-Fc, a novel antiarthritic biologic agent that targets proinflammatory cytokine expression in the joints of mice with collagen-induced arthritis. Arthritis Rheum. 2008;58(4):1038–43.
Zhao C, Zhang L, Kong W, Liang J, Xu X, Wu H, Feng X, Hua B, Wang H, Sun L. Umbilical cord-derived mesenchymal stem cells inhibit cadherin-11 expression by fibroblast-like synoviocytes in rheumatoid arthritis. J Immunol Res. 2015;2015:137695.
Noda S, Hosoya T, Komiya Y, Tagawa Y, Endo K, Komori K, Koga H, Takahara Y, Sugimoto K, Sekiya I, et al. CD34(+)THY1(+) synovial fibroblast subset in arthritic joints has high osteoblastic and chondrogenic potentials in vitro. Arthritis Res Ther. 2022;24(1):45.
Bouffi C, Bony C, Jorgensen C, Noel D. Skin fibroblasts are potent suppressors of inflammation in experimental arthritis. Ann Rheum Dis. 2011;70(9):1671–6.
Liu Y, Rao P, Qian H, Shi Y, Chen S, Lan J, Mu D, Chen R, Zhang X, Deng C, et al. Regulatory Fibroblast-Like synoviocytes cell membrane coated nanoparticles: a Novel targeted therapy for rheumatoid arthritis. Adv Sci (Weinh). 2023;10(4):e2204998.
Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, Collier AR, Turley SJ. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol. 2011;12(11):1096–104.
Nitta T, Tsutsumi M, Nitta S, Muro R, Suzuki EC, Nakano K, Tomofuji Y, Sawa S, Okamura T, Penninger JM, et al. Fibroblasts as a source of self-antigens for central immune tolerance. Nat Immunol. 2020;21(10):1172–80.
Talbott HE, Mascharak S, Griffin M, Wan DC, Longaker MT. Wound healing, fibroblast heterogeneity, and fibrosis. Cell Stem Cell. 2022;29(8):1161–80.
Zhu GQ, Tang Z, Huang R, Qu WF, Fang Y, Yang R, Tao CY, Gao J, Wu XL, Sun HX, et al. CD36(+) cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023;9(1):25.
Lavie D, Ben-Shmuel A, Erez N, Scherz-Shouval R. Cancer-associated fibroblasts in the single-cell era. Nat Cancer. 2022;3(7):793–807.
Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals Antigen-Presenting Cancer-Associated fibroblasts. Cancer Discov. 2019;9(8):1102–23.
Hong WC, Lee DE, Kang HW, Kim MJ, Kim M, Kim JH, Fang S, Kim HJ, Park JS. CD74 promotes a pro-inflammatory tumor microenvironment by inducing S100A8 and S100A9 secretion in pancreatic cancer. Int J Mol Sci. 2023;24(16):12993.
Dart A. Presenting fibroblasts. Nat Rev Cancer. 2022;22(4):193.
Kerdidani D, Aerakis E, Verrou KM, Angelidis I, Douka K, Maniou MA, Stamoulis P, Goudevenou K, Prados A, Tzaferis C, et al. Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J Exp Med. 2022;219(2):e20210815.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 81971496, 82171779, and 81971536), the Science Foundation of Fujian Province (grant numbers 2023J06055, 2021CXB021, 2021J05292, 2020J05309 and 2020J011250), and Scientific and Technological Projects of Xiamen City (grant numbers 3502Z20209004).
Author information
Authors and Affiliations
Contributions
HY. Q contributed to the literature review, initial draft and manuscript editing. CQ. D contributed to figure preparation and manuscript editing. SJ. C contributed to the literature review and manuscript editing. XW. Z contributed to the literature review and figure preparation. Y.H. contributed to the summary of the table in the manuscript and manuscript editing. JY. L contributed to manuscript editing. ADW contributed to the figure preparation. GX. S contributed to the literature review and drafted and edited the manuscript. Y.L. contributed to the literature review and drafted and edited the manuscript. All the authors provided final approval.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agreed to the publication of this manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qian, H., Deng, C., Chen, S. et al. Targeting pathogenic fibroblast-like synoviocyte subsets in rheumatoid arthritis. Arthritis Res Ther 26, 103 (2024). https://doi.org/10.1186/s13075-024-03343-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-024-03343-4